Abstract
FADD was initially described as an adaptor molecule for death receptor-mediated apoptosis, but subsequently it has been implicated in nonapoptotic cellular processes such as proliferation and cell cycle control. During the last decade, FADD has been shown to play a pivotal role in most of the signalosome complexes, such as the necroptosome and the inflammasome. Interestingly, various mechanisms involved in regulating FADD functions have been identified, essentially posttranslational modifications and secretion. All these aspects have been thoroughly addressed in previous reviews. However, FADD implication in cancer is complex, due to pleiotropic effects. It has been reported either as anti- or protumorigenic, depending on the cell type. Regulation of FADD expression in cancer is a complex issue since both overexpression and downregulation have been reported, but the mechanisms underlying such alterations have not been fully unveiled. Posttranslational modifications also constitute a relevant mechanism controlling FADD levels and functions in tumor cells. In this review, we aim to provide detailed, updated information on alterations leading to changes in FADD expression and function in cancer. The participation of FADD in various biological processes is recapitulated, with a mention of interesting novel functions recently proposed for FADD, such as regulation of gene expression and control of metabolic pathways. Finally, we gather all the available evidence regarding the clinical implications of FADD alterations in cancer, especially as it has been proposed as a potential biomarker with prognostic value.
1. Regulation of FADD Gene Expression
1.1. Genetic and Chromosomal Alterations Affecting FADD
The FADD gene is located on chromosome 11q13.3 in humans, and consists of two exons separated by a 2-kb intron and only one isoform. FADD is expressed in every adult and embryonic tissue in mice and humans [1,2] (Figure 1).
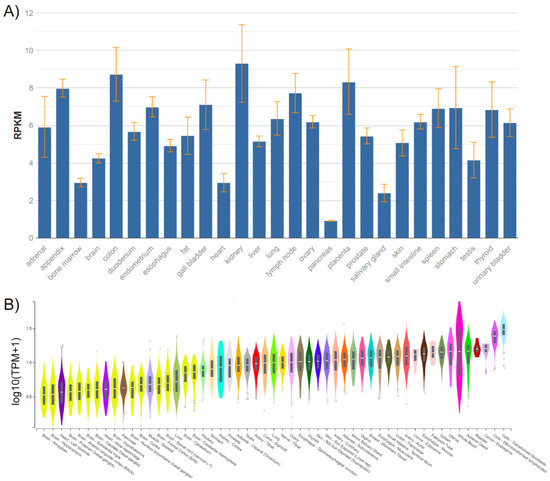
Figure 1.
FADD expression in healthy human tissues, performed by RNA sequencing. (A) Data obtained from BioProject PRJEB4337 of samples from 95 individuals representing 27 different healthy tissues [3]. RPKM, Reads Per Kilobase Million. (B) Data obtained from GTEx Analysis Release V7 (dbGaP Accession phs000424.v7.p2). Expression values are shown in transcripts per million (TPM), calculated from a gene model with isoforms collapsed to a single gene and no other normalization steps applied. Box plots are shown as median and 25th and 75th percentiles; points are displayed as outliers if they are above or below 1.5 times the interquartile range.
FADD expression is altered in many cancer types. This is, however, a controversial issue, since both overexpression [4,5,6,7,8,9,10] and downregulation [11,12,13,14,15] have been observed, depending on the cancer type. According to The Human Protein Atlas, FADD protein levels are low in most normal tissues. In cancer, FADD is detected in all tumor types analyzed by RNA sequencing according to The Cancer Genome Atlas (TCGA), and FADD protein levels obtained by immunohistochemistry with two different antibodies reveal results that are consistent with RNA-seq and/or protein/gene characterization data (Figure 2). A variety of FADD protein levels can be observed across the different tumor types; the mechanisms responsible for altered expression are, however, not always elucidated.
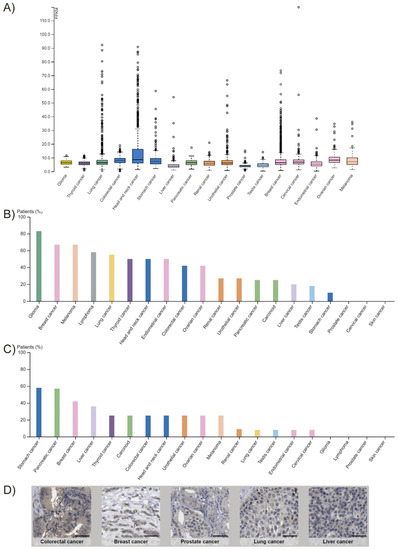
Figure 2.
FADD expression in cancer. (A) RNA-sequencing data from The Cancer Genome Atlas (TCGA) project of Genomic Data Commons (GDC). Seventeen cancer types representing 21 cancer subtypes with a corresponding major cancer type in The Human Pathology Atlas were included to allow for comparisons with the protein staining data from The Human Protein Atlas. The FPKMs (number fragments per kilobase of exon per million reads) were used for quantification of expression with a detection threshold of 1 FPKM. (B,C) FADD protein levels from The Human Protein Atlas. For each cancer, color-coded bars indicate the percentage of patients (maximum: 12) with high and medium protein expression level. Low or not detected protein expression results in a white bar. (B) Results obtained using HPA001464 antibody. Cases of colorectal, breast, ovarian, urothelial, gastric, pancreatic, and liver cancers, and melanomas showed weak to moderate cytoplasmic positivity. The remaining cancers were negative. (C) Results obtained using CAB010209 antibody. The majority of cancer cells showed weak to moderate cytoplasmic positivity. Nucleolar staining was observed in several cases. A few breast cancers were strongly stained. (D) Selection of five standard cancer tissue samples representative of the overall staining pattern. Scale bar: 50 µm.
