Spironolactone, a Classic Potassium-Sparing Diuretic, Reduces Survivin Expression and Chemosensitizes Cancer Cells to Non-DNA-Damaging Anticancer Drugs



Abstract
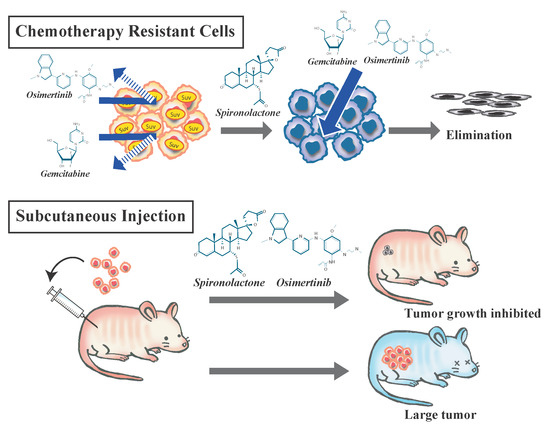
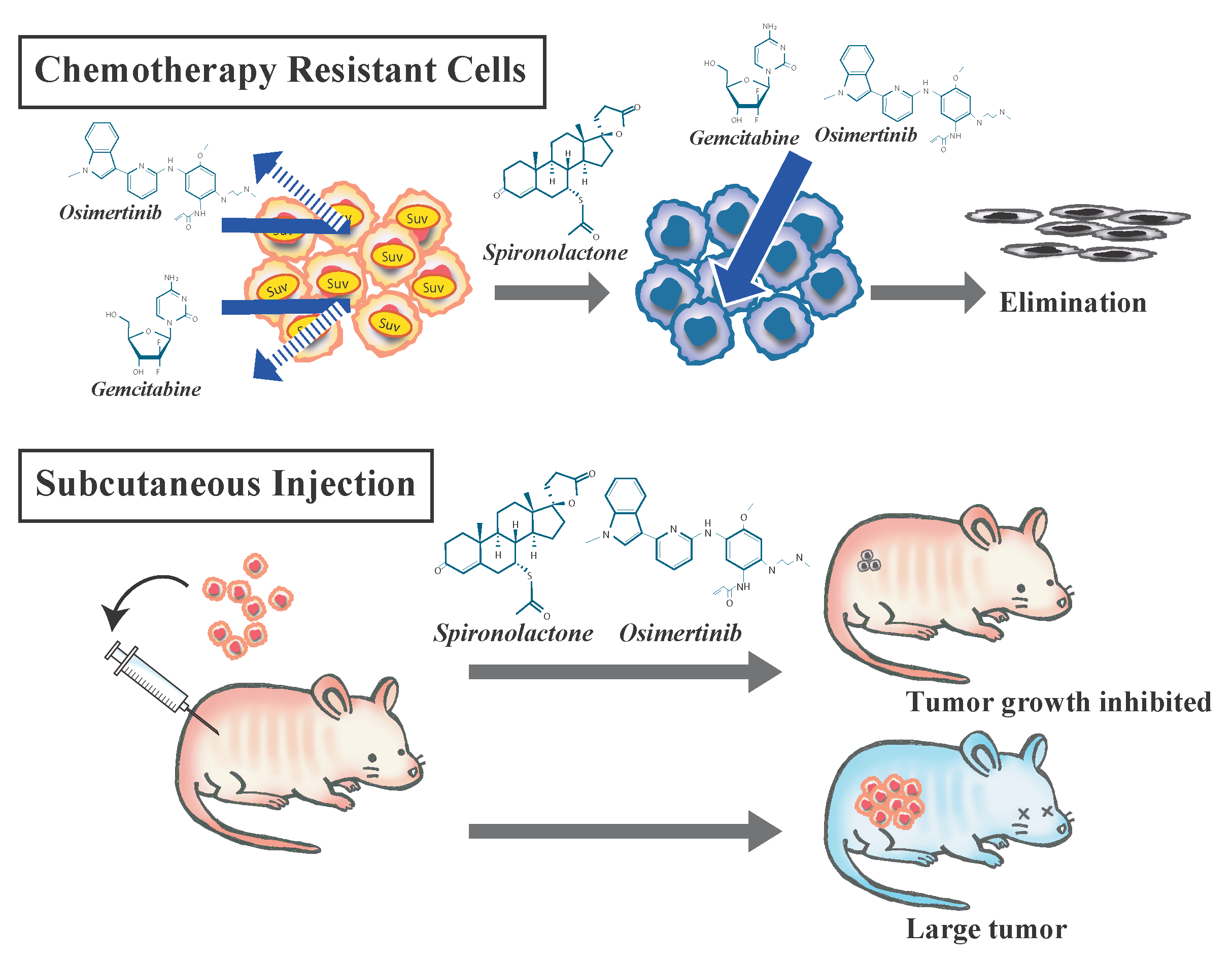{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
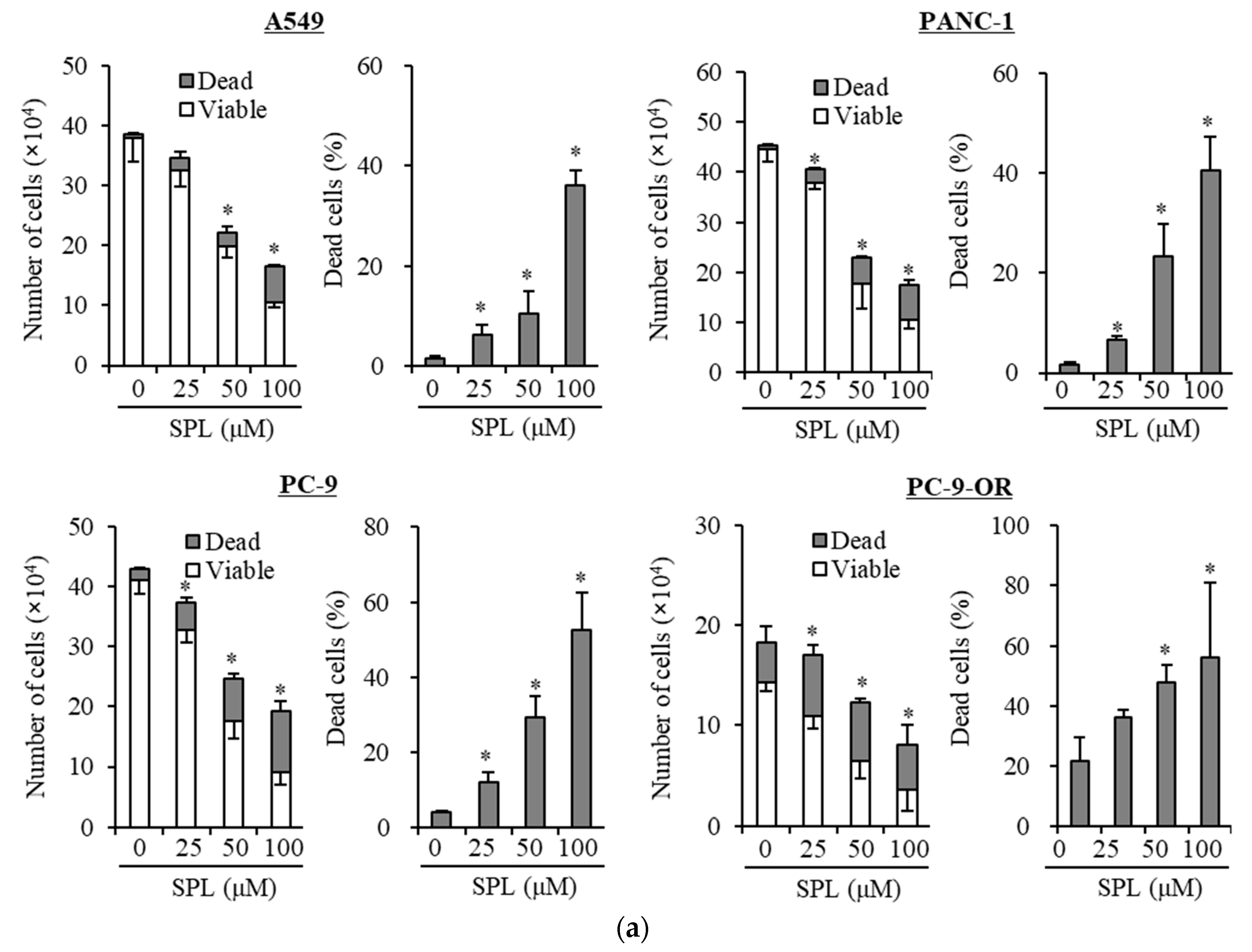
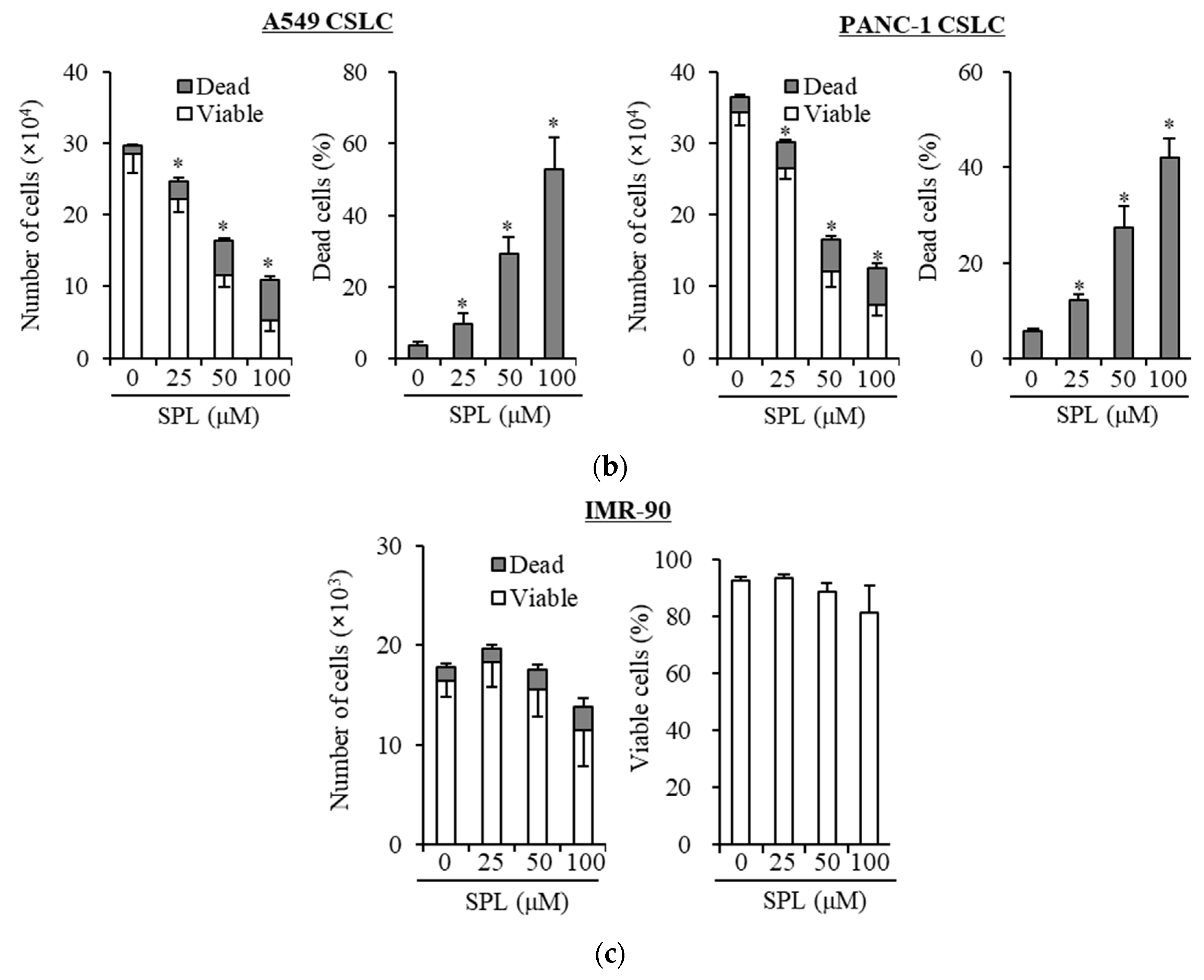
