Roles for Autophagy in Esophageal Carcinogenesis: Implications for Improving Patient Outcomes

Abstract
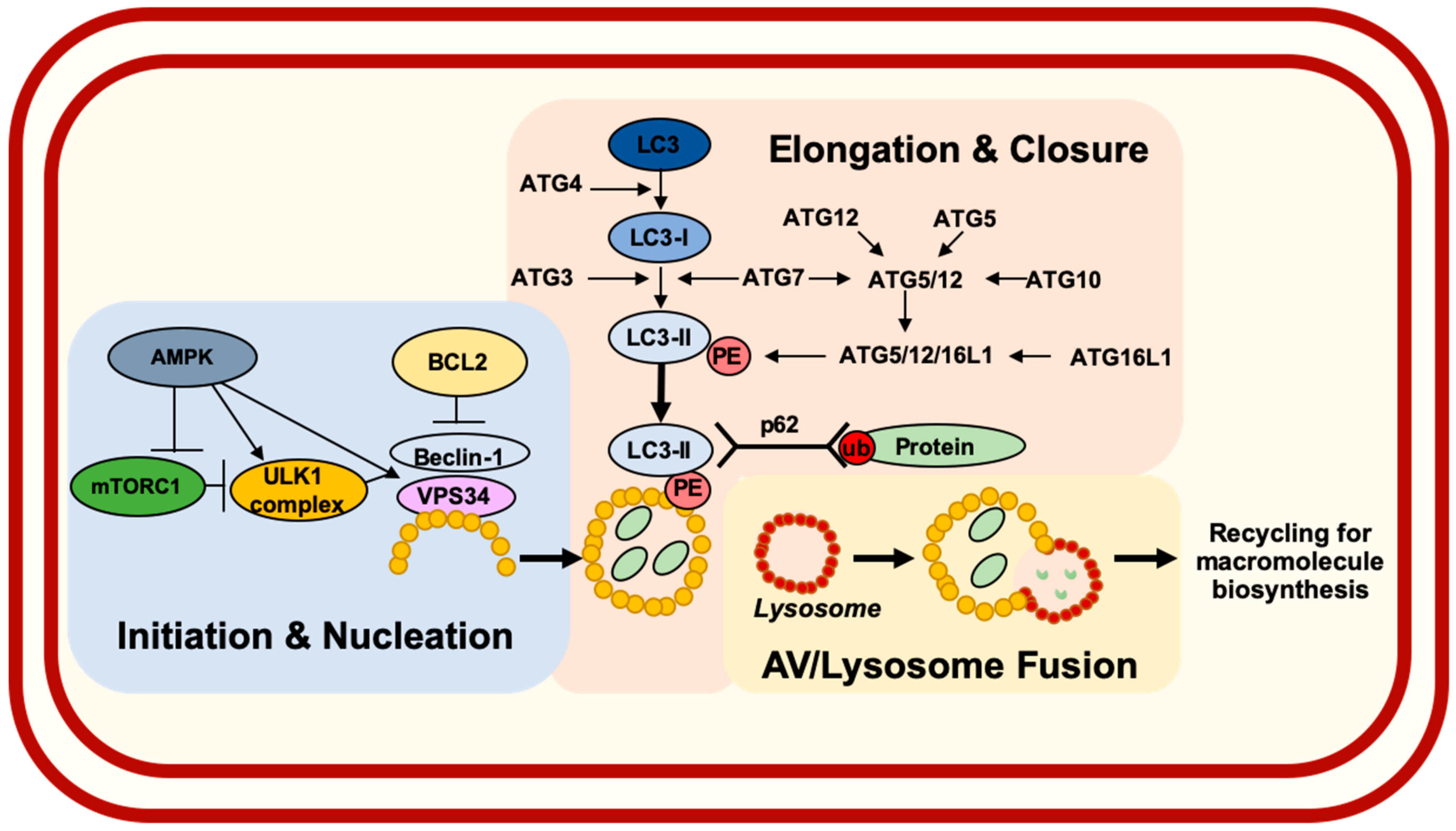
1. Introduction
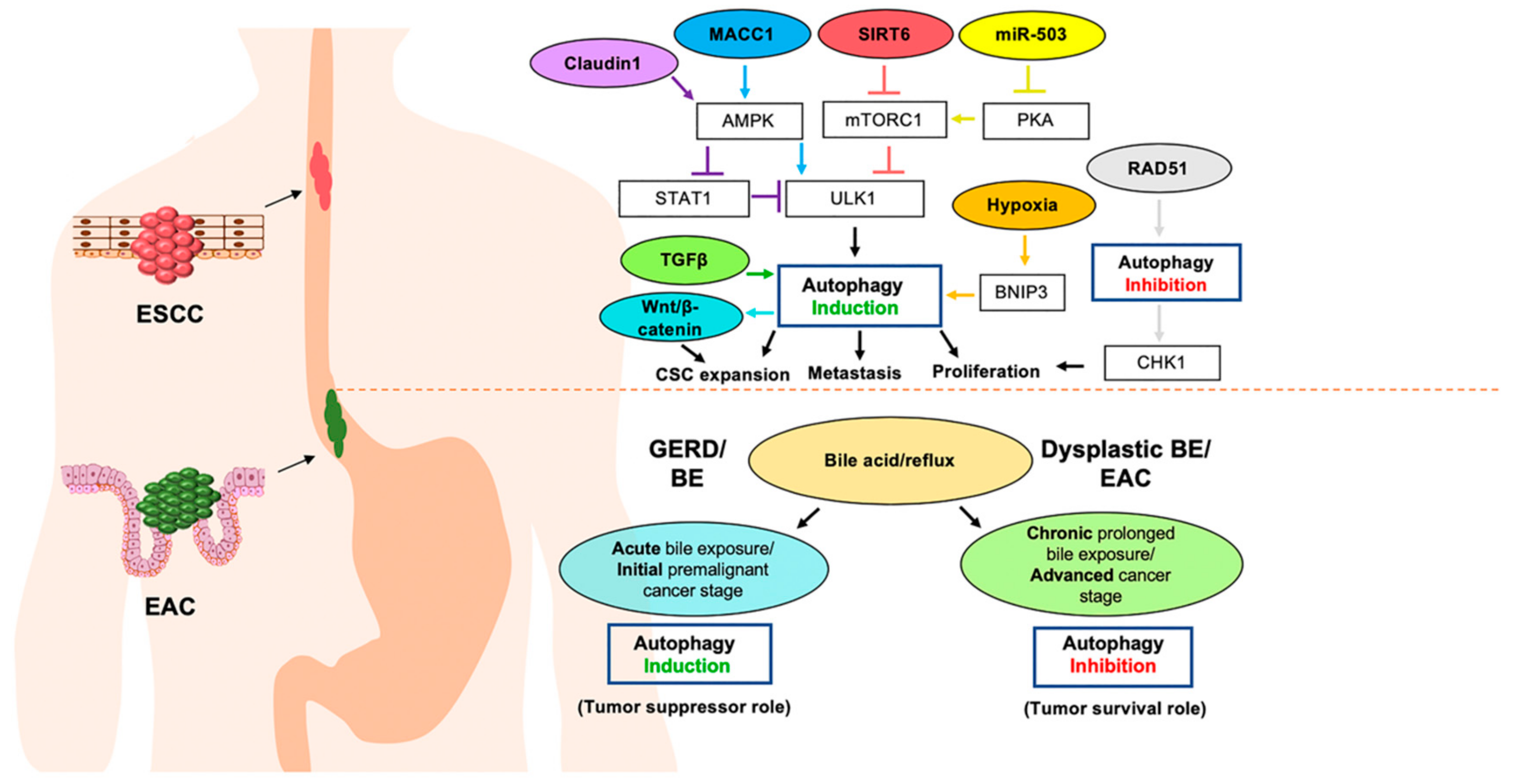
2. Roles for Autophagy in Esophageal Carcinogenesis
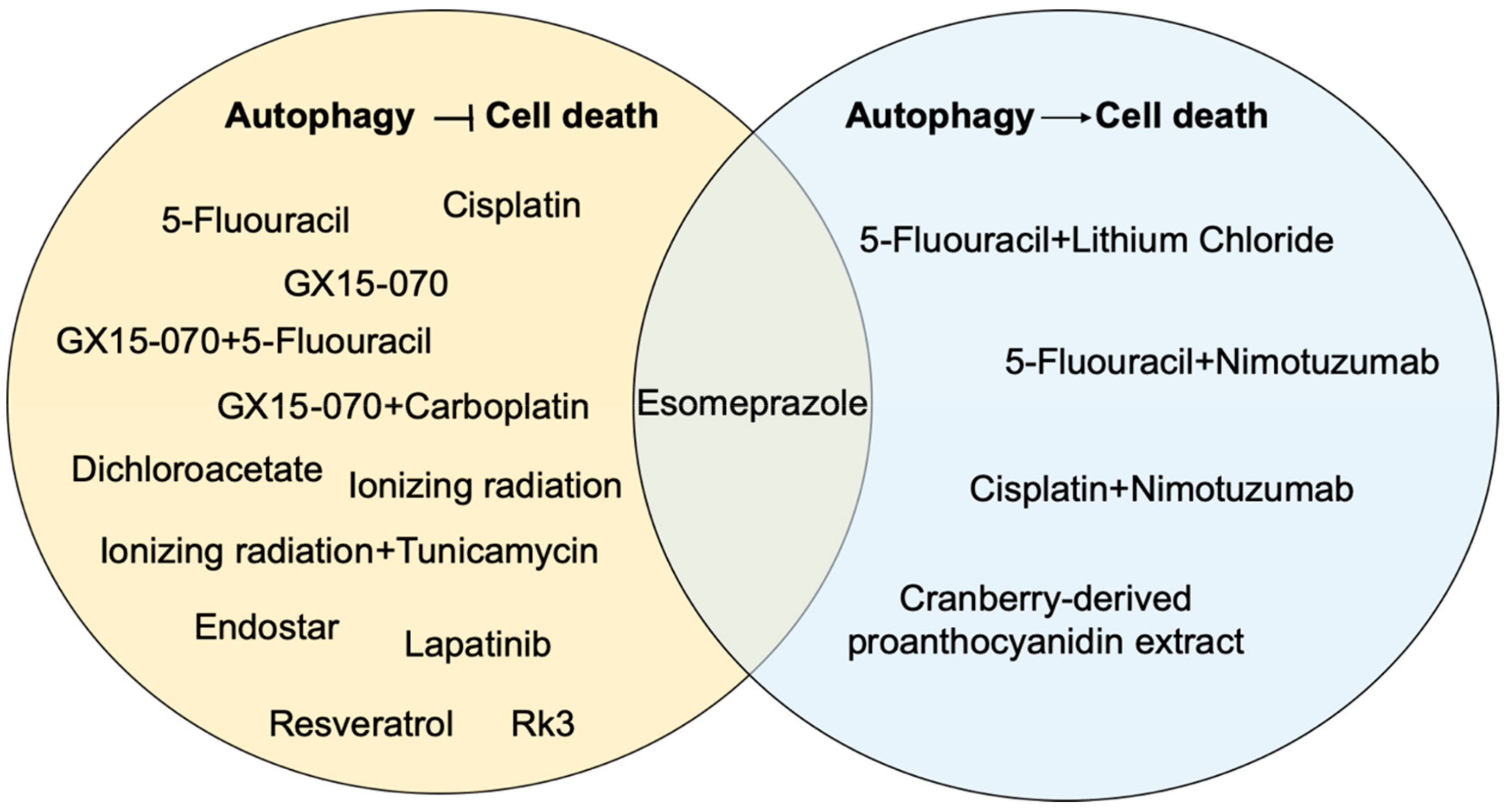
3. Roles for Autophagy in Esophageal Cancer Responses to Therapy
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Hamadeh, R.R.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, M.; Suzuki, S.; Dobashi, Y.; Yamane, T.; Kono, K.; Enomoto, N.; Ooi, A. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int. J. Cancer 2006, 118, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef]
- Hollstein, M.C.; Metcalf, R.A.; Welsh, J.A.; Montesano, R.; Harris, C.C. Frequent mutation of the p53 gene in human esophageal cancer. Proc. Natl. Acad. Sci. USA 1990, 87, 9958–9961. [Google Scholar] [CrossRef]
- Agrawal, N.; Jiao, Y.; Bettegowda, C.; Hutfless, S.M.; Wang, Y.; David, S.; Cheng, Y.; Twaddell, W.S.; Latt, N.L.; Shin, E.J.; et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012, 2, 899–905. [Google Scholar] [CrossRef]
- Cook, M.B.; Corley, D.A.; Murray, L.J.; Liao, L.M.; Kamangar, F.; Ye, W.; Gammon, M.D.; Risch, H.A.; Casson, A.G.; Freedman, N.D.; et al. Gastroesophageal reflux in relation to adenocarcinomas of the esophagus: A pooled analysis from the Barrett’s and Esophageal Adenocarcinoma Consortium (BEACON). PLoS ONE 2014, 9, e103508. [Google Scholar] [CrossRef]
- Reichenbach, Z.W.; Murray, M.G.; Saxena, R.; Farkas, D.; Karassik, E.G.; Klochkova, A.; Patel, K.; Tice, C.; Hall, T.M.; Gang, J.; et al. Clinical and translational advances in esophageal squamous cell carcinoma. Adv. Cancer Res. 2019, 144, 95–135. [Google Scholar] [CrossRef]
- Rustgi, A.K.; El-Serag, H.B. Esophageal carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Kong, J.; Whelan, K.A.; Laczkó, D.; Dang, B.; Caro Monroig, A.; Soroush, A.; Falcone, J.; Amaravadi, R.K.; Rustgi, A.K.; Ginsberg, G.G.; et al. Autophagy levels are elevated in barrett’s esophagus and promote cell survival from acid and oxidative stress. Mol. Carcinog. 2016, 55, 1526–1541. [Google Scholar] [CrossRef] [PubMed]
- Whelan, K.A.; Merves, J.F.; Giroux, V.; Tanaka, K.; Guo, A.; Chandramouleeswaran, P.M.; Benitez, A.J.; Dods, K.; Que, J.; Masterson, J.C.; et al. Autophagy mediates epithelial cytoprotection in eosinophilic oesophagitis. Gut 2017, 66, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; Longatti, A.; McKnight, N.C.; Tooze, S.A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 2009, 29, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef]
- Zhang, L.; Ouyang, L.; Guo, Y.; Zhang, J.; Liu, B. UNC-51-like Kinase 1: From an Autophagic Initiator to Multifunctional Drug Target. J. Med. Chem. 2018, 61, 6491–6500. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, D.; Li, J.; Deng, X.; Liang, G.; Long, Y.; He, X.; Dai, T.; Ren, D. MACC1 induces autophagy to regulate proliferation, apoptosis, migration and invasion of squamous cell carcinoma. Oncol. Rep. 2017, 38, 2369–2377. [Google Scholar] [CrossRef]