A major event leading to overexpression is DNA amplification. Chromosome 11q13.3 is a 1.7 Mb-region that contains 12 other genes apart from FADD [7]. Amplification of this region has been frequently found in human cancers and is associated with poor prognosis. Among the candidate genes within the region, FADD has attracted interest since it was described as the only gene in the minimum region of overlap within the amplification in a series of squamous cell carcinomas of the head and neck (HNSCC) [16]. Furthermore, this group and others have demonstrated the association between FADD amplification and high FADD levels with poor overall survival and disease-free survival, especially if both events occur together [6,17,18]. This supports the notion that FADD is a driver of the tumorigenic effects of the 11q13.3 amplification. Based on the results from the recent TCGA PanCancer Atlas Studies (https://www.cbioportal.org/results/cancerTypesSummary?case_set_id=all&gene_list=FADD&cancer_study_list=5c8a7d55e4b046111fee2296), with 10,953 patients and 10,967 tumor samples from various cancer types, DNA amplification is by far the most frequent event affecting FADD and is most prominent in esophageal squamous cell carcinoma and HNSCC, with nearly 60% and 30% of cases harboring the amplification, respectively [19,20,21,22,23,24]. Other cancer types showing FADD-containing chromosomal amplification of 11q13 region are breast cancer [25,26,27], bladder cancer [28,29,30,31], lung squamous cell carcinoma [32], and, less frequently, ovarian cancer, where the region commonly involved is more distal [33,34,35].
Nevertheless, increased FADD levels do not always coexist with FADD gene amplification, as reported in acute myelogenous leukemia [36] or lung adenocarcinoma [5,37], suggesting the occurrence of additional mechanisms capable of modulating FADD levels in cancer.
Allelic losses or chromosomal deletions might account for FADD downregulation. However, the results compiled in databases (cBioPortal) point to this being a very infrequent event affecting FADD. For a long time, allelic losses within 11q13 have been observed in several cancer types, but the analyses have focused on a more proximal region of the chromosome [38,39,40,41,42]. An association between isolated familial somatotropinoma (IFS) and loss of heterozygosity (LOH) on a 2.21 Mb-region of chromosome 11q13 has been well established, although the driver gene has not been identified [43]. Deletions and LOH encompassing FADD gene within 11q13 have been found in 25% (9/36) of cervical cancer samples [44], although FADD was not identified as a driver of the potential LOH-derived tumorigenic effects. In a cohort of 60 cases of non-small-cell lung cancer (NSCLC), LOH at chromosome 11q13.3 was found in around 20% of the samples [45]. Hemizygosity affecting FADD has also been reported in ocular coloboma [46], a manifestation of otodental syndrome or, as more recently re-named, the chromosome 11q13 deletion syndrome [47].
Data from TCGA PanCancer Atlas Studies show a correlation between FADD expression, as determined by RNA sequencing, and copy number alterations (Figure 3).
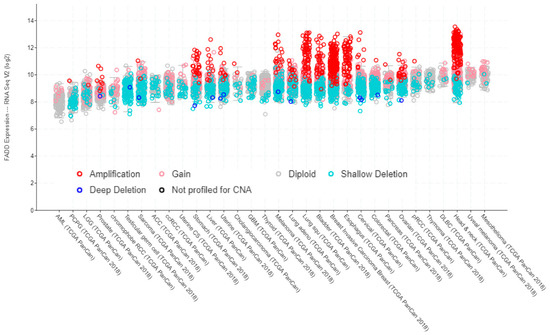
Figure 3.
FADD expression in human cancer, indicating the occurrence of copy number alterations. Screenshot image modified from cBioPortal. RNA-sequencing data are obtained from TCGA PanCancer Atlas Studies in 10,967 tumor samples from various origins.
Apart from cancer, allelic losses or gains involving FADD have been associated with developmental disabilities or congenital anomalies (specifically, global developmental delay for ClinVar Variation ID 153941; global developmental delay and abnormal heart morphology for ClinVar Variation ID 442080; abnormality of the ear, obsolete malformation of the heart and great vessels, short stature and polydactyly for ClinVar Variation ID 441904; and micrognathia, syndactyly, intrauterine growth retardation, ventricular septal defect and abnormal facial shape for ClinVar Variation ID 441903) [48].
Mutations in FADD might also account for altered expression. However, FADD mutations are not frequent in cancer. According to the COSMIC v89 (Catalogue of Somatic Mutations in Cancer) database, only 48 gene variants have been found in 56 out of 36187 (0.15%) tested tumor samples to date. Of them, 11 are synonymous and 37 affect the protein sequence either by nucleotide substitution or deletion. This concurs with the results gathered by the aforementioned TCGA PanCancer Atlas Studies, which showed 0.4% of somatic mutations in FADD and a total number of 47 different mutations, 41 of them affecting the protein sequence and six involving FADD in fusion proteins (FADD mutations, TCGA PanCancer Atlas, cBioPortal) (Figure 4A). The IntOGen-mutations platform (http://www.intogen.org/mutations/) identifies cancer drivers according to specific criteria [49]. To date, it has only compiled 12 FADD mutations, two of them synonymous (Figure 4B), and none of them is catalogued as a driver.
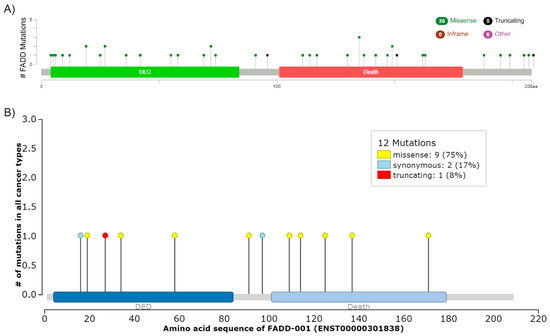
Figure 4.
