A Role for the Biological Clock in Liver Cancer

 ,
,  ,
, 
 ,
,  and
and 
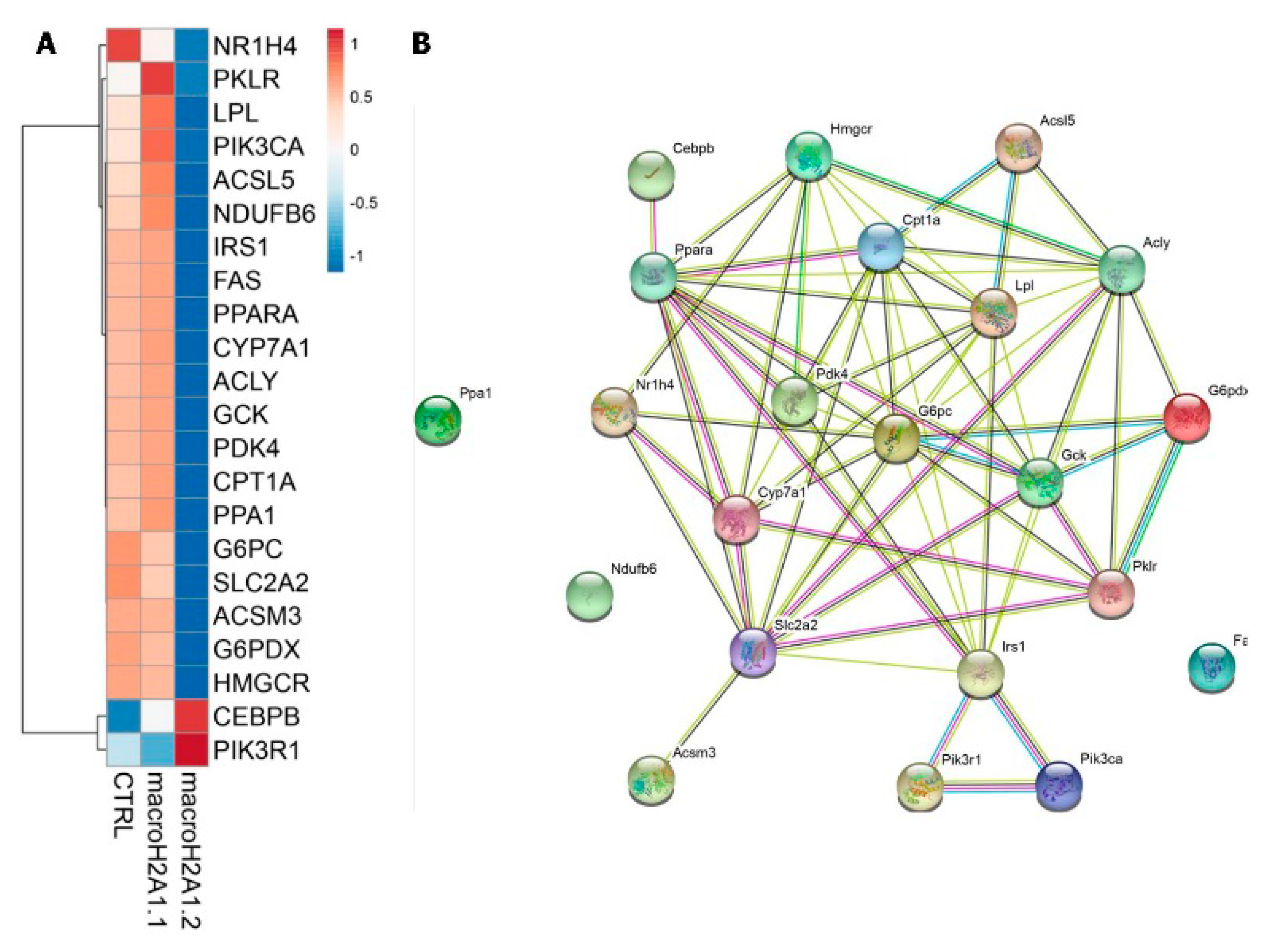{kind=link}
Abstract
:1. Introduction
2. The Circadian Clock Circuitry and the Molecular Mechanisms of Hepatocellular Carcinogenesis
3. The Biological Clock and the Oncogenic Pathways Involved in Hepatocarcinogenesis
3.1. WNT/β-Catenin Signaling Pathway
3.2. Hedgehog Signaling Pathway
3.3. MAP Kinase Signaling Pathways
3.4. PI3K/AKT/mTOR Signaling Pathway
3.5. Epigenetics
4. The Biological Clock and Systemic Therapy in Hepatocellular Carcinoma
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies from 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, U. Timing to perfection: The biology of central and peripheral circadian clocks. Neuron 2012, 74, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Lowrey, P.L.; Takahashi, J.S. Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet. 2011, 74, 175–230. [Google Scholar] [PubMed]
- Anderson, G.; Beischlag, T.V.; Vinciguerra, M.; Mazzoccoli, G. The circadian clock circuitry and the AHR signaling pathway in physiology and pathology. Biochem. Pharmacol. 2013, 85, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Laukkanen, M.O.; Vinciguerra, M.; Colangelo, T.; Colantuoni, V. A Timeless Link between Circadian Patterns and Disease. Trends Mol. Med. 2016, 22, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, J.A.; Albrecht, U. REV-ERB-erating nuclear receptor functions in circadian metabolism and physiology. Cell Res. 2012, 22, 1319–1321. [Google Scholar] [CrossRef] [PubMed]
- Partch, C.L.; Green, C.B.; Takahashi, J.S. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014, 24, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Koike, N.; Yoo, S.H.; Huang, H.C.; Kumar, V.; Lee, C.; Kim, T.K.; Takahashi, J.S. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 2012, 338, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Zocchi, L.; Kinoshita, C.; Borrelli, E.; Sassone-Corsi, P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS ONE 2010, 5, e8561. [Google Scholar] [CrossRef] [PubMed]
- Cardone, L.; Hirayama, J.; Giordano, F.; Tamaru, T.; Palvimo, J.J.; Sassone-Corsi, P. Circadian clock control by SUMOylation of BMAL1. Science 2005, 309, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, Y.; Lee, M.J.; Park, E.; Kang, S.H.; Chung, C.H.; Lee, K.H.; Kim, K. Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the CLOCK/BMAL1 complex. Mol. Cell. Biol. 2008, 28, 6056–6065. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Bozek, K.; Relogio, A.; Kielbasa, S.M.; Heine, M.; Dame, C.; Kramer, A.; Herzel, H. Regulation of clock-controlled genes in mammals. PLoS ONE 2009, 4, e4882. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.E.; DiTacchio, L.; Hayes, K.R.; Vollmers, C.; Pulivarthy, S.; Baggs, J.E.; Panda, S.; Hogenesch, J.B. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009, 5, e1000442. