Protecting Tumors by Preventing Human Papilloma Virus Antigen Presentation: Insights from Emerging Bioinformatics Algorithms


Abstract
1. Introduction
2. Normal MHC Class I Activity
3. Neoantigen Identification Using Bioinformatics Methods
4. Neoantigens in HNSCC
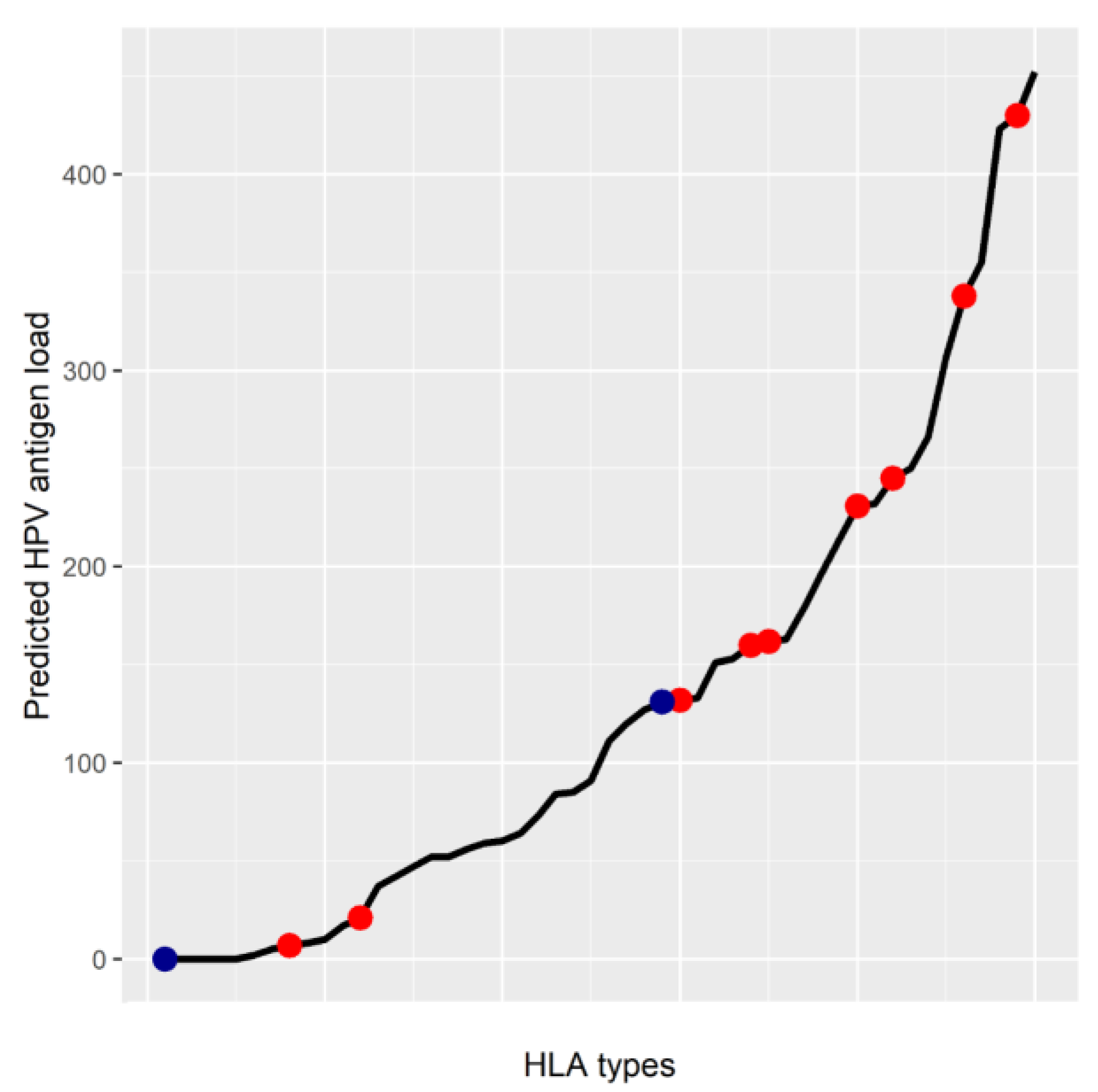
5. MHC Class I Genotype May Affect HPV Antigen Presentation
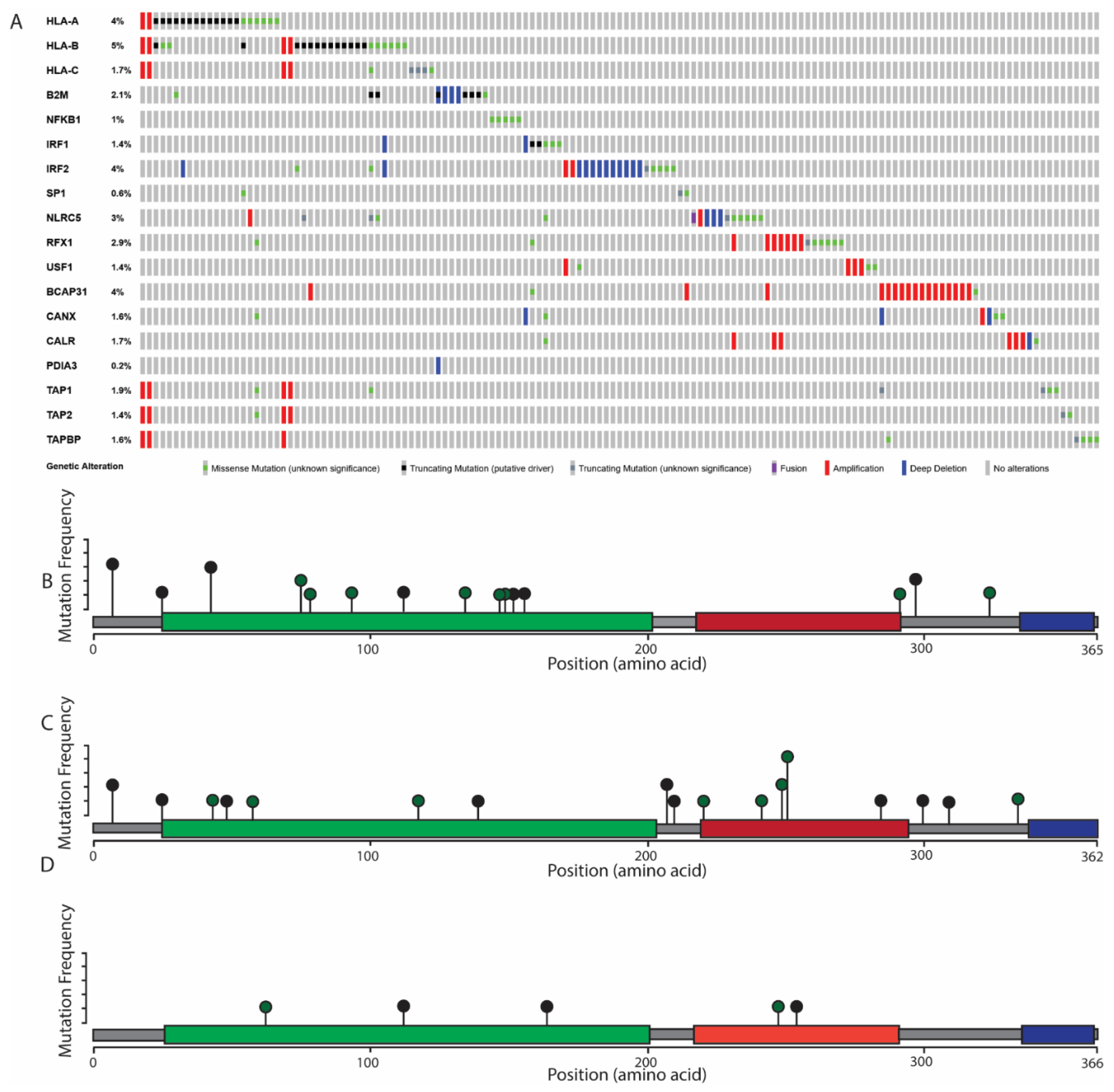
6. The Role of MHC Class I Changes in HNSCC
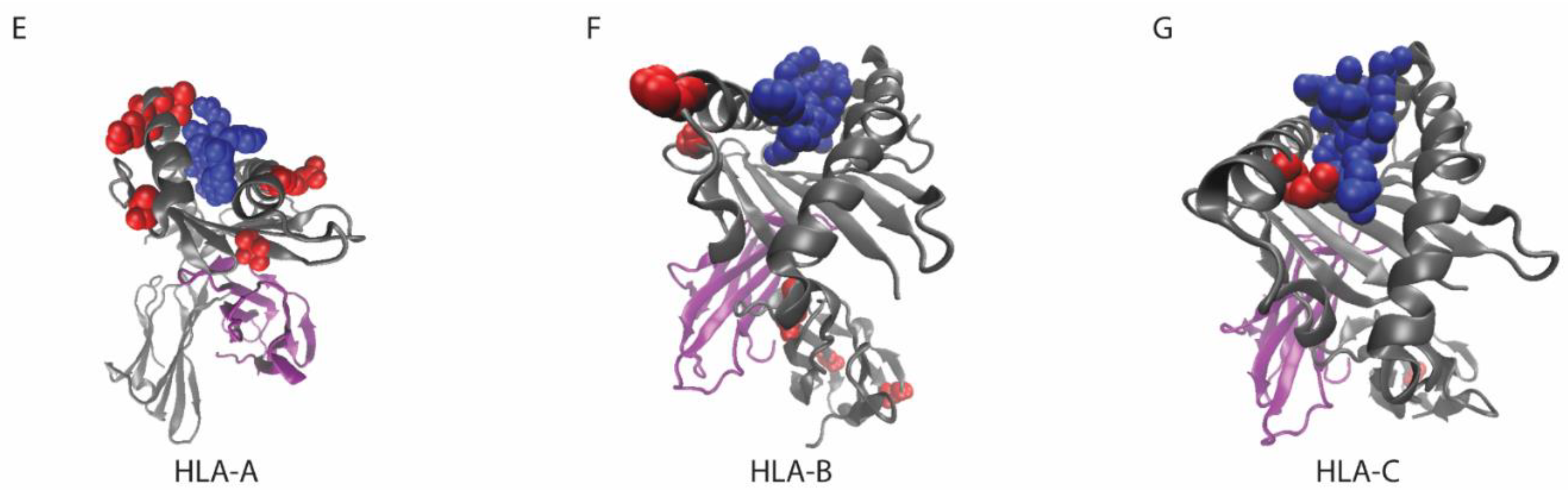
7. Considerations for MHC Class I Mutations
8. HPV E7 Inhibits MHC Class I Gene Expression
9. The Next Steps of HPV-Targeting Therapies
Funding
Conflicts of Interest
Abbreviations
| HLA | human leukocyte antigen. |
| B2M | β-2 microglobulin. |
| NFKB | nuclear factor κ b. |
| IRF | interferon regulatory factor. |
| SP | specificity protein. |
| NLRC | NLR Family CARD Domain Containing. |
| RFX | Regulator factor X. |
| USF | upstream transcription factor. |
| BCAP | B cell receptor associated protein. |
| CANX | calnexin. |
| CALR | calreticulin. |
| PDIA | Protein Disulfide Isomerase Family A. |
| TAP | Transporter associated with antigen processing. |
| TAPBP | Tapasin. Mutation data retrieved from the TCGA PanCancer Atlas. |
References
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Michmerhuizen, N.L.; Birkeland, A.C.; Bradford, C.R.; Brenner, J.C. Genetic determinants in head and neck squamous cell carcinoma and their influence on global personalized medicine. Genes Cancer 2016, 7, 182–200. [Google Scholar] [PubMed]
- Gingerich, M.A.; Smith, J.D.; Michmerhuizen, N.L.; Ludwig, M.; Devenport, S.; Matovina, C.; Brenner, C.; Chinn, S.B. Comprehensive review of genetic factors contributing to head and neck squamous cell carcinoma development in low-risk, nontraditional patients. Head Neck 2018, 40, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Tillman, B.N.; Yanik, M.; Birkeland, A.C.; Liu, C.-J.; Hovelson, D.H.; Cani, A.K.; Palanisamy, N.; Carskadon, S.; Carey, T.E.; Bradford, C.R.; et al. Fibroblast growth factor family aberrations as a putative driver of head and neck squamous cell carcinoma in an epidemiologically low-risk patient as defined by targeted sequencing. Head Neck 2016, 38, E1646–E1652. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef]
- D’Souza, G.; Fakhry, C.; Gillison, M.L.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Koch, W.M.; Westra, W.H. Case–Control Study of Human Papillomavirus and Oropharyngeal Cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef]