FADD mutations. (A) Screenshot modified from cBioPortal. Mutation diagram circles are colored with respect to the corresponding mutation types. In case of different mutation types at a single position, the color of the circle is determined with respect to the most frequent mutation type. (B) Screenshot image modified from IntOGen. The mutations needle plot shows the distribution of the observed cancer mutations along the protein sequence and its possible mutational clusters and hotspots. The needles’ height and head size represent mutational recurrence. Needles of different categories that fall in the same amino acid residues are stacked.
Particular studies confirm that FADD mutation is indeed a rare event in cancer. In NSCLC, four missense mutations of FADD were detected in 80 samples (5%) [50], whereas only one FADD mutation was found in 98 colorectal adenocarcinomas (1%) and no FADD mutation was detected in 116 advanced gastric adenocarcinomas [51]. Moreover, no mutations in FADD were found either in 92 samples from diverse hematological malignancies [52], in 24 lung adenocarcinomas with high FADD expression [5], in 15 osteosarcoma tumor samples [53], or in murine (14 samples) [12] and human (22 samples) [13] T-cell lymphoblastic lymphoma (T-LBL).
Contrary to the correlation observed between FADD expression and copy number alterations (Figure 3), no specific relationship between FADD expression and mutations can be deduced according to the data from TCGA PanCancer Atlas Studies (Figure 5).
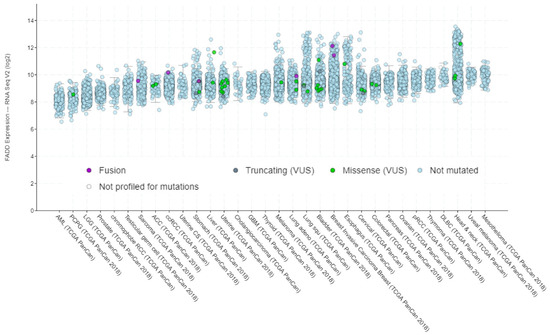
Figure 5.
FADD expression in human cancer, indicating the occurrence of mutations. Screenshot image modified from cBioPortal. RNA-sequencing data are obtained from TCGA PanCancer Atlas Studies in 10967 tumor samples from various origins.
Polymorphisms in FADD are numerous. To date, 816 variants have been described (Ensembl Release 97). However, only seven of them have had clinical consequences attributed, and these are not specifically related to any tumor type (Table 1) [54,55].

Table 1.
ClinVar search results for FADD [gene] and “single gene” [properties].
Among the 816 variants, the specific consequence of 135 of these variants is to affect the regulatory region of FADD, although none of them is catalogued in ClinVar due to reported clinical significance. Global minor allele frequency (MAF), defined in humans by the 1000 Genomes Project phase 3, is only available for 17 of them. According to this information, only two variants can be considered as common (MAF ≥ 5%), specifically rs12295430 (global MAF = 0.053) and rs41268205 (global MAF = 0.051). Interestingly, no publication has reported any functional impact associated with their occurrence. The remaining 15 are rare variants, with very low global MAF values (<0.001 for 13 of them). Among the 816 variants, 63 are reported as affecting transcription factor binding sites; only one of them (rs10898853) exhibits a minor allele frequency above rare variance (5%), specifically of 0.318. In particular, this variant has been associated with susceptibility to papillary thyroid cancer [56].
Polymorphisms reported in murine Fadd [57] did not show evident functionality. Regarding humans, the recent GTEx Project (GTEx Analysis Release V8, dbGaP Accession phs000424.v8.p2) identifies variations in gene expression that are highly correlated with genetic variation (the so-called eQTL, expression quantitative trait loci). To date, eighty-two functional polymorphisms predicted to influence tissue-specific FADD expression have been identified. However, according to the interpretation of the data, a limited number of variants are predicted to have an eQTL effect, specifically those with m-values > 0.9 (e.g., the three dots on the right in Figure 6).
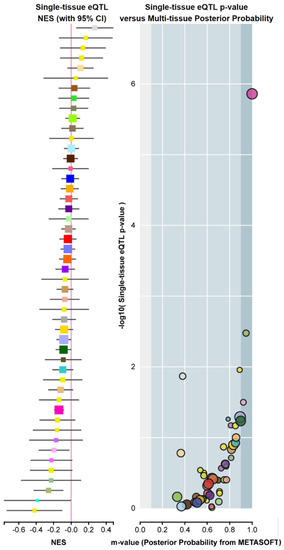
Figure 6.
Example of FADD expression quantitative trait loci (eQTLs), or FADD genetic variants exhibiting high correlation with changes in gene expression. Screenshot from GTEx Portal showing, for variant ID chr11_70241122_G_GCTT_b38, SNP rs559543475, single-tissue eQTL normalized effect size (NES) with 95% confidence interval (left) and Single-tissue eQTL p-value vs. Multi-tissue posterior probability (right). The Y axis indicates the −log10 of p-value (obtained from a t-test that compares observed beta from single-tissue eQTL analysis to a null beta of 0). The X axis indicates the m-value, which indicates the posterior probability that an eQTL effect exists in each tissue tested in the cross-tissue meta-analysis. The m-value ranges between 0 and 1 and is interpreted as follows: m-value < 0.1 indicates that the tissue is predicted to not have an eQTL effect; m-value > 0.9 indicates that the tissue is predicted to have an eQTL effect; otherwise, the prediction of the existence of an eQTL effect is ambiguous [58]. Normalized effect size (NES): the slope of the linear regression of normalized expression data versus the three genotype categories using single-tissue eQTL analysis, representing eQTL effect size. The normalized expression values are based on quantile normalization within each tissue, followed by inverse quantile normalization for each gene across samples. Colors represent distinct tissue categories.
In summary, FADD expression is frequently altered in cancer and its increase is most often related to DNA amplification. However, although allelic losses affecting FADD usually lead to decreased expression, the latter is not always explained by copy number alteration, nor is it by mutations or functional polymorphisms since these are infrequent.