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 302, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.; Sassone-Corsi, P. Riding tandem: Circadian clocks and the cell cycle. Cell 2007, 129, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Filipski, E.; King, V.M.; Li, X.; Granda, T.G.; Mormont, M.C.; Liu, X.; Claustrat, B.; Hastings, M.H.; Levi, F. Host circadian clock as a control point in tumor progression. J. Natl. Cancer Inst. 2002, 94, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Santoni-Rugiu, E.; Jensen, M.R.; Thorgeirsson, S.S. Disruption of the pRb/E2F pathway and inhibition of apoptosis are major oncogenic events in liver constitutively expressing c-myc and transforming growth factor alpha. Cancer Res. 1998, 58, 123–134. [Google Scholar] [PubMed]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D.F.; Asara, G.; Casabona, D.; Frau, M.; Seddaiu, M.A.; Feo, F. Cell cycle deregulation in liver lesions of rats with and without genetic predisposition to hepatocarcinogenesis. Hepatology 2002, 35, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, L.E. Cell cycle regulation and hepatocarcinogenesis. Cancer Biol. Ther. 2004, 3, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Filipski, E.; Subramanian, P.; Carriere, J.; Guettier, C.; Barbason, H.; Levi, F. Circadian disruption accelerates liver carcinogenesis in mice. Mutat. Res. 2009, 680, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Fleet, T.; Stashi, E.; Zhu, B.; Rajapakshe, K.; Marcelo, K.L.; Kettner, N.M.; Gorman, B.K.; Coarfa, C.; Fu, L.; O'Malley, B.W.; et al. Genetic and Environmental Models of Circadian Disruption Link SRC-2 Function to Hepatic Pathology. J. Biol. Rhythm. 2016, 31, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Chang, J.H.; Yeh, K.T.; Yang, M.Y.; Liu, T.C.; Lin, S.F.; Su, W.W.; Chang, J.G. Disturbance of circadian gene expression in hepatocellular carcinoma. Mol. Carcinog. 2008, 47, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Yu, C.; Jiang, J.X.; Liu, L.P.; Fang, X.; Wu, C. Hepatitis B virus X protein disrupts the balance of the expression of circadian rhythm genes in hepatocellular carcinoma. Oncol. Lett. 2014, 8, 2715–2720. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Yang, S.L.; Fang, X.; Jiang, J.X.; Sun, C.Y.; Huang, T. Hypoxia disrupts the expression levels of circadian rhythm genes in hepatocellular carcinoma. Mol. Med. Rep. 2015, 11, 4002–4008. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Miele, L.; Oben, J.; Grieco, A.; Vinciguerra, M. Biology, Epidemiology, Clinical Aspects of Hepatocellular Carcinoma and the Role of Sorafenib. Curr. Drug Targ. 2016, 17, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; De Cosmo, S.; Mazza, T. The Biological Clock: A Pivotal Hub in Non-alcoholic Fatty Liver Disease Pathogenesis. Front. Physiol. 2018, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Rey, G.; Cesbron, F.; Rougemont, J.; Reinke, H.; Brunner, M.; Naef, F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011, 9, e1000595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Chatterjee, S.; Li, L.; Kim, J.M.; Lee, J.; Yechoor, V.K.; Minze, L.J.; Hsueh, W.; Ma, K. The clock gene, brain and muscle Arnt-like 1, regulates adipogenesis via Wnt signaling pathway. FASEB J. 2012, 26, 3453–3463. [Google Scholar] [CrossRef] [PubMed]
- Boucsein, A.; Benzler, J.; Hempp, C.; Stohr, S.; Helfer, G.; Tups, A. Photoperiodic and Diurnal Regulation of WNT Signaling in the Arcuate Nucleus of the Female Djungarian Hamster, Phodopus sungorus. Endocrinology 2016, 157, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsu-Ura, T.; Moore, S.R.; Hong, C.I. WNT Takes Two to Tango: Molecular Links between the Circadian Clock and the Cell Cycle in Adult Stem Cells. J. Biol. Rhythm. 