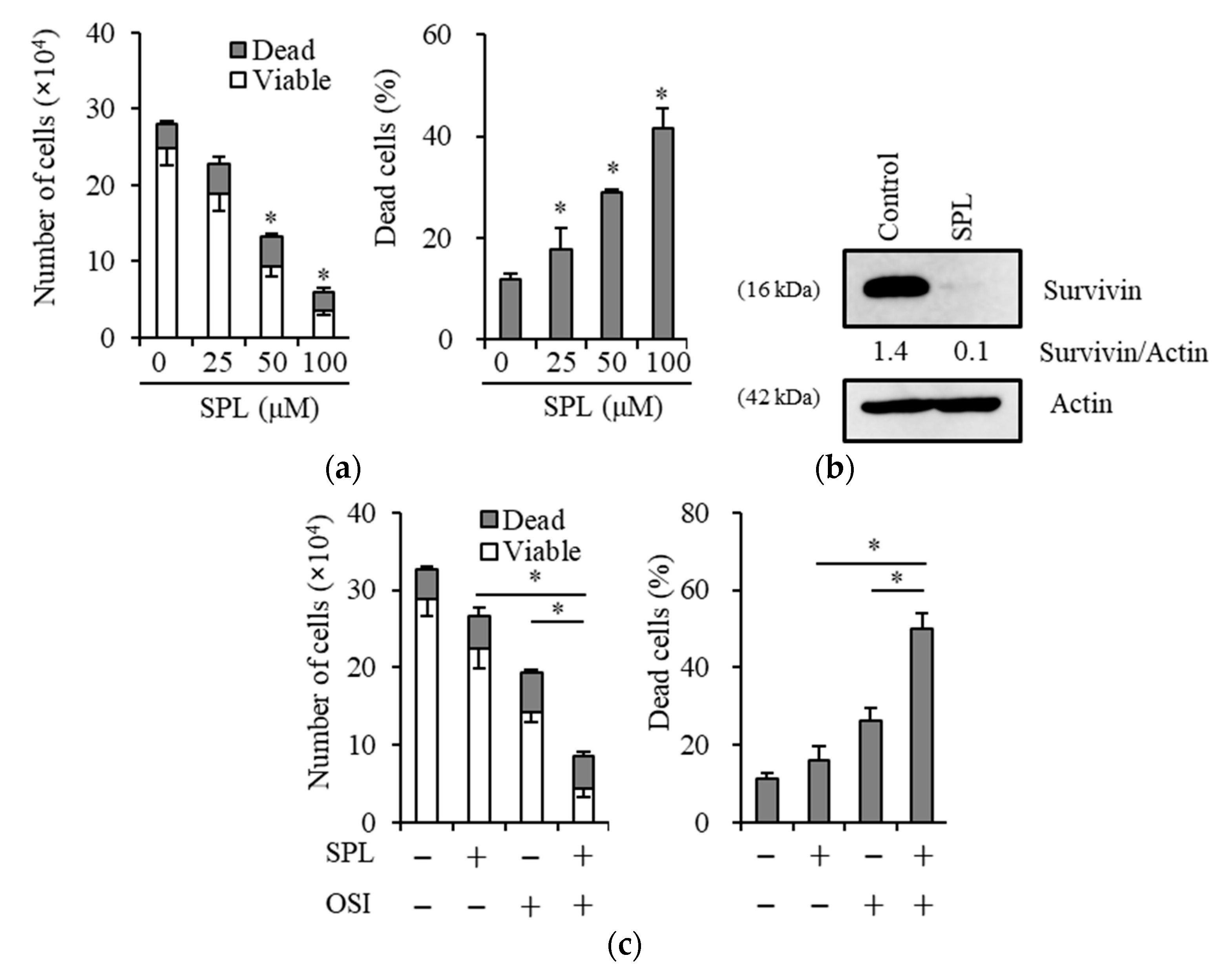
2.1. Spironolactone Inhibits Cancer Cell Growth and Is Cytotoxic to Cancer Cells, including CSCs, but not to Normal Cells