1.2. Transcription Factors Affecting FADD
The 5’ terminus of FADD gene was first studied by Kim and colleagues [59], who reported a 1-kb region containing consensus sequences for regulatory elements such as Lyf-1, a reverse copy of the core element of the insulin enhancer (EI), AP-1, N-Myc and SP-1; a TATA-box was identified 620 bp upstream of the translation start site. Much more recently, the signals of DNase hypersensitivity together with histone modification H3K4me3 have been used to predict promoter-like regions by ENCODE 4 consortium. This method has been applied to 107 human cell types and 14 mouse cell types with both DNase and H3K4me3 data generated by the ENCODE and Roadmap Epigenomic consortia. Transcription factor binding sites within promoter-like regions have been proposed based on of ChIP-seq data, generating peaks for 161 transcription factors in 91 cell types [60]. Regarding FADD, these transcription factor binding sites have been identified in the promoter region (UCSC Genome Browser); however, very few publications have so far reported the role of specific transcription factors on regulating FADD expression. One of them demonstrates that HIF-1α binds to and represses the FADD promoter [61]. Since HIF-1α is overexpressed in many cancers and has been associated with tumor aggressiveness and poor prognosis [62], it could be speculated that FADD downregulation would be involved. Another study [63] reports the interaction with and activation of FADD promoter by BRCA1, suggesting that BRCA1 loss or inactivation in a tumor can cause reduced levels of FADD that in turn desensitize cells to apoptosis. Hence, the authors propose FADD as a candidate biomarker for BRCA1-associated breast cancer. In addition, the PAX2 and VAX2 transcription factors have been reported to co-regulate fadd transcriptional activation in zebrafish [64]. As mentioned before, LOH affecting FADD was reported in ocular coloboma [46]; since PAX2 mutations have been identified as one of the most common causes of coloboma [65], it could be speculated that reduced FADD expression—either due to LOH or to mutations in PAX2 causing regulation of transcription—would play a role in coloboma.
1.3. Epigenetic Regulation of FADD Expression
Epigenetic regulation of gene expression is without a doubt a relevant mechanism. Changes in promoter methylation, histone modification, or the action of microRNAs (miRNAs) have been reported with variable frequency as regulatory processes affecting FADD levels.
1.3.1. Methylation of FADD Promoter Region
DNA methylation in cytosines within CpG islands of gene promoters is a prominent mechanism of gene expression regulation, since hypermethylation is related to inactivation of transcription and the opposite is true for hypomethylation [66,67]. Evidence for such mechanism affecting FADD expression is very limited. To date, FADD reduction due to hypermethylation of its promoter has been reported in myelodysplastic syndrome [68] and inflammatory processes like apical periodontitis [69]. Regarding cancer, one study reported an association between significant hypermethylation of FADD promoter in oral squamous cell carcinoma and reduced FADD expression [70,71]. Therefore, further studies on whether FADD promoter hypermethylation is a frequent event leading to FADD reduction, particularly in cancer, where resistance to apoptosis is a hallmark, would be of interest. If confirmed, FADD promoter hypermethylation could be tested as a prognostic factor.
1.3.2. Histone Modification of FADD
Histones undergo reversible, dynamic and multiple modifications at their basic amino acids, such as methylation, acetylation and phosphorylation. These modifications constitute the histone code, which can be implicated in the regulation of gene expression among other functions. As they can change the structure of chromatin, some epigenetic modifications are associated with active transcription, such as acetylation, while others are related with repression of transcription [67,72]. During recent years, interesting bioinformatics tools for prediction of histone modification and related function have been developed [73]. An in silico search for predicted histones marks affecting FADD using CHIP-seq data deposited in ENCODE (SCREEN hg19) revealed 12 candidate cis-regulatory elements (ccREs) located between the first and last transcription start sites of FADD plus 50kb upstream (Figure 7) [74,75]. In addition to the four core epigenomic marks used to generate the registry (representative DNase hypersensitivity sites, H3K4me3, H3K27ac, and CTCF ChIP-seq signals) all available histone marks and transcription factor ChIP-seq peaks are annotated, identified using ENCODE uniform processing pipelines. Each of the 12 candidate elements for FADD shows significant signals for many different histone marks (up to 31 in the candidate with the highest scores), but these data are not supported by any experimental evidence in the literature. Moreover, scarce evidence of histone modifications directly affecting FADD expression is available. It has been proposed that the inhibitor of growth 1 (ING1) may contribute to regulate acetylation levels of different histones, thus promoting transcriptional activation of components of the apoptotic pathway, such as FADD [76]. The authors propose that ING1 downregulation in glioblastoma would result in FADD suppression, leading to apoptosis resistance in cancer cells upon treatment with histone deacetylase inhibitors (HDACi). The expression of Fadd was found to be suppressed in murine leukemia expressing the aberrant RUNX1-EVI1 generated by t(3;21) [77]. RUNX1-EVI1 recruits histone deacetylase, thus resulting in transcriptional dysregulation of wild-type RUNX1-target genes. Fadd bears binding sites for RUNX1, so RUNX1-EVI1-driven Fadd deacetylation is a plausible explanation for Fadd repression in these tumors. Interestingly, the authors demonstrate the restoration of Fadd expression upon treatment with HDACi, proposing that such inhibitors could be useful for leukemia patients expressing RUNX1-EVI1.
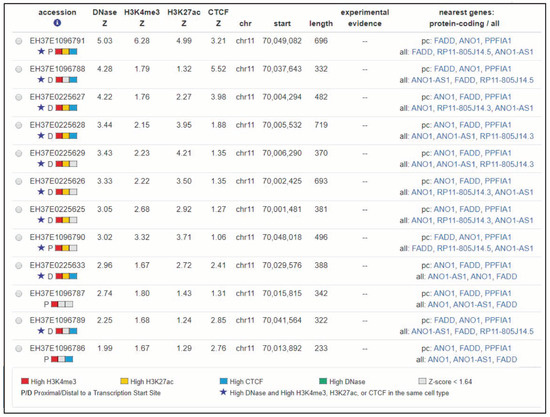
Figure 7.
