Novel Antrodia cinnamomea Extract Reduced Cancer Stem-Like Phenotype Changes and Resensitized KRAS-Mutant Colorectal Cancer via a MicroRNA-27a Pathway

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
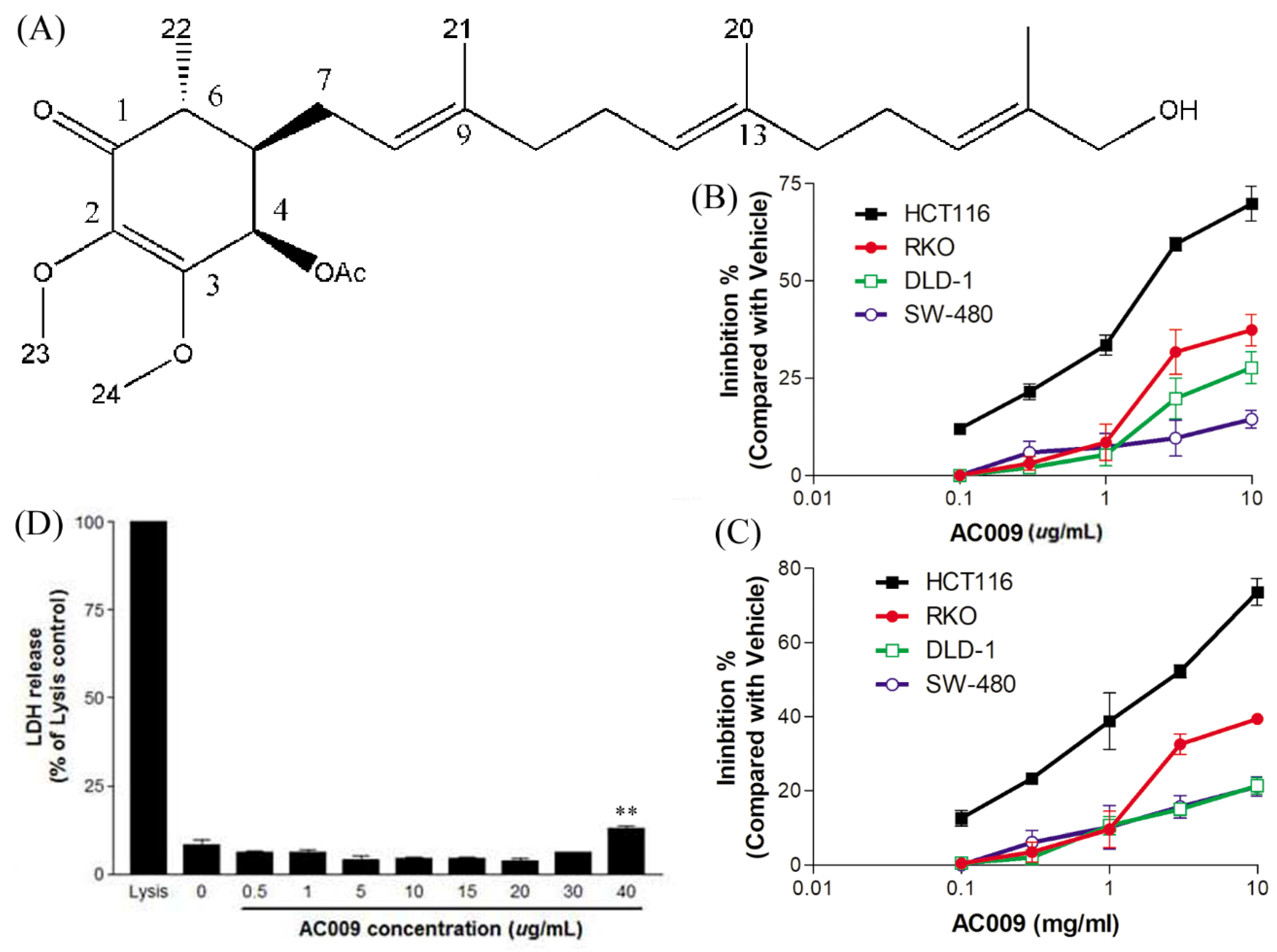
2.1. Effects of AC009 on CRC Cell Proliferation
2.2. Effects of AC009 on HCT116 Cell-Cycle Arrest and Apoptosis
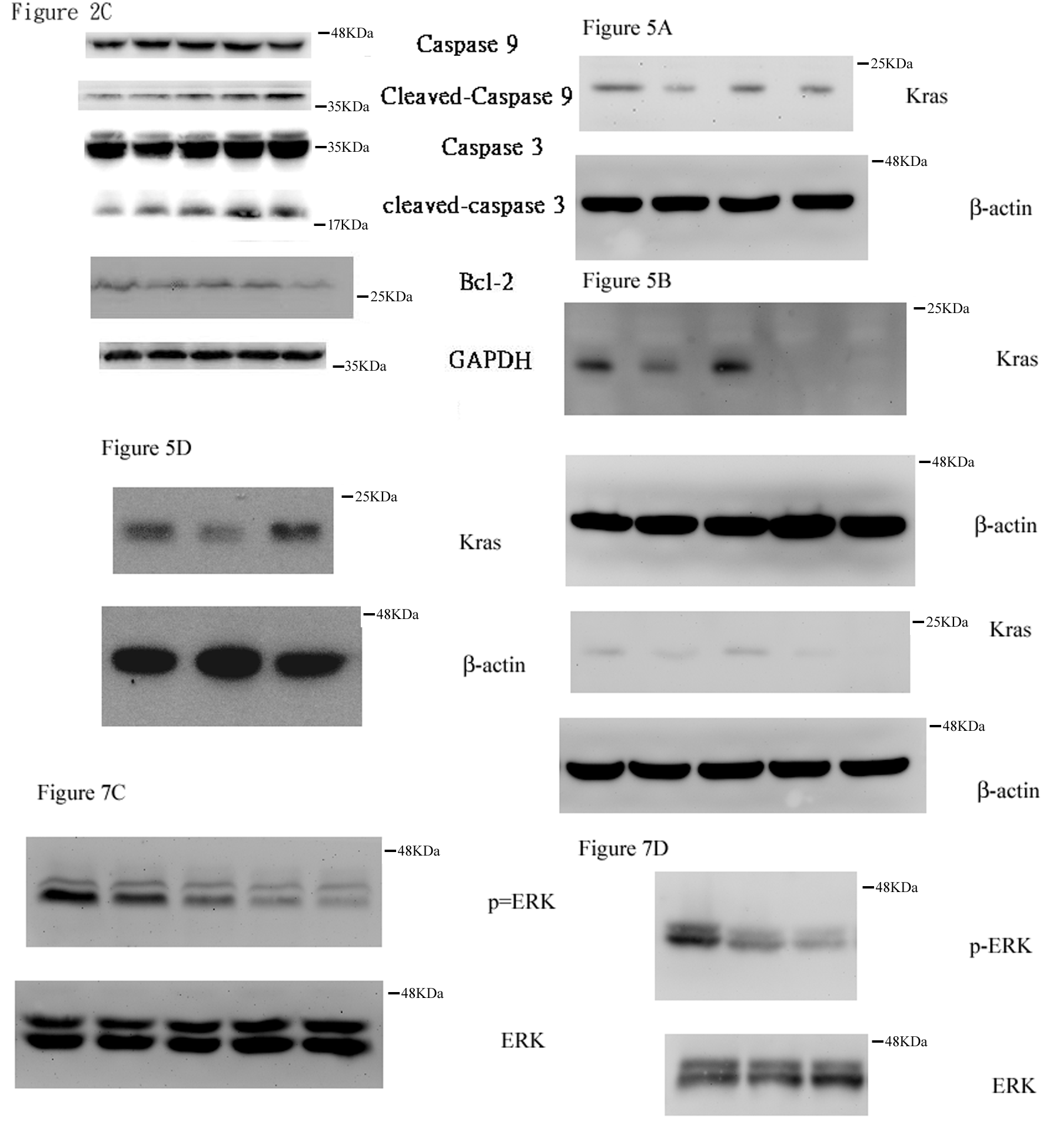
2.3. Effect of AC009 on Caspase Activation and Bcl-2 Inhibition
2.4. In Vivo Antitumor Effect of AC009
2.5. MiRNA and mRNA Microarray Analysis
2.6. Effect of AC009 on KRAS Protein Expression and Restoration of Cetuximab Antitumor Ability
2.7. Effect of AC009 on Restoring Cetuximab Antitumor Ability in In Vivo Models
2.8. Effect of miR-27a on Restoring Cetuximab Antitumor Ability
2.9. Effect of AC009 on Cancer Cell Stemness
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Culture
4.3. Cell Viability Sulforhodamine (SRB) Assay
4.4. Crystal Violet Assay
4.5. Lactate Dehydrogenase Releasing (LDH) Assay
4.6. Cell-Cycle Assay
4.7. Apoptosis Assay
4.8. Western Blotting Assay
4.9. Microarray Analysis
4.10. MiRNA Retrotranscription and Quantitative PCR
4.11. MiRNA and MiRNA Inhibitor Transfection
4.12. In Vivo Xenograft Model
4.13. Immunohistochemistry Study
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.; Barzi, A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.; Collins, P.D. Colon cancer: A civilization disorder. Dig. Dis. 2011, 29, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Fedirko, V.; Tramacere, I.; Bagnardi, V.; Rota, M.; Scotti, L.; Islami, F.; Negri, E.; Straif, K.; Romieu, I.; La Vecchia, C.; et al. Alcohol drinking and colorectal cancer risk: An overall and dose-response meta-analysis of published studies. Ann. Oncol. 2011, 22, 1958–1972. [Google Scholar] [CrossRef]