- Braaten, K.P.; Laufer, M.R. Human Papillomavirus (HPV), HPV-Related Disease, and the HPV Vaccine. Rev. Obstet. Gynecol. 2008, 1, 2–10. [Google Scholar]
- Nulton, T.J.; Olex, A.L.; Dozmorov, M.; Morgan, I.M.; Windle, B. Analysis of The Cancer Genome Atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 17684–17699. [Google Scholar] [CrossRef]
- Louvanto, K.; Rautava, J.; Willberg, J.; Wideman, L.; Syrjanen, K.; Grenman, S.; Syrjanen, S. Genotype-Specific Incidence and Clearance of Human Papillomavirus in Oral Mucosa of Women: A Six-Year Follow-Up Study. PLoS ONE 2013, 8, e53413. [Google Scholar] [CrossRef]
- Ou, D.; Adam, J.; Garberis, I.; Blanchard, P.; Nguyen, F.; Levy, A.; Casiraghi, O.; Gorphe, P.; Breuskin, I.; Janot, F.; et al. Influence of tumor-associated macrophages and HLA class I expression according to HPV status in head and neck cancer patients receiving chemo/bioradiotherapy. Radiother. Oncol. 2019, 130, 89–96. [Google Scholar] [CrossRef]
- Campo, M.; Graham, S.; Cortese, M.; Ashrafi, G.; Araibi, E.; Dornan, E.; Miners, K.; Nunes, C.; Man, S.; Graham, S. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 2010, 407, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Bottley, G.; Watherston, O.G.; Hiew, Y.L.; Norrild, B.; Cook, G.P.; Blair, G.E. High-risk human papillomavirus E7 expression reduces cell-surface MHC class I molecules and increases susceptibility to natural killer cells. Oncogene 2008, 27, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Partlová, S.; Boucek, J.; Kloudova, K.; Lukesova, E.; Zabrodsky, M.; Grega, M.; Fučíková, J.; Truxova, I.; Tachezy, R.; Spisek, R.; et al. Distinct patterns of intratumoral immune cell infiltrates in patients with HPV-associated compared to non-virally induced head and neck squamous cell carcinoma. OncoImmunology 2015, 4, e965570. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Ulrich, P.; Wilson, E.; Parikh, F.; Narang, P.; Yang, S.; Read, A.K.; Kim-Schulze, S.; Park, J.G.; Posner, M.; et al. Human papilloma virus specific immunogenicity and dysfunction of CD8+ T cells in head and neck cancer. Cancer Res. 2018, 78, 6159–6170. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Angell, T.; Lechner, M.; Liebertz, D.; Correa, A.; Sinha, U.; Kokot, N.; Epstein, A. Immune cell infiltration patterns and survival in head and neck squamous cell carcinoma. Head Neck Oncol. 2013, 5, 24. [Google Scholar]
- Hoesli, R.; Birkeland, A.C.; Rosko, A.J.; Issa, M.; Chow, K.L.; Michmerhuizen, N.L.; Mann, J.E.; Chinn, S.B.; Shuman, A.G.; Prince, M.E.; et al. Proportion of CD4 and CD8 tumor infiltrating lymphocytes predicts survival in persistent/recurrent laryngeal squamous cell carcinoma. Oral Oncol. 2018, 77, 83–89. [Google Scholar] [CrossRef]