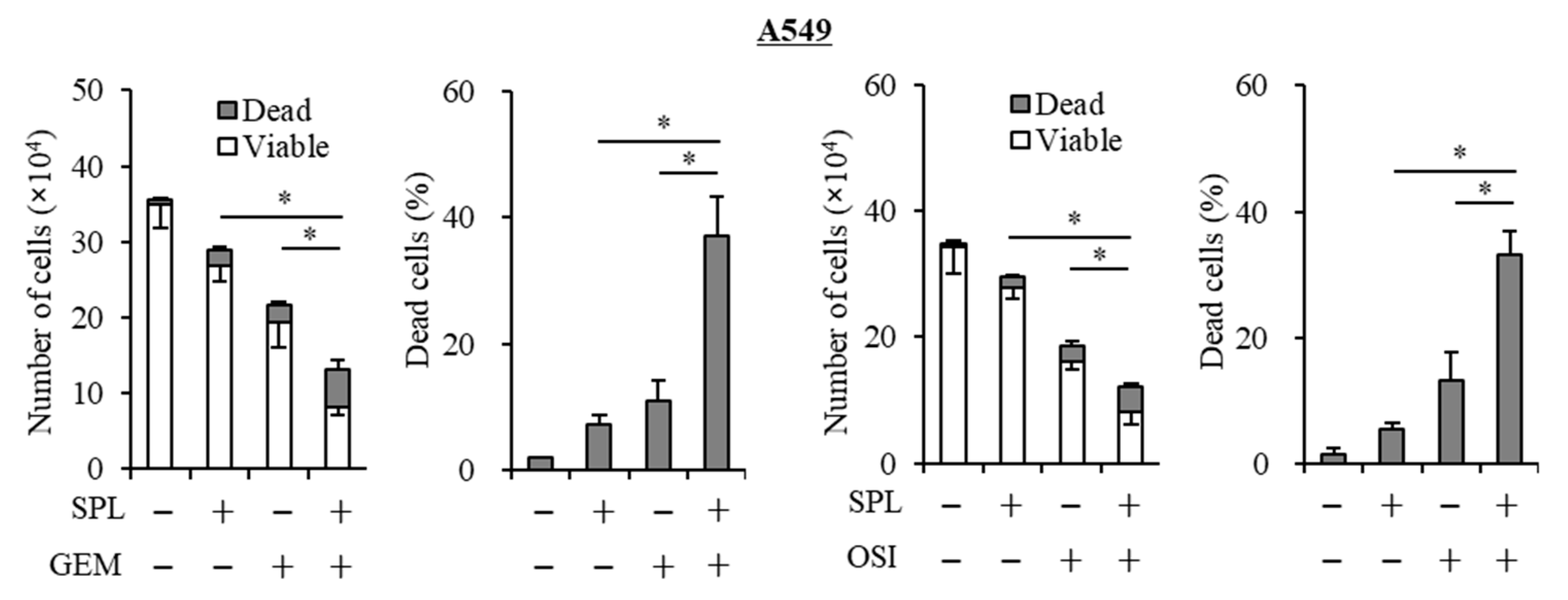
2.2. Spironolactone Decreases Resistance to Gemcitabine and Osimertinib
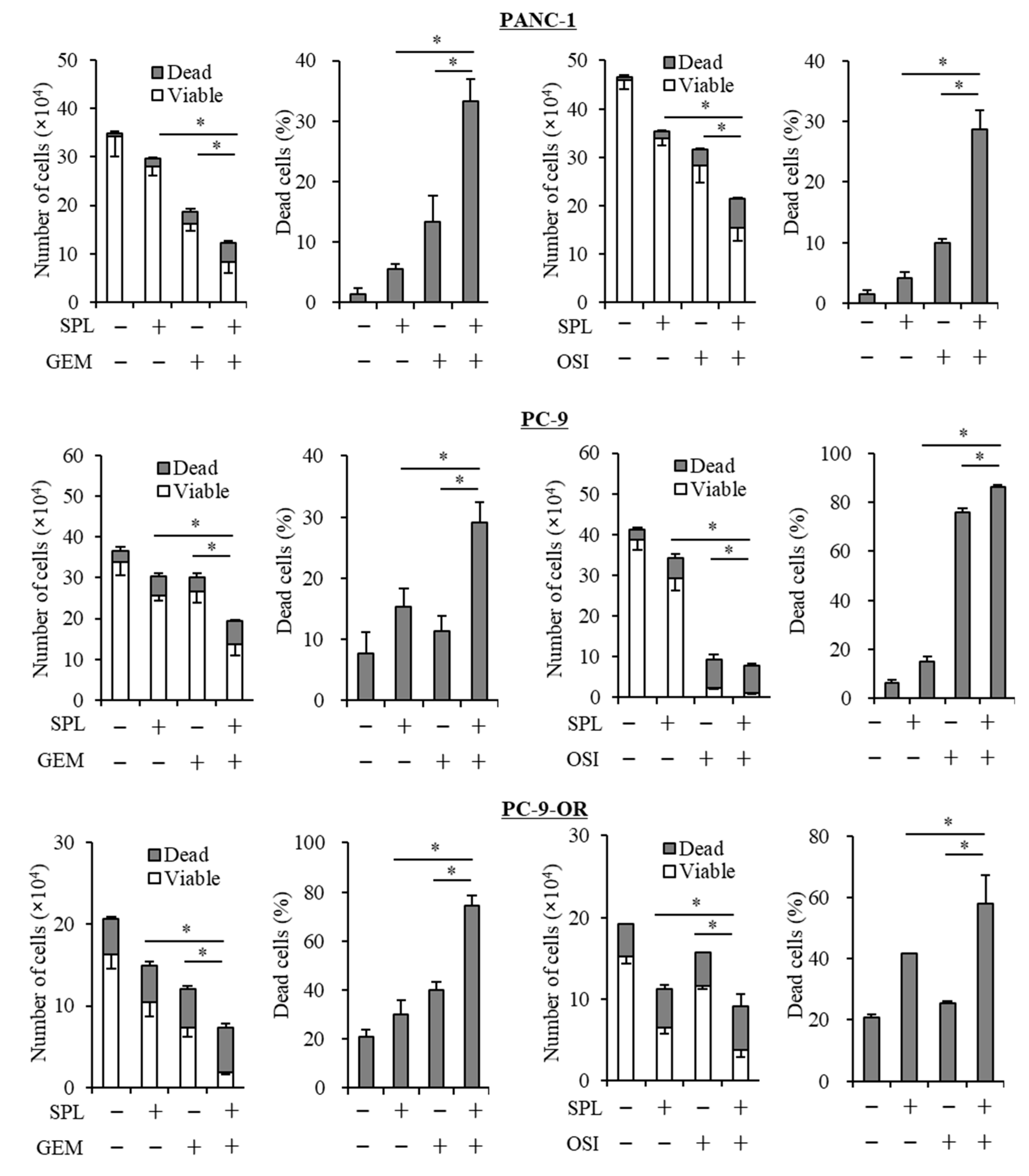
2.3. Spironolactone Reduces Resistance to Gemcitabine and Osimertinib in CSCs.
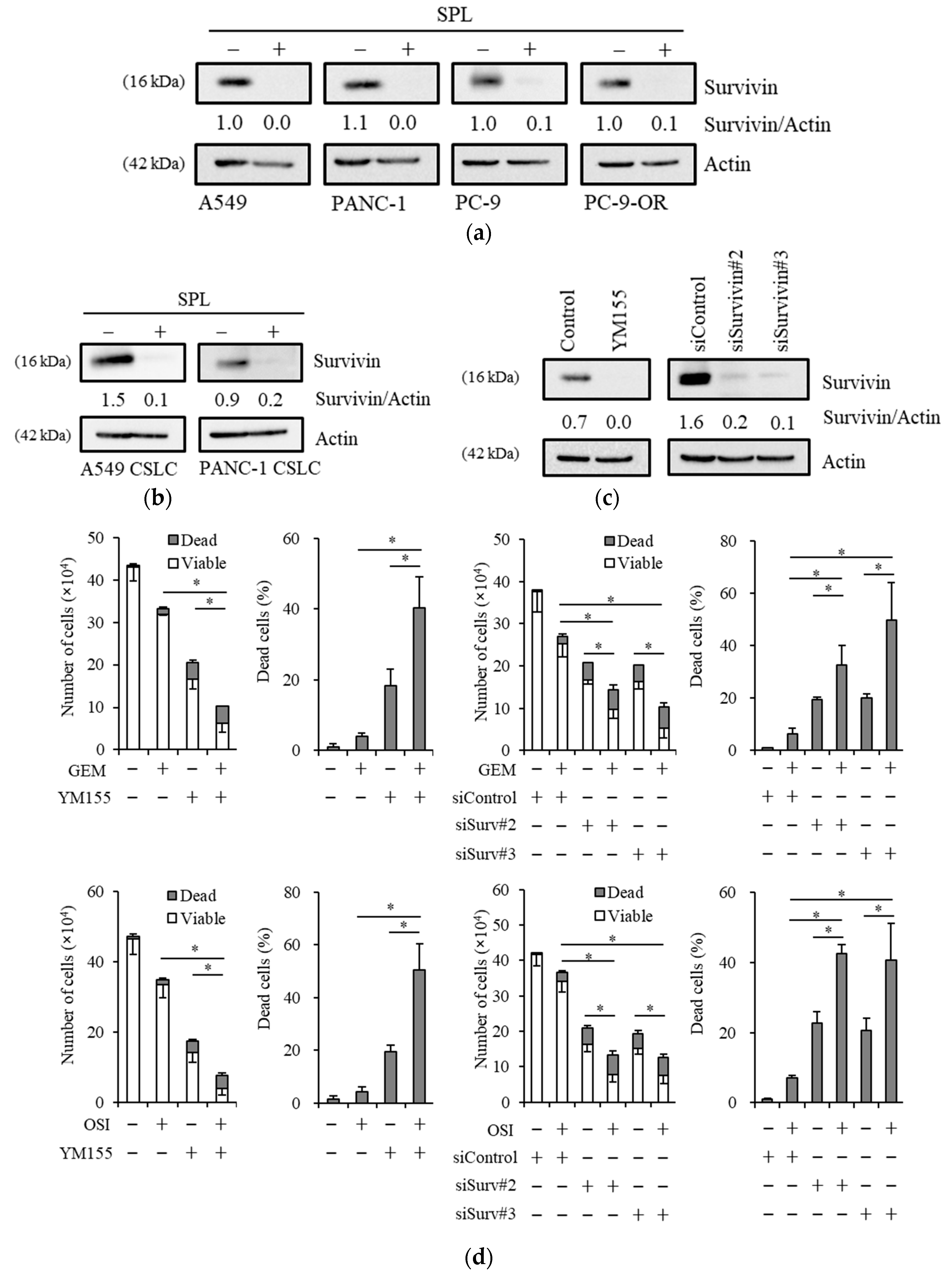
2.4. Reductions in Survivin Expression Are Involved in the Potency of Spironolactone to Decrease Resistance to Gemcitabine and Osimertinib
2.5. Spironolactone Exerts Anticancer, Chemosensitizing, and EGFR-TKI Sensitizing Effects with Survivin Reductions in Glioma Stem Cells
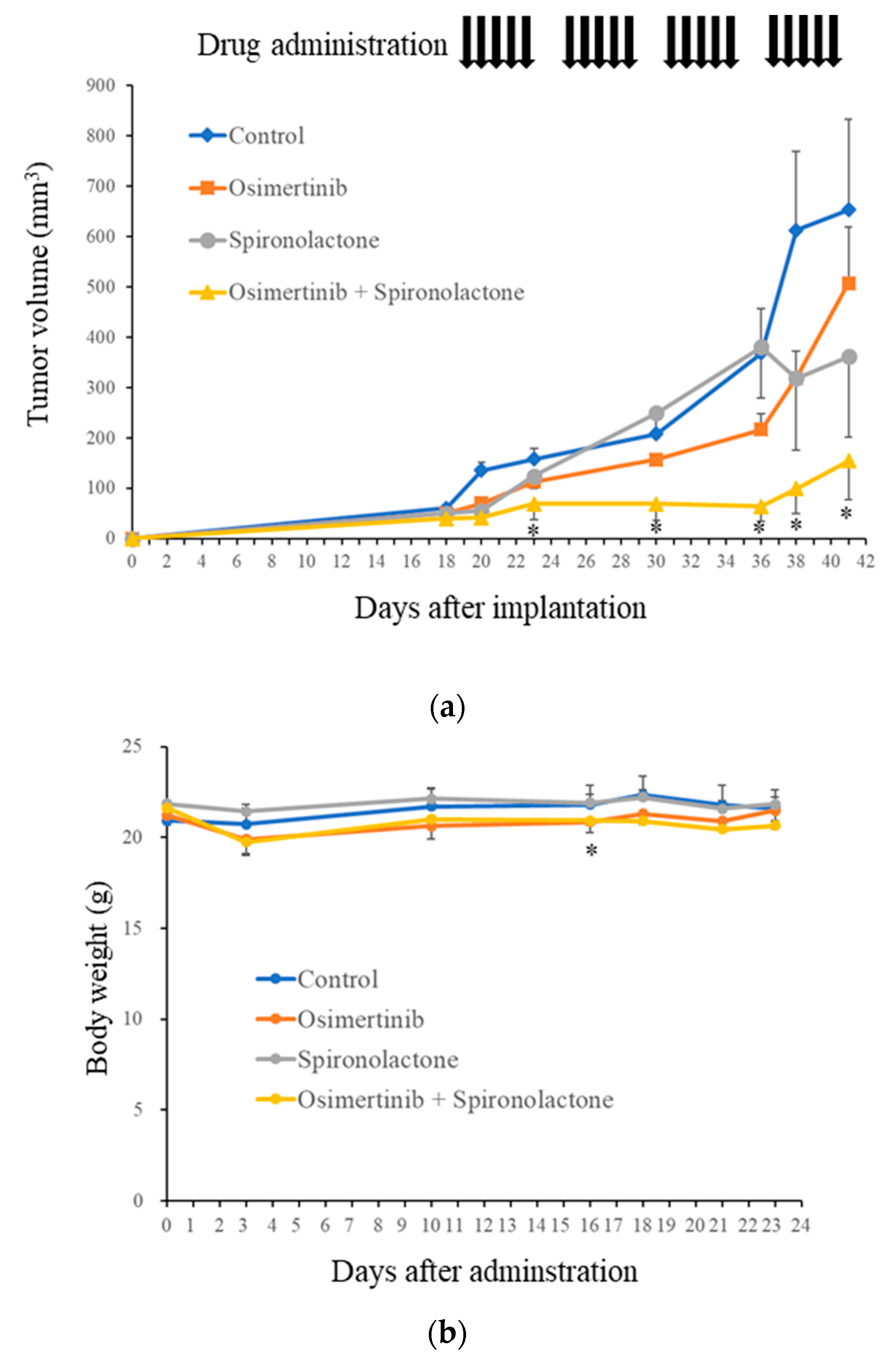
2.6. Spironolactone Augments Anti-Tumor Effects of Osimertinib In Vivo
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Cell Viability Assays
4.4. Gene Silencing with siRNA
4.5. Mouse Study
4.6. Immunoblot Analysis
4.7. RNA Extraction and Reverse Transcription-PCR
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar] [CrossRef] [PubMed]
- Kachalaki, S.; Ebrahimi, M.; Khosroshahi, L.M.; Mohammadinejad, S.; Baradaran, B. Cancer Chemoresistance; Biochemical and Molecular Aspects: A brief overview. Eur. J. Pharm. Sci. 2016, 89, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA. Available online: https://seer.cancer.gov/csr/1975_2016/ (accessed on 20 April 2019).
- Van Moorsel, C.J.; Peters, G.J.; Pinedo, H.M. Gemcitabine: Future Prospects of Single-Agent and Combination Studies. Oncologist 1997, 2, 127–134. [Google Scholar]
- Morabito, A.; Gebbia, V.; Di Maio, M.; Cinieri, S.; Vigano, M.G.; Bianco, R.; Barbera, S.; Cavanna, L.; De Marinis, F.; Montesarchio, V.; et al. Randomized phase III trial of gemcitabine and cisplatin vs. gemcitabine alone in patients with advanced non-small cell lung cancer and a performance status of 2: The CAPPA-2 study. Lung Cancer 2013, 81, 77–83. [Google Scholar] [CrossRef]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef]
- Dyawanapelly, S.; Kumar, A.; Chourasia, M.K. Lessons Learned from Gemcitabine: Impact of Therapeutic Carrier Systems and Gemcitabine’s Drug Conjugates on Cancer Therapy. Crit. Rev. Ther. Drug Carr. Syst. 2017, 34, 63–96. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Okusaka, T.; Furuse, J.; Funakoshi, A.; Ioka, T.; Yamao, K.; Ohkawa, S.; Boku, N.; Komatsu, Y.; Nakamori, S.; Iguchi, H.; et al. Phase II study of erlotinib plus gemcitabine in Japanese patients with unresectable pancreatic cancer. Cancer Sci. 2011, 102, 425–431. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Yuan, J.Q.; Di, M.Y.; Zheng, D.Y.; Chen, J.Z.; Ding, H.; Wu, X.Y.; Huang, Y.F.; Mao, C.; Tang, J.L. Gemcitabine plus erlotinib for advanced pancreatic cancer: A systematic review with meta-analysis. PLoS ONE 2013, 8, e57528. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef]