- Valtin, H. “Drink at least eight glasses of water a day.” Really? Is there scientific evidence for “8 × 8”? Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2002, 283, R993–R1004. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.-M.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef]
- Smith, G.; Carey, F.A.; Beattie, J.; Wilkie, M.J.V.; Lightfoot, T.J.; Coxhead, J.; Garner, R.C.; Steele, R.J.C.; Wolf, C.R. Mutations in APC, Kirsten-ras, and p53—Alternative genetic pathways to colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 9433–9438. [Google Scholar] [CrossRef]
- Brink, M.; De Goeij, A.F.; Weijenberg, M.P.; Roemen, G.M.; Lentjes, M.H.; Pachen, M.M.; Smits, K.M.; De Bruïne, A.P.; Goldbohm, R.A.; Brandt, P.A.V.D. K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 2003, 24, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. Systemic Therapy for Colon Cancer. Surg. Oncol. Clin. 2018, 27, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-L.; Kuo, Y.-H.; Tsai, C.-T.; Huang, Y.-T.; Chen, S.-C.; Chang, H.-W.; Lin, E.; Lin, W.-H.; Hseu, Y.-C. Anti-metastatic activities of Antrodia camphorata against human breast cancer cells mediated through suppression of the MAPK signaling pathway. Food Chem. Toxicol. 2011, 49, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Pan, J.H.; Liu, R.H.; Kuo, Y.H.; Sheen, L.Y.; Chiang, B.H. The 4-acetylantroquinonol B isolated from mycelium of Antrodia cinnamomea inhibits proliferation of hepatoma cells. J. Sci. Food Agric. 2010, 90, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Chen, S.-C.; Chen, H.-C.; Liao, J.-W.; Yang, H.-L. Antrodia camphorata inhibits proliferation of human breast cancer cells in vitro and in vivo. Food Chem. Toxicol. 2008, 46, 2680–2688. [Google Scholar] [CrossRef]
- Chen, K.C.; Peng, C.C.; Peng, R.Y.; Su, C.-H.; Chiang, H.-S.; Yan, J.-H.; Hsieh-Li, H.M. Unique Formosan MushroomAntrodia camphorataDifferentially Inhibits Androgen-Responsive LNCaP and -Independent PC-3 Prostate Cancer Cells. Nutr. Cancer 2007, 57, 111–121. [Google Scholar] [CrossRef]