- Mann, J.E.; Smith, J.D.; Birkeland, A.C.; Bellile, E.; Swiecicki, P.; Mierzwa, M.; Chinn, S.B.; Shuman, A.G.; Malloy, K.M.; Casper, K.A.; et al. Analysis of tumor-infiltrating CD103 resident memory T-cell content in recurrent laryngeal squamous cell carcinoma. Cancer Immunol. Immunother. 2019, 68, 213–220. [Google Scholar] [CrossRef]
- Lechner, A.; Schlößer, H.A.; Thelen, M.; Wennhold, K.; Rothschild, S.I.; Gilles, R.; Quaas, A.; Siefer, O.G.; Huebbers, C.U.; Cukuroglu, E.; et al. Tumor-associated B cells and humoral immune response in head and neck squamous cell carcinoma. OncoImmunology 2019, 8, 1535293. [Google Scholar] [CrossRef]
- Adiko, A.C.; Babdor, J.; Gutiérrez-Martínez, E.; Guermonprez, P.; Saveanu, L. Intracellular Transport Routes for MHC I and Their Relevance for Antigen Cross-Presentation. Front. Immunol. 2015, 6, 335. [Google Scholar] [CrossRef]
- Doherty, P.C.; Zinkernagel, R.M. Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 1975, 256, 50–52. [Google Scholar] [CrossRef]
- Gruener, M.; Bravo, I.G.; Momburg, F.; Alonso, A.; Tomakidi, P. The E5 protein of the human papillomavirus type 16 down-regulates HLA-I surface expression in calnexin-expressing but not in calnexin-deficient cells. Virol. J. 2007, 4, 116. [Google Scholar] [CrossRef] [PubMed]
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumour–immune cell interactions. Nat. Rev. Genet. 2016, 17, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, S.; Pritchard, A.L. Identifying neoantigens for use in immunotherapy. Mamm. Genome 2018, 29, 714–730. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef]
- Lundegaard, C.; Hoof, I.; Lund, O.; Nielsen, M. State of the art and challenges in sequence based T-cell epitope prediction. Immunome Res. 2010, 6, S3. [Google Scholar] [CrossRef]
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.O.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J. Biomed. Inform. 2015, 53, 405–414. [Google Scholar] [CrossRef]
- Snyder, A.; Chan, T.A. Immunogenic peptide discovery in cancer genomes. Curr. Opin. Genet. Dev. 2015, 30, 7–16. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Kesmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef]
- Bhasin, M.; Raghava, G.P.S. Pcleavage: An SVM based method for prediction of constitutive proteasome and immunoproteasome cleavage sites in antigenic sequences. Nucleic Acids Res. 2005, 33, W202–W207. [Google Scholar] [CrossRef]