- Wu, J.; Gao, F.; Xu, T.; Li, J.; Hu, Z.; Wang, C.; Long, Y.; He, X.; Deng, X.; Ren, D.; et al. CLDN1 induces autophagy to promote proliferation and metastasis of esophageal squamous carcinoma through AMPK/STAT1/ULK1 signaling. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Huang, N.; Liu, Z.; Zhu, J.; Cui, Z.; Li, Y.; Yu, Y.; Sun, F.; Pan, Q.; Yang, Q. Sirtuin 6 plays an oncogenic role and induces cell autophagy in esophageal cancer cells. Tumour Biol. 2017, 39, 1010428317708532. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.; Jain, A.; Khan, D.; Ahamed, F.; Mishra, S.; Giri, M.; Inbaraj, M.; Krishna, S.; Sarikhani, M.; Maity, S.; et al. SIRT6 transcriptionally regulates global protein synthesis through transcription factor Sp1 independent of its deacetylase activity. Nucleic Acids Res. 2019, 47, 9115–9131. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gao, F.; Xu, T.; Deng, X.; Wang, C.; Yang, X.; Hu, Z.; Long, Y.; He, X.; Liang, G.; et al. miR-503 suppresses the proliferation and metastasis of esophageal squamous cell carcinoma by triggering autophagy via PKA/mTOR signaling. Int. J. Oncol. 2018, 52, 1427–1442. [Google Scholar] [CrossRef]
- Ma, Z.; Chen, C.; Tang, P.; Zhang, H.; Yue, J.; Yu, Z. BNIP3 induces apoptosis and protective autophagy under hypoxia in esophageal squamous cell carcinoma cell lines: BNIP3 regulates cell death. Dis. Esophagus 2017, 30, 1–8. [Google Scholar] [CrossRef]
- Whelan, K.A.; Chandramouleeswaran, P.M.; Tanaka, K.; Natsuizaka, M.; Guha, M.; Srinivasan, S.; Darling, D.S.; Kita, Y.; Natsugoe, S.; Winkler, J.D.; et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene 2017, 36, 4843–4858. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, G.; Hua, X.; Li, Y.; Yan, H.; Che, X.; Tian, Z.; Liufu, H.; Huang, C.; Li, J.; et al. CD44s is a crucial ATG7 downstream regulator for stem-like property, invasion, and lung metastasis of human bladder cancer (BC) cells. Oncogene 2019, 38, 3301–3315. [Google Scholar] [CrossRef]
- Smit, J.K.; Faber, H.; Niemantsverdriet, M.; Baanstra, M.; Bussink, J.; Hollema, H.; van Os, R.P.; Plukker, J.T.; Coppes, R.P. Prediction of response to radiotherapy in the treatment of esophageal cancer using stem cell markers. Radiother. Oncol. 2013, 107, 434–441. [Google Scholar] [CrossRef]
- Wang, C.; Yan, F.H.; Zhang, J.J.; Huang, H.; Cui, Q.S.; Dong, W.; Zhang, W.W.; Zhao, Y.; Chen, H.Z.; Zhao, T.J. OV6 (+) cancer stem cells drive esophageal squamous cell carcinoma progression through ATG7-dependent beta-catenin stabilization. Cancer Lett. 2017, 391, 100–113. [Google Scholar] [CrossRef]