- Chang, C.W.; Chen, C.C.; Wu, M.J.; Chen, Y.S.; Chen, C.C.; Sheu, S.J.; Lin, T.W.; Chou, S.H.; Lin, S.C.; Liu, C.J.; et al. Active Component of Antrodia cinnamomea Mycelia Targeting Head and Neck Cancer Initiating Cells through Exaggerated Autophagic Cell Death. Evid.-Based Complement. Altern. Med. 2013, 2013, 1–15. [Google Scholar]
- Wang, Z.; Jiang, C.; Chen, W.; Zhang, G.; Luo, D.; Cao, Y.; Wu, J.; Ding, Y.; Liu, B. Baicalein Induces Apoptosis and Autophagy via Endoplasmic Reticulum Stress in Hepatocellular Carcinoma Cells. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Chang, T.C.; Yeh, C.T.; Adebayo, B.O.; Lin, Y.C.; Deng, L.; Rao, Y.K.; Huang, C.C.; Lee, W.H.; Wu, A.T.; Hsiao, M.; et al. 4-Acetylantroquinonol B inhibits colorectal cancer tumorigenesis and suppresses cancer stem-like phenotype. Toxicol. Appl. Pharmacol. 2015, 288, 258–268. [Google Scholar] [CrossRef]
- Yeung, T.M.; Gandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. USA 2010, 107, 3722–3727. [Google Scholar] [CrossRef]
- Jung, Y.-C.; Han, S.; Hua, L.; Ahn, Y.-H.; Cho, H.; Lee, C.-J.; Lee, H.; Cho, Y.-Y.; Ryu, J.-H.; Jeon, R.; et al. Kazinol-E is a specific inhibitor of ERK that suppresses the enrichment of a breast cancer stem-like cell population. Biochem. Biophys. Res. Commun. 2016, 470, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Tor, Y.S.; Yazan, L.S.; Foo, J.B.; Wibowo, A.; Ismail, N.; Cheah, Y.K.; Abdullah, R.; Ismail, M.; Ismail, I.S.; Yeap, S.K. Induction of Apoptosis in MCF-7 Cells via Oxidative Stress Generation, Mitochondria-Dependent and Caspase-Independent Pathway by Ethyl Acetate Extract of Dillenia suffruticosa and Its Chemical Profile. PLoS ONE 2015, 10, e0127441. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. VEGF-targeted cancer therapeutics—paradoxical effects in endocrine organs. Nat. Rev. Endocrinol. 