Screenshot image from SCREEN hg19 (search candidate cis-regulatory elements by ENCODE). This search is showing candidate cis-regulatory elements located between the first and last transcription start sites (TSSs) of FADD and up to 50 kb upstream.
1.3.3. microRNAs Controlling FADD
Gene silencing by microRNAs is another prominent mechanism of regulation, combining translational repression and mRNA destabilization of target genes [78]. According to miRTarBase, there are 20 miRNAs that putatively target murine Fadd. Of them, only mmu-miR-134-5p and mmu-miR-155-5p have been reported with strong evidence [79,80,81]. In humans, miRTarBase only contains seven putative miRNAs targeting FADD. Again, only two of them have been proven with strong evidence, hsa-miR-155-5p and hsa-miR-128-3p [82,83,84]. Thus, miR-155-5p is the only microRNA strongly demonstrated to regulate FADD both in mice and humans. In mice, mmu-miR-155-5p is upregulated in response to lipopolysaccharide and subsequent silencing of Fadd promotes an anti-apoptotic effect [80]. A similar effect has been observed in RAW264.7 cells, where mmu-miR-155-5p upregulation leads to Fadd silencing and, in consequence, to reduced apoptosis [81]. In the human gastric epithelium and mucosa, FADD silencing due to hsa-miR-155-5p overexpression induced upon Helicobacter pylori infection promotes the activation of an inflammatory response [83]. In contrast, hsa-miR-155-5p is downregulated in degenerative nucleus pulposus, leading to FADD increase, which promotes high levels of apoptosis and eventually intervertebral disc degeneration [82]. Its alterations in cancer are reviewed in [85]: miR-155 is upregulated in most hematological malignancies, such as B cell lymphomas, acute myelomonocytic leukemia, and acute monocytic leukemia, as well as in solid tumors, such as breast, colon, and lung cancers. However, a direct link between miR-155 overexpression and FADD downregulation in those tumor samples has not been demonstrated. Interestingly, the level of miR-155 expression can be used to distinguish between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma, so its convenience for diagnosis has been suggested.
2. Posttranslational Modifications of FADD Protein
The discrepancy between genetic alterations affecting FADD and protein levels suggests that the latter would be regulated by mechanisms affecting the stability of the protein, such as posttranslational modifications (PTMs) [6]. For example, it was demonstrated that transcription was not altered in samples from acute myelogenous leukemia exhibiting reduced FADD protein [36]. Previously, the same authors had proposed in the adenomatous or adenocarcinomatous mouse thyroid gland a non-proteasome mediated loss of FADD protein [15].
Different PTMs have been reported to affect the functions of FADD, but the most important and well-studied is phosphorylation [86,87,88]. Several phosphorylation sites have been identified in FADD [89,90,91], but the most relevant site conserved between mice and humans is serine 191 and serine 194, respectively [90]. The impact of FADD phosphorylation on apoptosis is controversial [5,90,92,93]. Our data indicate that the phosphorylation status of FADD does not affect apoptosis [13], but FADD phosphorylation could affect apoptosis indirectly, as a consequence of protein availability in the cell. It has been suggested that phosphorylated FADD normally resides in the nucleus and that activation with Fas or TNFα/ActD reduces its phosphorylation and redistributes unphosphorylated FADD to the cytoplasm, contributing to the efficient propagation of cell death signaling [94,95,96,97]. Phosphorylation in this serine has been reported to play a very important role in regulating cell growth and proliferation [37,88,90,98,99]. Moreover, the deregulation of FADD phosphorylation at serine 194 has been identified as a relevant clinical issue in several types of hematological [13,100,101] and solid cancers [5,7,37,88,90,98,99,102,103]. A molecular mechanism associating the increase in phosphorylated FADD with tumorigenesis has been hinted at only in limited studies. In lung cancer, it has been demonstrated that increased levels of nuclear phosphorylated FADD induce NF-κB, which is suggested to induce the expression of cyclin D1 (CCND1) at the transcriptional level, ultimately resulting in cell cycle deregulation [5]. Also in lung cancer, the induction of mitosis seems to depend on activation of KRAS and CK1α phosphorylation of FADD during G2–M, where FADD interacted with G2–M cell-cycle regulatory components PLK1, AURKA, and BUB1 [102]. In breast cancer, AK2 downregulation results in reduced interaction of AK2-DUSP26 complex with FADD and, subsequently, increased phosphorylated FADD levels in the nuclei of tumor cells, correlating with increased proliferation [98].
There is limited information regarding FADD regulation by PTMs other than phosphorylation, but the available evidence indicates that ubiquitination regulates FADD stability and, consequently, the ability of FADD to mediate apoptosis [104]. Interestingly, both modifications could be related. Very recently, we proposed that FADD phosphorylation stabilizes the protein and that its degradation is mediated by the proteasome [13]. However, a direct link between FADD phosphorylation status, FADD ubiquitination, and proteasome-mediated degradation has not been established so far. It has been described that E3-ubiquitin ligase Makorin RING Finger Protein 1 (MKRN1) induces K48-linked polyubiquitination of FADD and, therefore, its degradation by the proteasome [105]. However, it cannot be ruled out that other E3 ligases could also ubiquitinate FADD [104]. Eight lysines are susceptible to ubiquitination in FADD (K24, K33, K35, K110, K120, K125, K149, and K153). K6-linked polyubiquitination at residues K149 and K153 by C terminus HSC70-interacting protein (CHIP) has been reported to prevent the formation of the death-inducing signaling complex (DISC) and therefore to suppress the apoptosis mediated by Fas or DR4/DR5, although it does not affect FADD degradation [106]. On the other hand, the linear ubiquitin chain assembly complex (LUBAC), which is composed of HOIP, HOIL-1L, and SHARPIN subunits, specifically generates M1-linked linear polyubiquitination of FADD and NEMO upon NF-κB activation, also suppressing cell death [107]. Recently, it has been demonstrated that FADD is ubiquitinated by speckle-type POZ protein (SPOP), which causes proteasome-mediated degradation [108] and impairs the FADD-mediated activation of NF-κB signaling [108,109]. SPOP has been reported to suppress tumorigenesis by inhibiting the NF-κB pathway in certain tumor types [109,110], indicating that this SPOP‒FADD‒NF-κB axis might represent a molecular mechanism underlying the role of FADD alteration in non-small-cell lung cancer [109]. In summary, E3-ubiquitin ligases constitute a potential therapeutic target with regards to FADD availability and function in the tumor cell. The combined treatment with E3-ubiquitin ligase inhibitors and agents able to activate extrinsic apoptosis such as TRAIL or TNFα might be beneficial for certain tumor types.