- Van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Murtuza, A.; Bulbul, A.; Shen, J.P.; Keshavarzian, P.; Woodward, B.D.; Lopez-Diaz, F.J.; Lippman, S.M.; Husain, H. Novel Third-Generation EGFR Tyrosine Kinase Inhibitors and Strategies to Overcome Therapeutic Resistance in Lung Cancer. Cancer Res. 2019, 79, 689–698. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.H. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef]
- Wu, M.; Yu, Q.; Li, Q. Differences in reproductive toxicology between alopecia drugs: An analysis on adverse events among female and male cases. Oncotarget 2016, 7, 82074–82084. [Google Scholar] [CrossRef]
- Schwab, C.; Jagannath, S. The role of thalidomide in multiple myeloma. Clin. Lymphoma Myeloma 2006, 7, 26–29. [Google Scholar] [CrossRef]
- Eisen, H.J.; Tuzcu, E.M.; Dorent, R.; Kobashigawa, J.; Mancini, D.; Valantine-von Kaeppler, H.A.; Starling, R.C.; Sorensen, K.; Hummel, M.; Lind, J.M.; et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N. Engl. J. Med. 2003, 349, 847–858. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grunwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Greenway, B.; Johnson, P.J.; Williams, R. Control of malignant ascites with spironolactone. Br. J. Surg. 1982, 69, 441–442. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Y.; Zheng, Y. Incidence and risk of hypertension with bevacizumab in non-small-cell lung cancer patients: A meta-analysis of randomized controlled trials. Drug Des. Dev. Ther. 2015, 9, 4751–4760. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, J.; Nie, W.; Huang, G.; Tang, J.; Guan, X. Risk of hypertension with regorafenib in cancer patients: A systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2014, 70, 225–231. [Google Scholar] [CrossRef]
- Esquenazi, Y.; Lo, V.P.; Lee, K. Critical Care Management of Cerebral Edema in Brain Tumors. J. Intensiv. Care Med. 2017, 32, 15–24. [Google Scholar] [CrossRef]
- Gold, A.; Eini, L.; Nissim-Rafinia, M.; Viner, R.; Ezer, S.; Erez, K.; Aqaqe, N.; Hanania, R.; Milyavsky, M.; Meshorer, E.; et al. Spironolactone inhibits the growth of cancer stem cells by impairing DNA damage response. Oncogene 2019, 38, 3103–3118. [Google Scholar] [CrossRef]