- Zhang, G.L.; Petrovsky, N.; Kwoh, C.K.; August, J.T.; Brusic, V. PRED (TAP): A system for prediction of peptide binding to the human transporter associated with antigen processing. Immunome Res. 2006, 2, 3. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trolle, T.; Nielsen, M. NetTepi: An integrated method for the prediction of T cell epitopes. Immunogenetics 2014, 66, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R. NeoPredPipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinform. 2019, 20, 264. [Google Scholar] [CrossRef] [PubMed]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef]
- Bais, P.; Namburi, S.; Gatti, D.M.; Zhang, X.; Chuang, J.H. CloudNeo: A cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics 2017, 33, 3110–3112. [Google Scholar] [CrossRef]
- Tappeiner, E.; Finotello, F.; Charoentong, P.; Mayer, C.; Rieder, D.; Trajanoski, Z. TIminer: NGS data mining pipeline for cancer immunology and immunotherapy. Bioinformatics 2017, 33, 3140–3141. [Google Scholar] [CrossRef]
- Wang, T.-Y.; Wang, L.; Alam, S.K.; Hoeppner, L.H.; Yang, R. ScanNeo: Identifying indel derived neoantigens using RNA-Seq data. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Zhang, J.; Mardis, E.R.; Maher, C.A. INTEGRATE-neo: A pipeline for personalized gene fusion neoantigen discovery. Bioinformatics 2017, 33, 555–557. [Google Scholar] [CrossRef]
- Chen, Y.P.; Wang, Y.Q.; Lv, J.W.; Li, Y.Q.; Chua, M.L.K.; Le, Q.T.; Lee, N.; Colevas, A.D.; Seiwert, T.; Hayes, D.N.; et al. Identification and validation of novel microenvironment-based immune molecular subgroups of head and neck squamous cell carcinoma: Implications for immunotherapy. Ann. Oncol. 2019, 30, 68–75. [Google Scholar] [CrossRef]
- Karosiene, E.; Lundegaard, C.; Lund, O.; Nielsen, M. NetMHCcons: A consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012, 64, 177–186. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Liu, Q.; Wei, J.; Liu, B. Personalized cancer neoantigen vaccines come of age. Theranostics 2018, 8, 4238–4246. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, A.C.; Yanik, M.; Tillman, B.N.; Scott, M.V.; Foltin, S.K.; Mann, J.E.; Michmerhuizen, N.L.; Ludwig, M.L.; Sandelski, M.M.; Komarck, C.M.; et al. Identification of Targetable ERBB2 Aberrations in Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol. Neck Surg. 2016, 142, 559–567. [Google Scholar] [CrossRef]