2014, 10, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Ho, C.L.; Wang, J.L.; Lee, C.C.; Cheng, H.Y.; Wen, W.C.; Cheng, H.H.Y.; Chen, M.C.M. Antroquinonol blocks Ras and Rho signaling via the inhibition of protein isoprenyltransferase activity in cancer cells. Biomed. Pharmacother. 2014, 68, 1007–1014. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Sun, X.; Chen, J.; Sun, X.; Zheng, J.; Chen, R. MicroRNA-27a functions as a tumor suppressor in renal cell carcinoma by targeting epidermal growth factor receptor. Oncol. Lett. 2016, 11, 4217–4223. [Google Scholar] [CrossRef]
- Jiang, Y.; Duan, Y.; Zhou, H. MicroRNA-27a directly targets KRAS to inhibit cell proliferation in esophageal squamous cell carcinoma. Oncol. Lett. 2015, 9, 471–477. [Google Scholar] [CrossRef]
- Bao, Y.; Chen, Z.; Guo, Y.; Feng, Y.; Li, Z.; Han, W.; Wang, J.; Zhao, W.; Jiao, Y.; Li, K.; et al. Tumor Suppressor MicroRNA-27a in Colorectal Carcinogenesis and Progression by Targeting SGPP1 and Smad2. PLoS ONE 2014, 9, e105991. [Google Scholar] [CrossRef]
- Zhao, N.; Sun, H.; Sun, B.; Zhu, D.; Zhao, X.; Wang, Y.; Gu, Q.; Dong, X.; Liu, F.; Zhang, Y.; et al. miR-27a-3p suppresses tumor metastasis and VM by down-regulating VE-cadherin expression and inhibiting EMT: An essential role for Twist-1 in HCC. Sci. Rep. 2016, 6, 23091. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.L.; Houghton, P.J. Establishment of human tumor xenografts in immunodeficient mice. Nat. Protoc. 2007, 2, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Dragu, D.L.; Necula, L.G.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Therapies targeting cancer stem cells: Current trends and future challenges. World J. Stem Cells 2015, 7, 1185–1201. [Google Scholar] [PubMed]