3. The Roles of FADD and Their Participation in Cancer
As a bimodular protein, FADD is a well-known adapter for receptor-induced cell death. Through homotypic interaction of its death domain (DD), FADD can be recruited to death receptors (DRs) belonging to the family of tumor necrosis factor (TNF) receptors including TNF-R1, FAS (CD95/APO-1), DR3, TRAIL (TNF-related apoptosis-inducing ligand)-R1 (DR4), and TRAIL-R2 (DR5), transmitting apoptosis initiating signals by their specific death ligands [1,111,112,113]. The death-effector domain (DED) of FADD recruits DED-only proteins (procaspase-8, procaspase-10, or c-FLIP) to form an active DISC. Mediated both by proximity-induced dimerization and proteolytic cleavage [114], initiator procaspases activate in the DISC and, once activated, caspase-8/caspase-10 initiate the caspase cascade, directly through cleavage of procaspase-3 or indirectly via Bid cleavage, eventually inducing apoptosis. Alternatively, under certain stimuli resulting in the blockade of apoptosis, such as the triggering of viral defense mechanisms or under caspase inhibitory conditions, death receptors signaling may lead to necroptosis, which occurs through a RIPK1‒RIPK3‒MLKL axis [115]. FADD acts as a negative regulator of this process, as it adapts the recruitment of caspase-8/cFLIP to RIPK1/3, which leads to cleavage of their kinase domain and subsequent inactivation [116,117]. The so-called ripoptosome comprises RIPK1/FADD/caspase-8 and induces apoptosis in a ligand/receptor-independent manner as it can be induced upon genotoxic stresses such as etoposide or removal/inhibition of the inhibitor of apoptosis (IAP) protein family; recruitment of RIPK3 to the complex switches the ripoptosome towards necroptosis [105,118]. In cancer, apoptosis impairment appears as a plausible consequence of FADD reduction [87,119]. Moreover, as FADD can prevent necroptosis, FADD reduction in cancer would also induce a switch from apoptotic signaling to necroptosis [117].
Additionally, FADD has been proposed to participate in cell cycle progression, T-cell proliferation, survival, or genome surveillance in the nucleus [87,96,120,121]. Several studies have evidenced defects in proliferation and cell cycle progression in FADD-deficient cells [87,101,122,123,124]. Although these effects could be simply attributed to the enhancement of RIPK1-dependent cell death in the absence of FADD, many studies using phosphorylation mutants highlight the relevance of FADD phosphorylation in G2/M phase and its interaction with proteins that are important for cell cycle progression [5,90,92,102,120,124,125,126,127]. Whether FADD participation in these functions is an indirect consequence of being sheltered from death receptors in the nucleus or an actively regulated role remains unclear. FADD can recruit proteins that regulate the NF-κB and MAPK pathways, which promote proliferation and cell cycle progression [5]. Our group has reported a reduction of FADD together with reduced apoptosis in T-LBL samples, but the accumulation of phosphorylated FADD in the nuclei of tumor cells [12,13]. Interestingly, we found that levels of phosphorylated FADD correlated with the proliferation capacity of tumor cells, supporting previous evidence in different cancer types [5,102].
As reviewed in [86], FADD has also emerged as a component of various signalosomes apart from the DISC and the necrosome, such as the FADDosome, the innateosome and the inflammasome. These can be initiated by death receptors, Toll-like receptors (TLR) or pathogen-induced signaling, and they can lead to pro-inflammatory signaling instead of cell death. The participation of FADD in signaling complexes eventually leading to gene expression changes has long been reported, particularly in relationship with innate immunity in mammals [128], Drosophila [129], or fish [130]. At the molecular level, this pathway seems to involve NF-κB activation. This pleiotropic transcription factor is present in almost all cell types and regulates the expression—either as an activator or as a repressor—of genes involved in many biological processes such as inflammation, immunity, differentiation, cell growth, and apoptosis. In consequence, aberrant or constitutive NF-κB activation impacts on various hallmarks of cancer as proliferation, migration or apoptosis and it has been detected in many tumor types both of hematological and solid nature [131]. Such hyperactivation of NF-κB signaling in cancer may be achieved by chromosomal alterations directly affecting members of the NF-κB pathway, as amplification of c-Rel on chromosome 2p14–15, chromosomal rearrangements or deletions affecting the NF-κB2 locus on chromosome 10q24, t(14;19)(q32;q13) translocation resulting in increased expression of the transcriptional coactivator of p50 or p52 homodimers Bcl-3, and loss-of-function mutations in IkBα (reviewed in [131]). However, since RelA, RelB and NF-kB1 alterations are rare in human cancer, different mechanisms might provide the source for NF-κB hyperactivation. This is the case with the tumor suppressor PTEN and RAS proto-oncogene, which are frequently mutated in cancer and whose abnormalities may in turn aberrantly activate NF-κB. However, it is also the case with NF-κB activation induced by death receptors or Toll-like receptors signaling. The formation of a “FADDosome” complex upon TRAIL-R signaling that promotes NF-κB activation and pro-inflammatory cytokine/chemokine production [132], operating both in nontransformed and transformed cells, has been demonstrated. In such a complex, FADD is the necessary adapter between caspase-8 and RIPK1. The transduction is critically dependent on caspase-8 in a protease-independent manner as a scaffold. Ubiquitin-modified RIPK1 would recruit proteins directly involved in activating NF-κB signaling, leading to the expression of inflammatory genes and eventually to the production of cytokines and chemokines. A similar “FADDosome” complex is, according to the literature, likely to occur upon CD95 signaling [133] and TLR signaling [134,135] in certain conditions. A frequent scenario in certain tumor types is the occurrence of CASP-8 mutations that abolish the protease activity of the protein, rendering it unable to promote apoptosis. However, as they can bind to FADD, they can promote NF-κB activation upon appropriate death receptor or TLR stimuli [136,137]. This may act synergistically with the FADD overexpression observed in certain cancers. Related to this, increased levels of phosphorylated FADD have been linked to NF-κB hyperactivation in lung adenocarcinomas [5]. In this context, tumor cells would not only benefit from apoptosis resistance, but would also trigger tumorigenic pathways downstream NF-κB-mediated gene expression, such as inflammation or proliferation.