- Giefing, M.; Wierzbicka, M.; Szyfter, K.; Brenner, J.; Braakhuis, B.; Brakenhoff, R.; Bradford, C.; Sorensen, J.; Rinaldo, A.; Rodrigo, J.; et al. Moving towards personalised therapy in head and neck squamous cell carcinoma through analysis of next generation sequencing data. Eur. J. Cancer 2016, 55, 147–157. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Ren, L.; Leisegang, M.; Deng, B.; Matsuda, T.; Kiyotani, K.; Kato, T.; Harada, M.; Park, J.-H.; Saloura, V.; Seiwert, T.; et al. Identification of neoantigen-specific T cells and their targets: Implications for immunotherapy of head and neck squamous cell carcinoma. OncoImmunology 2019, 8, e1568813. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Davidson, E.J.; A Davidson, J.; Sterling, J.C.; Baldwin, P.J.W.; Kitchener, H.C.; Stern, P.L. Association between human leukocyte antigen polymorphism and human papillomavirus 16-positive vulval intraepithelial neoplasia in British women. Cancer Res. 2003, 63, 400–403. [Google Scholar]
- Qiu, X.; Zhang, F.; Chen, D.; Azad, A.K.; Zhang, L.; Yuan, Y.; Jiang, Z.; Liu, W.; Tan, Y.; Tao, N. HLA-B*07 is a high risk allele for familial cervical cancer. Asian Pac. J. Cancer Prev. 2011, 12, 2597–2600. [Google Scholar] [PubMed]
- Hildesheim, A.; Schiffman, M.; Scott, D.R.; Marti, D.; Kissner, T.; E Sherman, M.; Glass, A.G.; Manos, M.M.; Lorincz, A.T.; Kurman, R.J.; et al. Human leukocyte antigen class I/II alleles and development of human papillomavirus-related cervical neoplasia: Results from a case-control study conducted in the United States. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1035–1041. [Google Scholar]
- Vita, R.; Mahajan, S.; A Overton, J.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, K. A comprehensive review on host genetic susceptibility to human papillomavirus infection and progression to cervical cancer. Indian J. Hum. Genet. 2011, 17, 132–144. [Google Scholar] [CrossRef]
- Grandis, J.R.; Falkner, D.M.; Melhem, M.F.; E Gooding, W.; Drenning, S.D.; A Morel, P. Human leukocyte antigen class I allelic and haplotype loss in squamous cell carcinoma of the head and neck: Clinical and immunogenetic consequences. Clin. Cancer Res. 2000, 6, 2794–2802. [Google Scholar]
- Nasman, A.; Andersson, E.; Nordfors, C.; Grün, N.; Johansson, H.; Munck-Wikland, E.; Massucci, G.; Dalianis, T.; Ramqvist, T. MHC class I expression in HPV positive and negative tonsillar squamous cell carcinoma in correlation to clinical outcome. Int. J. Cancer 2013, 132, 72–81. [Google Scholar] [CrossRef]
- Girdlestone, J.; Isamat, M.; Gewert, D.; Milstein, C. Transcriptional regulation of HLA-A and -B: Differential binding of members of the Rel and IRF families of transcription factors. Proc. Natl. Acad. Sci. USA 1993, 90, 11568–11572. [Google Scholar] [CrossRef]