- Lin, H.C.; Lin, M.H.; Liao, J.H.; Wu, T.H.; Lee, T.H.; Mi, F.L.; Wu, C.H.; Chen, K.C.; Cheng, C.H.; Lin, C.W. Antroquinonol, a Ubiquinone Derivative from the Mushroom Antrodia camphorata, Inhibits Colon Cancer Stem Cell-like Properties: Insights into the Molecular Mechanism and Inhibitory Targets. J. Agric. Food Chem. 2017, 65, 51–59. [Google Scholar] [CrossRef]
- Jang, H.-J.; Bak, Y.; Pham, T.-H.; Kwon, S.-B.; Kim, B.-Y.; Hong, J.; Yoon, D.-Y. STK899704 inhibits stemness of cancer stem cells and migration via the FAK-MEK-ERK pathway in HT29 cells. BMB Rep. 2018, 51, 596–601. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, Y.L.; Qiu, F.M.; Zhang, T.; Chen, Z.G.; Zheng, S.; Huang, J. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis 2010, 31, 1376–1380. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-J.; Lai, K.-C.; Lee, A.-S.; Chang, C.-H.; Liu, C.-L.; Chung, C.-H. Novel Antrodia cinnamomea Extract Reduced Cancer Stem-Like Phenotype Changes and Resensitized KRAS-Mutant Colorectal Cancer via a MicroRNA-27a Pathway. Cancers 2019, 11, 1657. https://doi.org/10.3390/cancers11111657
Lin T-J, Lai K-C, Lee A-S, Chang C-H, Liu C-L, Chung C-H. Novel Antrodia cinnamomea Extract Reduced Cancer Stem-Like Phenotype Changes and Resensitized KRAS-Mutant Colorectal Cancer via a MicroRNA-27a Pathway. Cancers. 2019; 11(11):1657. https://doi.org/10.3390/cancers11111657
Chicago/Turabian StyleLin, Tsung-Jen, Kuo-Chu Lai, An-Sheng Lee, Chien-Hsin Chang, Chiung-Lin Liu, and Ching-Hu Chung. 2019. "Novel Antrodia cinnamomea Extract Reduced Cancer Stem-Like Phenotype Changes and Resensitized KRAS-Mutant Colorectal Cancer via a MicroRNA-27a Pathway" Cancers 11, no. 11: 1657. https://doi.org/10.3390/cancers11111657
APA StyleLin, T.-J., Lai, K.-C., Lee, A.-S., Chang, C.-H., Liu, C.-L., & Chung, C.-H. (2019). Novel Antrodia cinnamomea Extract Reduced Cancer Stem-Like Phenotype Changes and Resensitized KRAS-Mutant Colorectal Cancer via a MicroRNA-27a Pathway. Cancers, 11(11), 1657. https://doi.org/10.3390/cancers11111657