- Alekseev, S.; Ayadi, M.; Brino, L.; Egly, J.M.; Larsen, A.K.; Coin, F. A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem. Biol. 2014, 21, 398–407. [Google Scholar] [CrossRef]
- Shahar, O.D.; Kalousi, A.; Eini, L.; Fisher, B.; Weiss, A.; Darr, J.; Mazina, O.; Bramson, S.; Kupiec, M.; Eden, A.; et al. A high-throughput chemical screen with FDA approved drugs reveals that the antihypertensive drug Spironolactone impairs cancer cell survival by inhibiting homology directed repair. Nucleic Acids Res. 2014, 42, 5689–5701. [Google Scholar] [CrossRef]
- Carnero, A.; Garcia-Mayea, Y.; Mir, C.; Lorente, J.; Rubio, I.T.; LLeonart, M.L. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 2016, 49, 25–36. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. Brexpiprazole, a Serotonin-Dopamine Activity Modulator, Can Sensitize Glioma Stem Cells to Osimertinib, a Third-Generation EGFR-TKI, via Survivin Reduction. Cancers 2019, 11, 947. [Google Scholar] [CrossRef]
- Sanomachi, T.; Suzuki, S.; Togashi, K.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M.; Yamamoto, M. Brexpiprazole Reduces Survivin and Reverses EGFR Tyrosine Kinase Inhibitor Resistance in Lung and Pancreatic Cancer. Anticancer Res. 2019, 39, 4817–4828. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamamoto, M.; Togashi, K.; Sanomachi, T.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. In vitro and in vivo anti-tumor effects of brexpiprazole, a newly-developed serotonin-dopamine activity modulator with an improved safety profile. Oncotarget 2019, 10, 3547–3558. [Google Scholar] [CrossRef]
- Suzuki, S.; Okada, M.; Kuramoto, K.; Takeda, H.; Sakaki, H.; Watarai, H.; Sanomachi, T.; Seino, S.; Yoshioka, T.; Kitanaka, C. Aripiprazole, an Antipsychotic and Partial Dopamine Agonist, Inhibits Cancer Stem Cells and Reverses Chemoresistance. Anticancer Res. 2016, 36, 5153–5161. [Google Scholar] [CrossRef]
- Ciccolini, J.; Serdjebi, C.; Peters, G.J.; Giovannetti, E. Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: An EORTC-PAMM perspective. Cancer Chemother. Pharmacol. 2016, 78, 1–12. [Google Scholar] [CrossRef]
- De Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16. [Google Scholar] [CrossRef]
- Okamoto, K.; Okamoto, I.; Hatashita, E.; Kuwata, K.; Yamaguchi, H.; Kita, A.; Yamanaka, K.; Ono, M.; Nakagawa, K. Overcoming erlotinib resistance in EGFR mutation-positive non-small cell lung cancer cells by targeting survivin. Mol. Cancer Ther. 2012, 11, 204–213. [Google Scholar] [CrossRef]