- Meissner, T.B.; Liu, Y.-J.; Lee, K.-H.; Li, A.; Biswas, A.; Van Eggermond, M.C.J.A.; van den Elsen, P.J.; Kobayashi, K.S. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J. Immunol. 2012, 188, 4951–4958. [Google Scholar] [CrossRef]
- Gobin, S.J.; Keijsers, V.; Van Zutphen, M.; van den Elsen, P.J. The role of enhancer A in the locus-specific transactivation of classical and nonclassical HLA class I genes by nuclear factor kappa B. J. Immunol. 1998, 161, 2276–2283. [Google Scholar]
- Ladasky, J.J.; Boyle, S.; Seth, M.; Li, H.; Pentcheva, T.; Abe, F.; Steinberg, S.J.; Edidin, M. Bap31 enhances the endoplasmic reticulum export and quality control of human class I MHC molecules. J. Immunol. 2006, 177, 6172–6181. [Google Scholar] [CrossRef]
- Hewitt, E.W. The MHC class I antigen presentation pathway: Strategies for viral immune evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef]
- Rizvi, S.M.; Del Cid, N.; Lybarger, L.; Raghavan, M. Distinct functions for the glycans of tapasin and heavy chains in the assembly of MHC class I molecules. J. Immunol. 2011, 186, 2309–2320. [Google Scholar] [CrossRef]
- Rizvi, S.M.; Raghavan, M. Mechanisms of function of tapasin, a critical major histocompatibility complex class I assembly factor. Traffic 2010, 11, 332–347. [Google Scholar] [CrossRef]
- Fairfax, B.P.; Makino, S.; Radhakrishnan, J.; Plant, K.; Leslie, S.; Dilthey, A.; Ellis, P.; Langford, C.; Vannberg, F.O.; Knight, J.C. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet. 2012, 44, 502–510. [Google Scholar] [CrossRef]
- Shukla, S.A.; Rooney, M.S.; Rajasagi, M.; Tiao, G.; Dixon, P.M.; Lawrence, M.S.; Stevens, J.; Lane, W.J.; DellaGatta, J.L.; Steelman, S.; et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 2015, 33, 1152–1158. [Google Scholar] [CrossRef]
- Ludwig, M.L.; Kulkarni, A.; Birkeland, A.C.; Michmerhuizen, N.L.; Foltin, S.K.; Mann, J.E.; Hoesli, R.C.; Devenport, S.N.; Jewell, B.M.; Shuman, A.G.; et al. The genomic landscape of UM-SCC oral cavity squamous cell carcinoma cell lines. Oral Oncol. 2018, 87, 144–151. [Google Scholar] [CrossRef]
- Mann, J.E.; Kulkarni, A.; Birkeland, A.C.; Kafelghazal, J.; Eisenberg, J.; Jewell, B.M.; Ludwig, M.L.; Spector, M.E.; Jiang, H.; Carey, T.E.; et al. The molecular landscape of the University of Michigan laryngeal squamous cell carcinoma cell line panel. Head Neck 2019, 41, 3114–3124. [Google Scholar] [CrossRef]
- Robinson, J.; Soormally, A.R.; Hayhurst, J.D.; Marsh, S.G. The IPD-IMGT/HLA Database—New developments in reporting HLA variation. Hum. Immunol. 2016, 77, 233–237. [Google Scholar] [CrossRef]