- Zhu, X.; Pan, Q.; Huang, N.; Wu, J.; Zhen, N.; Sun, F.; Li, Z.; Yang, Q. RAD51 regulates CHK1 stability via autophagy to promote cell growth in esophageal squamous carcinoma cells. Tumour Biol. 2016, 37, 16151–16161. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef]
- Roesly, H.B.; Khan, M.R.; Chen, H.D.; Hill, K.A.; Narendran, N.; Watts, G.S.; Chen, X.; Dvorak, K. The decreased expression of Beclin-1 correlates with progression to esophageal adenocarcinoma: The role of deoxycholic acid. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Willet, S.G.; Lewis, M.A.; Miao, Z.F.; Liu, D.; Radyk, M.D.; Cunningham, R.L.; Burclaff, J.; Sibbel, G.; Lo, H.G.; Blanc, V.; et al. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J. 2018, 37, e98311. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.U.; Mills, J.C. The cyclical hit model: How paligenosis might establish the mutational landscape in Barrett’s esophagus and esophageal adenocarcinoma. Curr. Opin. Gastroenterol. 2019, 35, 363–370. [Google Scholar] [CrossRef]
- Stairs, D.B.; Bayne, L.J.; Rhoades, B.; Vega, M.E.; Waldron, T.J.; Kalabis, J.; Klein-Szanto, A.; Lee, J.S.; Katz, J.P.; Diehl, J.A.; et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell 2011, 19, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.H.; Knudsen, B.; Bemis, D.; Tickoo, S.; Gudas, L.J. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin. Cancer Res. 2004, 10, 301–313. [Google Scholar] [CrossRef]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Que, J.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Wu, X.; He, C.; Guo, L.; Zhang, S.; Xiao, Y.; Guo, W.; Tan, B. Autophagy-related proteins LC3 and Beclin-1 impact the efficacy of chemoradiation on esophageal squamous cell carcinoma. Pathol. Res. Pract. 2013, 209, 562–567. [Google Scholar] [CrossRef]
- Chen, H.I.; Tsai, H.P.; Chen, Y.T.; Tsao, S.C.; Chai, C.Y. Autophagy and Apoptosis Play Opposing Roles in Overall Survival of Esophageal Squamous Cell Carcinoma. Pathol. Oncol. Res. 2016, 22, 699–705. [Google Scholar] [CrossRef]
- Hao, C.L.; Li, Y.; Yang, H.X.; Luo, R.Z.; Zhang, Y.; Zhang, M.F.; Cheng, Y.F.; Wang, X. High level of microtubule-associated protein light chain 3 predicts poor prognosis in resectable esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 4213–4221. [Google Scholar]
- Sakurai, T.; Okumura, H.; Matsumoto, M.; Uchikado, Y.; Setoyama, T.; Omoto, I.; Owaki, T.; Maemura, K.; Ishigami, S.; Natsugoe, S. The expression of LC-3 is related to tumor suppression through angiogenesis in esophageal cancer. Med. Oncol. 2013, 30, 701. [Google Scholar] [CrossRef]