- Okamoto, K.; Okamoto, I.; Okamoto, W.; Tanaka, K.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res. 2010, 70, 10402–10410. [Google Scholar] [CrossRef]
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006, 66, 10100–10111. [Google Scholar] [CrossRef]
- Suzuki, S.; Okada, M.; Takeda, H.; Kuramoto, K.; Sanomachi, T.; Togashi, K.; Seino, S.; Yamamoto, M.; Yoshioka, T.; Kitanaka, C. Involvement of GLUT1-mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. Oncotarget 2018, 9, 32667–32679. [Google Scholar] [CrossRef]
- Liu, Y.; Lear, T.; Iannone, O.; Shiva, S.; Corey, C.; Rajbhandari, S.; Jerome, J.; Chen, B.B.; Mallampalli, R.K. The Proapoptotic F-box Protein Fbxl7 Regulates Mitochondrial Function by Mediating the Ubiquitylation and Proteasomal Degradation of Survivin. J. Biol. Chem. 2015, 290, 11843–11852. [Google Scholar] [CrossRef]
- Li, Z.; Pei, X.H.; Yan, J.; Yan, F.; Cappell, K.M.; Whitehurst, A.W.; Xiong, Y. CUL9 mediates the functions of the 3M complex and ubiquitylates survivin to maintain genome integrity. Mol. Cell 2014, 54, 805–819. [Google Scholar] [CrossRef]
- Chen, H.; Yang, F.; Li, X.; Gong, Z.J.; Wang, L.W. Long noncoding RNA LNC473 inhibits the ubiquitination of survivin via association with USP9X and enhances cell proliferation and invasion in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2018, 499, 702–710. [Google Scholar] [CrossRef]
- Delyani, J.A.; Rocha, R.; Cook, C.S.; Tobert, D.S.; Levin, S.; Roniker, B.; Workman, D.L.; Sing, Y.L.; Whelihan, B. Eplerenone: A selective aldosterone receptor antagonist (SARA). Cardiovasc. Drug Rev. 2001, 19, 185–200. [Google Scholar] [CrossRef]
- Fagart, J.; Hillisch, A.; Huyet, J.; Barfacker, L.; Fay, M.; Pleiss, U.; Pook, E.; Schafer, S.; Rafestin-Oblin, M.E.; Kolkhof, P. A new mode of mineralocorticoid receptor antagonism by a potent and selective nonsteroidal molecule. J. Biol. Chem. 2010, 285, 29932–29940. [Google Scholar] [CrossRef]
- Kolkhof, P.; Barfacker, L. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Mineralocorticoid receptor antagonists: 60 years of research and development. J. Endocrinol. 2017, 234, T125–T140. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Okada, M.; Shibuya, K.; Sato, A.; Seino, S.; Watanabe, E.; Suzuki, S.; Seino, M.; Kitanaka, C. Specific role of JNK in the maintenance of the tumor-initiating capacity of A549 human non-small cell lung cancer cells. Oncol. Rep. 2013, 30, 1957–1964. [Google Scholar] [CrossRef]