As discussed previously, the participation of FADD in the regulation of gene expression would be indirect, as a member of cytoplasmic protein complexes involved in transducing signals. Some authors have also proposed, however, that FADD may act in a more direct manner, as it is itself a member of the transcription factor complexes in the nucleus. In one study, it was speculated that FADD might be involved in regulating MyoD transcription [138]. The authors proposed that FADD would regulate the binding of SRF and/or MEF2 transcription factors to a MyoD enhancer, facilitating acetylation via recruitment of acetyltransferases such as CBP or p300, in turn activating gene transcription. Nevertheless, since FADD does not exhibit any DNA-binding motif, it might bind other proteins to form a functional transcription factor complex; in particular, the NF-κB activator NKAP has been proposed as a candidate to associate with FADD and regulate gene transcription in thymocytes [139]. NKAP has been shown to associate with HDAC3 as a part of a DNA-binding complex and, with CIR, as a part of the NOTCH co-repressor complex [140]. FADD association with NKAP in the nucleus would stabilize the latter, therefore contributing to its role as a transcriptional repressor able to hamper NOTCH-mediated transcriptional activation. High levels of NKAP have been observed in most tumor types (https://www.proteinatlas.org/ENSG00000101882-NKAP/pathology), and it has been described as an oncogene in hepatocellular carcinoma [141], glioma [142], and breast cancer [143], even as an unfavorable prognostic indicator in the latter (https://www.proteinatlas.org/ENSG00000101882-NKAP/pathology/tissue/breast+cancer). FADD downregulation in certain cancer types—such as T-LBL—might result in the reduced stability of NKAP complexes in the nucleus, contributing to NOTCH hyperactivation frequently found in T-cell lymphoblastic neoplasms [144]. However, there is no evidence for this association yet.
Besides its major role in cell death and the principal nonapoptotic functions described so far, comparative proteomics approaches have recently provided evidence for FADD’s involvement in energy metabolism [145,146,147,148]. In particular, the comparison of FADD-expressing with FADD-deficient mouse embryonic fibroblasts showed an enriched cluster of changed proteins involved in lipid metabolism, fatty acid metabolism, glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation [146,147]. The same authors proposed a mechanism whereby FADD-deficient thymocytes would exhibit reduced glucose uptake due to the accumulation of PKC-α, which may result in transcriptional repression of the glucose transporter gene Glut1 [145]. Finally, this group has demonstrated in vivo in a mouse model that FADD is relevant to glucose and fat metabolism [148]. If FADD involvement in these functions is confirmed in humans, it could be speculated that the alterations in FADD observed in tumor cells may also impact on energy metabolism. The link between cancer and altered metabolism is well recognized, as cancer cells often utilize aerobic glycolysis, the well-known Warburg effect, to meet the needs of rapidly dividing neoplastic cells [149]. How FADD alterations would contribute to either metabolic switch in cancer still needs to be addressed.
4. Clinical Implications of FADD Alterations in Cancer
Considering the various alterations that FADD has been reported to undergo in pathological processes like cancer, many authors support the notion that the FADD level is a relevant factor to consider for diagnosis, prognosis, or therapeutic strategies. According to the information collected by The Human Protein Atlas, the FADD level is an unfavorable prognostic marker in lung, head and neck, and cervical cancers, whereas it constitutes a favorable prognostic marker in thyroid cancer (Figure 8).
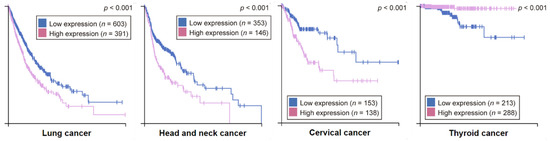
Figure 8.
Kaplan‒Meier plots for cancer types where high FADD expression has significant (p < 0.001) association with patient survival. Based on the FPKM value, patients were divided based on FADD mRNA level into one of the two groups “low” (under cutoff) or “high” (over cutoff). The X-axis shows time for survival (years) and the Y-axis shows the probability of survival, where 1.0 corresponds to 100 percent. The prognosis of each group of patients was examined by Kaplan‒Meier survival estimators, and the survival outcomes of the two groups were compared by log-rank tests. (From The Human Protein Atlas).