- Wang, Z.B.; Peng, X.Z.; Chen, S.S.; Ning, F.L.; Du, C.J.; Wang, K.; Ma, W.; Cheng, Y.F. High p53 and MAP1 light chain 3A co-expression predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mol. Med. Rep. 2013, 8, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Miyata, H.; Makino, T.; Masuike, Y.; Furukawa, H.; Tanaka, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. High Expression of the Mitophagy-Related Protein Pink1 is Associated with a Poor Response to Chemotherapy and a Poor Prognosis for Patients Treated with Neoadjuvant Chemotherapy for Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2017, 24, 4025–4032. [Google Scholar] [CrossRef]
- El-Mashed, S.; O’Donovan, T.R.; Kay, E.W.; Abdallah, A.R.; Cathcart, M.C.; O’Sullivan, J.; O’Grady, A.; Reynolds, J.; O’Reilly, S.; O’Sullivan, G.C.; et al. LC3B globular structures correlate with survival in esophageal adenocarcinoma. BMC Cancer 2015, 15, 582. [Google Scholar] [CrossRef] [PubMed]
- Adams, O.; Dislich, B.; Berezowska, S.; Schlafli, A.M.; Seiler, C.A.; Kroll, D.; Tschan, M.P.; Langer, R. Prognostic relevance of autophagy markers LC3B and p62 in esophageal adenocarcinomas. Oncotarget 2016, 7, 39241–39255. [Google Scholar] [CrossRef]
- Wu, N.; Zhu, Y.; Xu, X.; Zhu, Y.; Song, Y.; Pang, L.; Chen, Z. The anti-tumor effects of dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A on inducing autophagy in esophageal squamous cell carcinoma. J. Cancer 2018, 9, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, Y.; Lu, C.; Zhang, L. Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1alpha expression. Pathol. Oncol. Res. 2009, 15, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Duan, B.S.; Huang, J.X.; Jiao, X.; Zhu, X.W.; Sheng, H.H.; Gao, H.J.; Yu, H. Association of the expression of unc-51-Like kinase 1 with lymph node metastasis and survival in patients with esophageal squamous cell carcinoma. Int. J. Clin. Exp. Med. 2014, 7, 1349–1354. [Google Scholar] [PubMed]
- Jiang, S.; Li, Y.; Zhu, Y.H.; Wu, X.Q.; Tang, J.; Li, Z.; Feng, G.K.; Deng, R.; Li, D.D.; Luo, R.Z.; et al. Intensive expression of UNC-51-like kinase 1 is a novel biomarker of poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Sci. 2011, 102, 1568–1575. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Blum, A.E.; Venkitachalam, S.; Guo, Y.; Kieber-Emmons, A.M.; Ravi, L.; Chandar, A.K.; Iyer, P.G.; Canto, M.I.; Wang, J.S.; Shaheen, N.J.; et al. RNA Sequencing Identifies Transcriptionally Viable Gene Fusions in Esophageal Adenocarcinomas. Cancer Res. 2016, 76, 5628–5633. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, P.; Williams, P.A.; Whelan, K.A.; Samper, F.C.; Andres, S.F.; Simon, L.A.; Parham, L.R.; Mizuno, R.; Lundsmith, E.T.; Lee, D.S.; et al. Posttranscriptional regulation of colonic epithelial repair by RNA binding protein IMP1/IGF2BP1. EMBO Rep. 2019, 20, e47074. [Google Scholar] [CrossRef]