- Gensterblum-Miller, E.; Wu, W.; Sawalha, A.H.; Gensterblum, E. Novel Transcriptional Activity and Extensive Allelic Imbalance in the Human MHC Region. J. Immunol. 2018, 200, 1496–1503. [Google Scholar] [CrossRef]
- Hafner, N.; Driesch, C.; Gajda, M.; Jansen, L.; Kirchmayr, R.; Runnebaum, I.B.; Dürst, M. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts. Oncogene 2008, 27, 1610–1617. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- van Houten, V.M.; Snijders, P.J.; van den Brekel, M.W.; Kummer, J.A.; Meijer, C.J.; van Leeuwen, B.; Denkers, F.; Smeele, L.E.; Snow, G.B.; Brakenhoff, R.H. Biological evidence that human papillomaviruses are etiologically involved in a subgroup of head and neck squamous cell carcinomas. Int. J. Cancer 2001, 93, 232–235. [Google Scholar] [CrossRef]
- Dyson, N.; Guida, P.; Münger, K.; Harlow, E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol. 1992, 66, 6893–6902. [Google Scholar]
- Heller, C.; Weisser, T.; Mueller-Schickert, A.; Rufer, E.; Hoh, A.; Leonhardt, R.M.; Knittler, M.R. Identification of Key Amino Acid Residues That Determine the Ability of High Risk HPV16-E7 to Dysregulate Major Histocompatibility Complex Class I Expression. J. Boil. Chem. 2011, 286, 10983–10997. [Google Scholar] [CrossRef]
- Georgopoulos, N.T.; Proffitt, J.L.; Blair, G.E. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene 2000, 19, 4930–4935. [Google Scholar] [CrossRef]
- Li, H.; Zhan, T.; Li, C.; Liu, M.; Wang, Q.K. Repression of MHC class I transcription by HPV16E7 through interaction with a putative RXRbeta motif and NF-kappaB cytoplasmic sequestration. Biochem. Biophys. Res. Commun. 2009, 388, 383–388. [Google Scholar] [CrossRef]
- Šímová, J.; Polláková, V.; Indrová, M.; Mikyšková, R.; Bieblová, J.; Štěpánek, I.; Bubeník, J.; Reinis, M. Immunotherapy augments the effect of 5-azacytidine on HPV16-associated tumours with different MHC class I-expression status. Br. J. Cancer 2011, 105, 1533–1541. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Program Name | Input Data Type | Summary | Website |
|---|---|---|---|
| CloudNeo | WES or WGS | Integrates neoantigen peptide sequence calling, HLA typing, and peptide-MHC binding affinity predictions | https://github.com/TheJacksonLaboratory/CloudNeo |
| INTEGRATE-neo | RNA-seq | Integrates gene fusion identification | https://github.com/ChrisMaherLab/INTEGRATE-Neo |
| NeoPredPipe | variant call set | Integrates putative neoantigen peptide sequence identification and MHC binding affinity prediction | https://github.com/MathOnco/NeoPredPipe |