- Suzuki, S.; Okada, M.; Shibuya, K.; Seino, M.; Sato, A.; Takeda, H.; Seino, S.; Yoshioka, T.; Kitanaka, C. JNK suppression of chemotherapeutic agents-induced ROS confers chemoresistance on pancreatic cancer stem cells. Oncotarget 2015, 6, 458–470. [Google Scholar] [CrossRef]
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [PubMed]
- Sanomachi, T.; Suzuki, S.; Kuramoto, K.; Takeda, H.; Sakaki, H.; Togashi, K.; Seino, S.; Yoshioka, T.; Okada, M.; Kitanaka, C. Olanzapine, an Atypical Antipsychotic, Inhibits Survivin Expression and Sensitizes Cancer Cells to Chemotherapeutic Agents. Anticancer Res. 2017, 37, 6177–6188. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Okada, M.; Kuramoto, K.; Suzuki, S.; Sakaki, H.; Sanomachi, T.; Seino, S.; Yoshioka, T.; Hirano, H.; Arita, K.; et al. Antitumor activity of gemcitabine against high-grade meningioma in vitro and in vivo. Oncotarget 2017, 8, 90996–91008. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanomachi, T.; Suzuki, S.; Togashi, K.; Sugai, A.; Seino, S.; Okada, M.; Yoshioka, T.; Kitanaka, C.; Yamamoto, M. Spironolactone, a Classic Potassium-Sparing Diuretic, Reduces Survivin Expression and Chemosensitizes Cancer Cells to Non-DNA-Damaging Anticancer Drugs. Cancers 2019, 11, 1550. https://doi.org/10.3390/cancers11101550
Sanomachi T, Suzuki S, Togashi K, Sugai A, Seino S, Okada M, Yoshioka T, Kitanaka C, Yamamoto M. Spironolactone, a Classic Potassium-Sparing Diuretic, Reduces Survivin Expression and Chemosensitizes Cancer Cells to Non-DNA-Damaging Anticancer Drugs. Cancers. 2019; 11(10):1550. https://doi.org/10.3390/cancers11101550
Chicago/Turabian StyleSanomachi, Tomomi, Shuhei Suzuki, Keita Togashi, Asuka Sugai, Shizuka Seino, Masashi Okada, Takashi Yoshioka, Chifumi Kitanaka, and Masahiro Yamamoto. 2019. "Spironolactone, a Classic Potassium-Sparing Diuretic, Reduces Survivin Expression and Chemosensitizes Cancer Cells to Non-DNA-Damaging Anticancer Drugs" Cancers 11, no. 10: 1550. https://doi.org/10.3390/cancers11101550
APA StyleSanomachi, T., Suzuki, S., Togashi, K., Sugai, A., Seino, S., Okada, M., Yoshioka, T., Kitanaka, C., & Yamamoto, M. (2019). Spironolactone, a Classic Potassium-Sparing Diuretic, Reduces Survivin Expression and Chemosensitizes Cancer Cells to Non-DNA-Damaging Anticancer Drugs. Cancers, 11(10), 1550. https://doi.org/10.3390/cancers11101550