- Kijima, T.; Nakagawa, H.; Shimonosono, M.; Chandramouleeswaran, P.M.; Hara, T.; Sahu, V.; Kasagi, Y.; Kikuchi, O.; Tanaka, K.; Giroux, V.; et al. Three-Dimensional Organoids Reveal Therapy Resistance of Esophageal and Oropharyngeal Squamous Cell Carcinoma Cells. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Falvey, C.M.; O’Donovan, T.R.; El-Mashed, S.; Nyhan, M.J.; O’Reilly, S.; McKenna, S.L. UBE2L6/UBCH8 and ISG15 attenuate autophagy in esophageal cancer cells. Oncotarget 2017, 8, 23479–23491. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, T.R.; Rajendran, S.; O’Reilly, S.; O’Sullivan, G.C.; McKenna, S.L. Lithium Modulates Autophagy in Esophageal and Colorectal Cancer Cells and Enhances the Efficacy of Therapeutic Agents In Vitro and In Vivo. PLoS ONE 2015, 10, e0134676. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, M.J.; O’Donovan, T.R.; Elzinga, B.; Crowley, L.C.; O’Sullivan, G.C.; McKenna, S.L. The BH3 mimetic HA14-1 enhances 5-fluorouracil-induced autophagy and type II cell death in oesophageal cancer cells. Br. J. Cancer 2012, 106, 711–718. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Liu, J.C.; Wang, J.J.; Li, Y.H.; Pan, J.; Zhang, Y.R. Autophagy inhibition increased the anti-tumor effect of cisplatin on drug-resistant esophageal cancer cells. J. Biol. Regul. Homeost. Agents 2017, 31, 645–652. [Google Scholar]
- Yu, L.; Gu, C.; Zhong, D.; Shi, L.; Kong, Y.; Zhou, Z.; Liu, S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014, 355, 34–45. [Google Scholar] [CrossRef]
- Feng, Y.; Gao, Y.; Wang, D.; Xu, Z.; Sun, W.; Ren, P. Autophagy Inhibitor (LY294002) and 5-fluorouracil (5-FU) Combination-Based Nanoliposome for Enhanced Efficacy Against Esophageal Squamous Cell Carcinoma. Nanoscale Res. Lett. 2018, 13, 325. [Google Scholar] [CrossRef]
- Nyhan, M.J.; O’Donovan, T.R.; Boersma, A.W.; Wiemer, E.A.; McKenna, S.L. MiR-193b promotes autophagy and non-apoptotic cell death in oesophageal cancer cells. BMC Cancer 2016, 16, 101. [Google Scholar] [CrossRef] [PubMed]
- Adams, O.; Janser, F.A.; Dislich, B.; Berezowska, S.; Humbert, M.; Seiler, C.A.; Kroell, D.; Slotta-Huspenina, J.; Feith, M.; Ott, K.; et al. A specific expression profile of LC3B and p62 is associated with nonresponse to neoadjuvant chemotherapy in esophageal adenocarcinomas. PLoS ONE 2018, 13, e0197610. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xie, C. Radiation-induced autophagy promotes esophageal squamous cell carcinoma cell survival via the LKB1 pathway. Oncol. Rep. 2016, 35, 3559–3565. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Guo, L.; Wu, X.; He, C.; Zhang, S.; Xiao, Y.; Yang, Y.; Hao, D. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol. Med. Rep. 2015, 12, 1645–1652. [Google Scholar] [CrossRef]
- Tao, H.; Qian, P.; Lu, J.; Guo, Y.; Zhu, H.; Wang, F. Autophagy inhibition enhances radiosensitivity of Eca109 cells via the mitochondrial apoptosis pathway. Int. J. Oncol. 2018, 52, 1853–1862. [Google Scholar] [CrossRef]
- Chen, Y.S.; Song, H.X.; Lu, Y.; Li, X.; Chen, T.; Zhang, Y.; Xue, J.X.; Liu, H.; Kan, B.; Yang, G.; et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis. Esophagus 2011, 24, 437–443. [Google Scholar] [CrossRef]