| NetChop | Peptide sequences | Predicts peptide cleavage sites | http://www.cbs.dtu.dk/services/NetChop |
| NetMHC | Peptide sequences, MHC haplotype | Predicts neoantigen binding affinity in an MHC type-dependent manner | http://www.cbs.dtu.dk/services/NetMHC |
| NetMHCcons | Peptide sequences, MHC DNA sequence | Predicts antigen binding affinities of rare MHC haplotypes | http://www.cbs.dtu.dk/services/NetMHCcons |
| NetMHCpan | Peptide sequences, MHC haplotype | Similar to NetMHC, with more MHC types included in training data | http://www.cbs.dtu.dk/services/NetMHCpan |
| NetTepi | Peptide sequences, MHC haplotype | Predicts neoantigen activity by combining peptide-MHC binding affinity and stability, and T cell propensity | http://www.cbs.dtu.dk/services/NetTepi |
| Pcleavage | Protein sequences | Predicts peptide cleavage sites | http://crdd.osdd.net/raghava/pcleavage |
| PRED(TAP) | Peptide sequences | Predicts peptide-TAP binding patterns | antigen.i2r.a-star.edu.sg/predTAP (currently unavailable) |
| pVAC-Seq | WES or WGS and RNA-seq | Combines variant calling and RNA-seq to identify transcribed putative antigens | https://github.com/griffithlab/pVAC-Seq |
| ScanNeo | RNA-seq | Neoantigen sequence prediction, optimized for indel mutations | https://github.com/ylab-hi/ScanNeo |
| TIminer | RNA-seq, somatic mutation calling | Integrates RNA-seq and somatic mutations to predict expressed neoantigens | https://icbi.i-med.ac.at/software/timiner/timiner.shtml |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gensterblum-Miller, E.; Brenner, J.C. Protecting Tumors by Preventing Human Papilloma Virus Antigen Presentation: Insights from Emerging Bioinformatics Algorithms. Cancers 2019, 11, 1543. https://doi.org/10.3390/cancers11101543
Gensterblum-Miller E, Brenner JC. Protecting Tumors by Preventing Human Papilloma Virus Antigen Presentation: Insights from Emerging Bioinformatics Algorithms. Cancers. 2019; 11(10):1543. https://doi.org/10.3390/cancers11101543
Chicago/Turabian StyleGensterblum-Miller, Elizabeth, and J. Chad Brenner. 2019. "Protecting Tumors by Preventing Human Papilloma Virus Antigen Presentation: Insights from Emerging Bioinformatics Algorithms" Cancers 11, no. 10: 1543. https://doi.org/10.3390/cancers11101543
APA StyleGensterblum-Miller, E., & Brenner, J. C. (2019). Protecting Tumors by Preventing Human Papilloma Virus Antigen Presentation: Insights from Emerging Bioinformatics Algorithms. Cancers, 11(10), 1543. https://doi.org/10.3390/cancers11101543