2018, 33, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotak, M.; Sumova, A.; Pacha, J. Cross-talk between the circadian clock and the cell cycle in cancer. Ann. Med. 2014, 46, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Janich, P.; Pascual, G.; Merlos-Suarez, A.; Batlle, E.; Ripperger, J.; Albrecht, U.; Cheng, H.Y.; Obrietan, K.; Di Croce, L.; Benitah, S.A. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature 2011, 480, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Wang, Y.; Xie, J. The Hedgehog pathway: Role in cell differentiation, polarity and proliferation. Arch. Toxicol. 2015, 89, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicklick, J.K.; Li, Y.X.; Jayaraman, A.; Kannangai, R.; Qi, Y.; Vivekanandan, P.; Ludlow, J.W.; Owzar, K.; Chen, W.; Torbenson, M.S.; et al. Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis 2006, 27, 748–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marbach-Breitruck, E.; Matz-Soja, M.; Abraham, U.; Schmidt-Heck, W.; Sales, S.; Rennert, C.; Kern, M.; Aleithe, S.; Spormann, L.; Thiel, C.; et al. Tick-tock hedgehog-mutual crosstalk with liver circadian clock promotes liver steatosis. J. Hepatol. 2019, 70, 1192–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoccoli, G.; Keshavarzian, A.; Vinciguerra, M. Hedgehog signaling keeps liver clock in check. J. Hepatol. 2019, 70, 1054–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- De Paula, R.M.; Lamb, T.M.; Bennett, L.; Bell-Pedersen, D. A connection between MAPK pathways and circadian clocks. Cell Cycle 2008, 7, 2630–2634. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.S.; Bell-Pedersen, D. Diverse roles for MAPK signaling in circadian clocks. Adv. Genet. 2013, 84, 1–39. [Google Scholar] [PubMed] [Green Version]
- Bennett, L.D.; Beremand, P.; Thomas, T.L.; Bell-Pedersen, D. Circadian activation of the mitogen-activated protein kinase MAK-1 facilitates rhythms in clock-controlled genes in Neurospora crassa. Eukaryot. Cell 2013, 12, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caster, S.Z.; Castillo, K.; Sachs, M.S.; Bell-Pedersen, D. Circadian clock regulation of mRNA translation through eukaryotic elongation factor eEF-2. Proc. Natl. Acad. Sci. USA 2016, 113, 9605–9610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T. Mammalian target of rapamycin: A signaling kinase for every aspect of cellular life. Method. Mol. Biol. 2012, 821, 1–14. [Google Scholar]
- Cao, R. mTOR Signaling, Translational Control, and the Circadian Clock. Front. Genet. 2018, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Dang, F.; Li, P.; Wang, P.; Xu, Q.; Liu, Z.; Li, Y.; Wu, Y.; Chen, Y.; Liu, Y. The Circadian Protein Period2 Suppresses mTORC1 Activity via Recruiting Tsc1 to mTORC1 Complex. Cell Metab. 2019, 29, 653–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giallongo, S.; Lo Re, O.; Vinciguerra, M. Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers. Cancers 2019, 11, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F. Dysregulated Epigenetic Modifications in the Pathogenesis of NAFLD-HCC. Adv. Exp. Med. Biol. 2018, 1061, 79–93. [Google Scholar]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Deans, J.R.; Kwok, C.; Patel, P.; Fu, L.; Berdeaux, R.; Sun, K.; Kolonin, M.G.; et al. Incompatibility of the circadian protein BMAL1 and HNF4alpha in hepatocellular carcinoma. Nat. Commun. 2018, 9, 4349. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Vinciguerra, M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA Hypomethylation and Histone Variant macroH2A1 Synergistically Attenuate Chemotherapy-Induced Senescence to Promote Hepatocellular Carcinoma Progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Sceusi, E.L.; Loose, D.S.; Wray, C.J. Clinical implications of DNA methylation in hepatocellular carcinoma. HPB 2011, 13, 369–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jueliger, S.; Lyons, J.; Cannito, S.; Pata, I.; Pata, P.; Shkolnaya, M.; Lo Re, O.; Peyrou, M.; Villarroya, F.; Pazienza, V.; et al. Efficacy and epigenetic interactions of novel DNA hypomethylating agent guadecitabine (SGI-110) in preclinical models of hepatocellular carcinoma. Epigenetics 2016, 11, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; Leroy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Janich, P.; Cantarino, N.; Noguera, M.; Valero, V.; Musulen, E.; Douet, J.; Posavec, M.; Martin-Caballero, J.; Sumoy, L.; et al. MacroH2A1 regulates the balance between self-renewal and differentiation commitment in embryonic and adult stem cells. Mol. Cell. Biol. 2012, 32, 1442–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Rocha, S.W.; Pata, I.; Valcikova, B.; Uldrijan, S.; et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Douet, J.; Buschbeck, M.; Fusilli, C.; Pazienza, V.; Panebianco, C.; Castracani, C.C.; Mazza, T.; Li Volti, G.; Vinciguerra, M. Histone variant macroH2A1 rewires carbohydrate and lipid metabolism of hepatocellular carcinoma cells towards cancer stem cells. Epigenetics 2018, 13, 829–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, G.; Valekunja, U.K.; Feeney, K.A.; Wulund, L.; Milev, N.B.; Stangherlin, A.; Ansel-Bollepalli, L.; Velagapudi, V.; O'Neill, J.S.; Reddy, A.B. The Pentose Phosphate Pathway Regulates the Circadian Clock. Cell Metab. 2016, 24, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Martelot, G.; Claudel, T.; Gatfield, D.; Schaad, O.; Kornmann, B.; Lo Sasso, G.; Moschetta, A.; Schibler, U. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol. 2009, 7, e1000181. [Google Scholar]
- Zheng, Z.; Kim, H.; Qiu, Y.; Chen, X.; Mendez, R.; Dandekar, A.; Zhang, X.; Zhang, C.; Liu, A.C.; Yin, L.; et al. CREBH Couples Circadian Clock With Hepatic Lipid Metabolism. Diabetes 2016, 65, 3369–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghesan, M.; Mazzoccoli, G.; Sheedfar, F.; Oben, J.; Pazienza, V.; Vinciguerra, M. Histone variants and lipid metabolism. Biochem. Soc. Trans. 2014, 42, 1409–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedfar, F.; Vermeer, M.; Pazienza, V.; Villarroya, J.; Rappa, F.; Cappello, F.; Mazzoccoli, G.; Villarroya, F.; van der Molen, H.; Hofker, M.H.; et al. Genetic ablation of macrohistone H2A1 leads to increased leanness, glucose tolerance and energy expenditure in mice fed a high-fat diet. Int. J. Obes. 2015, 39, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazienza, V.; Borghesan, M.; Mazza, T.; Sheedfar, F.; Panebianco, C.; Williams, R.; Mazzoccoli, G.; Andriulli, A.; Nakanishi, T.; Vinciguerra, M. SIRT1-metabolite binding histone macroH2A1.1 protects hepatocytes against lipid accumulation. Aging 2014, 6, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Dang, Y.; Matsu-Ura, T.; He, Y.; He, Q.; Hong, C.I.; Liu, Y. DNA Replication Is Required for Circadian Clock Function by Regulating Rhythmic Nucleosome Composition. Mol. Cell 2017, 67, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Wu, B.; Delahaye, F.; Singer, R.H.; Greally, J.M. Retargeting of macroH2A following mitosis to cytogenetic-scale heterochromatic domains. J. Cell Biol. 2019, 218, 1810–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzily-Rokni, M.; Friedman, N.; Ron-Bigger, S.; Isaac, S.; Michlin, D.; Eden, A. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acid. Res. 2011, 39, 1326–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-Rodriguez, V.A.; de Groot, M.H.M.; Rijo-Ferreira, F.; Green, C.B.; Takahashi, J.S. Mice under Caloric Restriction Self-Impose a Temporal Restriction of Food Intake as Revealed by an Automated Feeder System. Cell Metab. 2017, 26, 267–277 e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaix, A.; Zarrinpar, A.; Miu, P.; Panda, S. Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges. Cell Metab. 2014, 20, 991–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Sassone-Corsi, P. Time for food: The intimate interplay between nutrition, metabolism, and the circadian clock. Cell 2015, 161, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, H.; Genzer, Y.; Cohen, R.; Chapnik, N.; Madar, Z.; Froy, O. Timed high-fat diet resets circadian metabolism and prevents obesity. FASEB J. 2012, 26, 3493–3502. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz, D.; Barnea, M.; Wainstein, J.; Froy, O. High caloric intake at breakfast vs. dinner differentially influences weight loss of overweight and obese women. Obesity 2013, 21, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz, D.; Wainstein, J.; Ahren, B.; Bar-Dayan, Y.; Landau, Z.; Rabinovitz, H.R.; Froy, O. High-energy breakfast with low-energy dinner decreases overall daily hyperglycaemia in type 2 diabetic patients: A randomised clinical trial. Diabetologia 2015, 58, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Garaulet, M.; Gomez-Abellan, P.; Alburquerque-Bejar, J.J.; Lee, Y.C.; Ordovas, J.M.; Scheer, F.A. Timing of food intake predicts weight loss effectiveness. Int. J. Obes. 2013, 37, 604–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, C.J.; Garcia, J.I.; Myers, S.; Yang, J.N.; Trienekens, N.; Scheer, F.A. The Human Circadian System Has a Dominating Role in Causing the Morning/Evening Difference in Diet-Induced Thermogenesis. Obesity 2015, 23, 2053–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.D.; Ordovas, J.M.; Scheer, F.A.; Turek, F.W. Circadian Rhythms, Metabolism, and Chrononutrition in Rodents and Humans. Adv. Nutr. 2016, 7, 399–406. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzoccoli, G.; Miele, L.; Marrone, G.; Mazza, T.; Vinciguerra, M.; Grieco, A. A Role for the Biological Clock in Liver Cancer. Cancers 2019, 11, 1778. https://doi.org/10.3390/cancers11111778
Mazzoccoli G, Miele L, Marrone G, Mazza T, Vinciguerra M, Grieco A. A Role for the Biological Clock in Liver Cancer. Cancers. 2019; 11(11):1778. https://doi.org/10.3390/cancers11111778
Chicago/Turabian StyleMazzoccoli, Gianluigi, Luca Miele, Giuseppe Marrone, Tommaso Mazza, Manlio Vinciguerra, and Antonio Grieco. 2019. "A Role for the Biological Clock in Liver Cancer" Cancers 11, no. 11: 1778. https://doi.org/10.3390/cancers11111778
APA StyleMazzoccoli, G., Miele, L., Marrone, G., Mazza, T., Vinciguerra, M., & Grieco, A. (2019). A Role for the Biological Clock in Liver Cancer. Cancers, 11(11), 1778. https://doi.org/10.3390/cancers11111778