- Pang, X.L.; He, G.; Liu, Y.B.; Wang, Y.; Zhang, B. Endoplasmic reticulum stress sensitizes human esophageal cancer cell to radiation. World J. Gastroenterol. 2013, 19, 1736–1748. [Google Scholar] [CrossRef]
- Zhu, H.; Song, H.; Chen, G.; Yang, X.; Liu, J.; Ge, Y.; Lu, J.; Qin, Q.; Zhang, C.; Xu, L.; et al. eEF2K promotes progression and radioresistance of esophageal squamous cell carcinoma. Radiother. Oncol. 2017, 124, 439–447. [Google Scholar] [CrossRef]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; He, G.; Chen, H.; Zhu, C.; Qin, Q.; Yan, J.; Zhang, X.; Sun, X. High-mobility group box 1 protein modulated proliferation and radioresistance in esophageal squamous cell carcinoma. J. Gastroenterol. Hepatol. 2019, 34, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zheng, S.; Zhang, X.; Gong, T.; Lv, X.; Fu, S.; Zhang, S.; Yin, X.; Hao, J.; Shan, C.; et al. High mobility group box 1 promotes radioresistance in esophageal squamous cell carcinoma cell lines by modulating autophagy. Cell Death Dis. 2019, 10, 136. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Pan, B.; Yi, J.; Chen, L. Featured article: Autophagic activation with nimotuzumab enhanced chemosensitivity and radiosensitivity of esophageal squamous cell carcinoma. Exp. Biol. Med. 2014, 239, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Janser, F.A.; Adams, O.; Butler, V.; Schlafli, A.M.; Dislich, B.; Seiler, C.A.; Kroll, D.; Langer, R.; Tschan, M.P. Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3069. [Google Scholar] [CrossRef]
- Suntharalingam, M.; Winter, K.; Ilson, D.; Dicker, A.P.; Kachnic, L.; Konski, A.; Chakravarthy, A.B.; Anker, C.J.; Thakrar, H.; Horiba, N.; et al. Effect of the Addition of Cetuximab to Paclitaxel, Cisplatin, and Radiation Therapy for Patients With Esophageal Cancer: The NRG Oncology RTOG 0436 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2017, 3, 1520–1528. [Google Scholar] [CrossRef]
- Crosby, T.; Hurt, C.N.; Falk, S.; Gollins, S.; Staffurth, J.; Ray, R.; Bridgewater, J.A.; Geh, J.I.; Cunningham, D.; Blazeby, J.; et al. Long-term results and recurrence patterns from SCOPE-1: A phase II/III randomised trial of definitive chemoradiotherapy +/− cetuximab in oesophageal cancer. Br. J. Cancer 2017, 116, 709–716. [Google Scholar] [CrossRef]
- Ruhstaller, T.; Thuss-Patience, P.; Hayoz, S.; Schacher, S.; Knorrenschild, J.R.; Schnider, A.; Plasswilm, L.; Budach, W.; Eisterer, W.; Hawle, H.; et al. Neoadjuvant chemotherapy followed by chemoradiation and surgery with and without cetuximab in patients with resectable esophageal cancer: A randomized, open-label, phase III trial (SAKK 75/08). Ann. Oncol. 2018, 29, 1386–1393. [Google Scholar] [CrossRef]
- Pan, J.; Cheng, C.; Verstovsek, S.; Chen, Q.; Jin, Y.; Cao, Q. The BH3-mimetic GX15-070 induces autophagy, potentiates the cytotoxicity of carboplatin and 5-fluorouracil in esophageal carcinoma cells. Cancer Lett. 2010, 293, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wu, W.K.; Gu, C.; Zhong, D.; Zhao, X.; Kong, Y.; Lin, Q.; Chan, M.T.; Zhou, Z.; Liu, S. Obatoclax impairs lysosomal function to block autophagy in cisplatin-sensitive and -resistant esophageal cancer cells. Oncotarget 2016, 7, 14693–14707. [Google Scholar] [CrossRef] [PubMed]
- Chueca, E.; Apostolova, N.; Esplugues, J.V.; Garcia-Gonzalez, M.A.; Lanas, A.; Piazuelo, E. Proton Pump Inhibitors Display Antitumor Effects in Barrett’s Adenocarcinoma Cells. Front. Pharmacol. 2016, 7, 452. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008, 68, 1223–1231. [Google Scholar] [CrossRef]
- Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Parish, C.R.; Board, P.G.; Blackburn, A.C. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 2010, 120, 253–260. [Google Scholar] [CrossRef]
- Jia, H.Y.; Wang, H.N.; Xia, F.Y.; Sun, Y.; Liu, H.L.; Yan, L.L.; Li, S.S.; Jiang, D.C.; Xu, M.M. Dichloroacetate induces protective autophagy in esophageal squamous carcinoma cells. Oncol. Lett. 2017, 14, 2765–2770. [Google Scholar] [CrossRef][Green Version]
- Xu, M.; Huang, H.; Xiong, Y.; Peng, B.; Zhou, Z.; Wang, D.; Yang, X. Combined chemotherapy plus endostar with sequential stereotactic radiotherapy as salvage treatment for recurrent esophageal cancer with severe dyspnea: A case report and review of the literature. Oncol. Lett. 2014, 8, 291–294. [Google Scholar] [CrossRef]
- Deng, W.Y.; Song, T.; Li, N.; Luo, S.X.; Li, X. Clinical observation and therapeutic evaluation of Rh-endostatin combined with DP regimen in treating patients with advanced esophageal cancer. Asian Pac. J. Cancer Prev. 2014, 15, 6565–6570. [Google Scholar] [CrossRef]
- Zhong, Z.; Gu, X.; Zhang, Z.; Wang, D.; Qing, Y.; Li, M.; Dai, N. Recombinant human endostatin combined with definitive chemoradiotherapy as primary treatment for patients with unresectable but without systemic metastatic squamous cell carcinoma of the oesophagus. Br. J. Radiol. 2012, 85, 1104–1109. [Google Scholar] [CrossRef]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, G.; Wei, X.; Zhang, J.; Chiu, J.F.; Hasenmayer, D.; Zhang, D.; Zhang, H. Resveratrol-induced apoptosis is enhanced by inhibition of autophagy in esophageal squamous cell carcinoma. Cancer Lett. 2013, 336, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, F.; Maffei, R.; Beau, I.; Codogno, P.; Ghidoni, R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008, 15, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, J.; Fu, R.; Zhu, C.; Fan, D. The ginsenoside Rk3 exerts anti-esophageal cancer activity in vitro and in vivo by mediating apoptosis and autophagy through regulation of the PI3K/Akt/mTOR pathway. PLoS ONE 2019, 14, e0216759. [Google Scholar] [CrossRef]
- Kresty, L.A.; Weh, K.M.; Zeyzus-Johns, B.; Perez, L.N.; Howell, A.B. Cranberry proanthocyanidins inhibit esophageal adenocarcinoma in vitro and in vivo through pleiotropic cell death induction and PI3K/AKT/mTOR inactivation. Oncotarget 2015, 6, 33438–33455. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Risk Factor | Expression Description | n | Prognostic Relevance | References |
|---|---|---|---|---|
| ESCC | ||||
| LC3 * | High/Postive | 150 | (M, OS) OR 1.657, 95% CI; 1.048–2.622 | [35] |
| 43 | Decreased overall survival (p = 0.032) | [36] | ||
| 129 | Decreased overall survival (p = 0.0382) | [26] | ||
| 142 | Increased overall survival (p = 0.04) | [38] | ||
| Negative | 118 | Decreased overall survial (p = 0.021) | [43] | |
| Beclin-1 | Negative | 54 | Decreased overall survival (p = 0.004) | [44,45] |
| 118 | (M, OS) HR 0.511, 95% CI; 0.299–0.874 | [43] | ||
| ULK1 | High | 248 | (M, OS) RR 2.220, 95% CI: 1.434–3.436 | [46] |
| Low | 86 | (U, OS) HR 1.754, 95% CI: 1.022–3.010 | [45] | |
| LC3, Beclin-1 | Positive, Positive | 150 | Decreased overall survival (p = 0.038) | [35] |
| LC3A, p53 | High, High | 114 | (M, OS) HR 2.8, 95% CI: 1.536–6.183 | [39] |
| EAC | ||||
| LC3 | Low (Diffuse Cytoplasmic) | 104 | Decreased overall survival (p < 0.001) | [41] |
| High (Crescent or Ring-like) | 104 | Decreased overall survival (p = 0.02) | ||
| High (Globular) | 104 | (M, OS NaN) HR 6.086, 95% CI: 3.179–11.653 | ||
| p62 (Cyto/Nuc) | Low/Low | 116 | (M, OS) HR 0.561, 95% CI: 0.329–0.956 | [42] |
| LC3B, p62 | Low, Low | 116 | (M, OS) HR 0.549, 95% CI: 0.330–0.914 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saxena, R.; Klochkova, A.; Murray, M.G.; Kabir, M.F.; Samad, S.; Beccari, T.; Gang, J.; Patel, K.; Hamilton, K.E.; Whelan, K.A. Roles for Autophagy in Esophageal Carcinogenesis: Implications for Improving Patient Outcomes. Cancers 2019, 11, 1697. https://doi.org/10.3390/cancers11111697
Saxena R, Klochkova A, Murray MG, Kabir MF, Samad S, Beccari T, Gang J, Patel K, Hamilton KE, Whelan KA. Roles for Autophagy in Esophageal Carcinogenesis: Implications for Improving Patient Outcomes. Cancers. 2019; 11(11):1697. https://doi.org/10.3390/cancers11111697
Chicago/Turabian StyleSaxena, Reshu, Alena Klochkova, Mary Grace Murray, Mohammad Faujul Kabir, Safiyah Samad, Tyler Beccari, Julie Gang, Kishan Patel, Kathryn E. Hamilton, and Kelly A. Whelan. 2019. "Roles for Autophagy in Esophageal Carcinogenesis: Implications for Improving Patient Outcomes" Cancers 11, no. 11: 1697. https://doi.org/10.3390/cancers11111697
APA StyleSaxena, R., Klochkova, A., Murray, M. G., Kabir, M. F., Samad, S., Beccari, T., Gang, J., Patel, K., Hamilton, K. E., & Whelan, K. A. (2019). Roles for Autophagy in Esophageal Carcinogenesis: Implications for Improving Patient Outcomes. Cancers, 11(11), 1697. https://doi.org/10.3390/cancers11111697