In the literature, an association of FADD increase with poor clinical outcome has been described in many solid tumors [4,5,6,7,8,9,10,18,109,150,151,152]. In contrast, a reduction of FADD has been reported in fewer tumor types, among them hematological malignancies [11,12,13,14,15], but also in pathologies different from cancer like dementia [153]. Altered levels of phosphorylated FADD have also been reported, either a reduction [12,13,154,155,156] or an increase [5,7,37,98,100,101,102], but in all cases associated with poor outcome. This apparent discrepancy can probably be explained by the fact that FADD can play, as explained before, various roles depending on the cell type and molecular context. For example, our group demonstrated that both murine [12] and human [13] T-LBL samples exhibiting overall reduction of FADD and impaired apoptosis could be stratified according to their levels of phosphorylated FADD. S191/194-P-FADD was identified as an unfavorable clinical factor in T-LBL, suggesting that FADD phosphorylation status may serve as a new biomarker with prognostic value. On the contrary, another group reported that phosphorylated FADD was not prognostic in laryngeal carcinoma treated with radiotherapy [157], confirming that the predictive value of phosphorylated FADD also depends on cellular type and context.
Regarding the improvement of therapy, some interesting pieces of evidence concerning FADD are also available. For example, it has been reported that overexpressing FADD in colorectal cancer [158] or malignant glioma [159,160] renders tumor cells more sensitive to apoptosis, thus improving the effect of chemotherapy. In tumor types with frequent FADD amplification, such as head and neck cancer, it has been suggested that combined treatment with SMAC mimetics (Birinapant) and radiation may be particularly useful as FADD is key in sensitization to cell death [16]. Also, some chemotherapeutic agents—such as Carboplatin in tongue carcinoma [161] or Nortriptyline in bladder cancer cells [162]—induce the expression of FADD, thus contributing to tumor cell sensitization to apoptosis. FADD induction was observed in ovarian cancer cells upon combined treatment with cisplatin and AT-101, a natural BH3-mimetic molecule [163]. Interestingly, such treatment inhibited both DNMT and HDAC enzyme activities, allowing us to speculate that histone deacetylation might underlie the observed induction of FADD transcription. In chronic lymphocytic leukemia, it has been observed that the inhibition of histone deacetylase with Romidepsin exerts an apoptosis sensitization effect molecularly mediated by enhanced FADD recruitment to the DISC, although the exact mechanism is not clear [164]. In KRAS-driven lung cancer, it has been indicated that the inhibition of FADD phosphorylation suppresses tumor development, suggesting that FADD kinase is a plausible therapeutic target [102]. A summary of the FADD alterations reported in different cancer types and therapeutic agents described to target tumor cells through mechanisms involving FADD is depicted in Figure 9 and Figure 10, respectively.
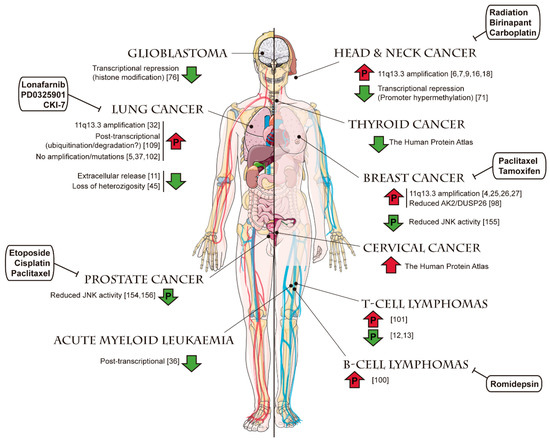
Figure 9.
Alterations of FADD levels reported in different cancer types. For each tumor type, reported evidence of an increase (upwards red arrow) or a decrease (downwards green arrow) of FADD levels is shown, indicating the underlying mechanism when this information is available (references indicated in brackets). Arrows including a “P” indicate changes also affecting the levels of phosphorylated FADD. Anticancer therapies reported to target FADD in certain tumor types are shown in white boxes, and their mechanisms of action are detailed in Figure 10. Templates to build this figure were obtained from SMART Servier Medical Art (Attribution 3.0 Unported, CC BY 3.0).
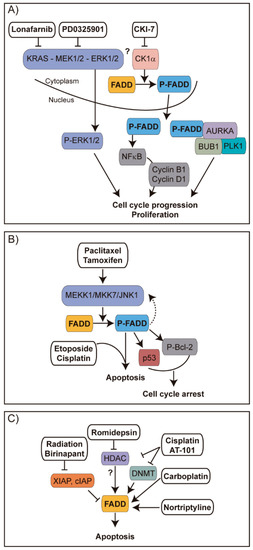
Figure 10.
Strategies for therapeutic intervention of cancer involving FADD. (A) In lung cancer, CKIα-mediated phosphorylation of FADD leads to the translocation of S194-P-FADD to the nucleus, where it can induce expression of cyclins B1 and D1 through NF-κB signaling [5], or it can interact with key G2/M transition proteins like AURKA, PLK1 or BUB1 [102], promoting cell cycle progression and proliferation. Inhibiting KRAS with Lonafarnib, MEK with PD0325901, or CK1α with CKI-7 decreased the abundance of phosphorylated FADD and decreased cell proliferation, apparently due to a loss of interaction between FADD and the G2/M transition proteins. As a result, tumor treated cells would fail to progress through G2/M. (B) Tamoxifen and paclitaxel in breast cancer [155] and paclitaxel in prostate cancer [165] have been reported to activate the MEKK1/MKK7/JNK pathway, which contributes to FADD phosphorylation. In breast cancer, this results in cell cycle arrest and suppression of cancer growth through p53 stabilization or Bcl-2 phosphorylation. In prostate cancer, phosphorylated FADD in turn upregulates MEKK1 and downstream JNK1 activation, which is essential for sensitization to apoptosis induced by etoposide or cisplatin combined with paclitaxel [165]. Thus, chemosensitization can be amplified through FADD phosphorylation and the MEKK1/MKK7/JNK1 pathway. (C) In head and neck cancer, the SMAC mimetic Birinapant plus radiation induces tumor regression [16]. Radiation induces DNA damage and in consequence the intrinsic cell death pathway through mitochondrial release of SMAC. The negative effect of SMAC on IAPs is enhanced by SMAC mimetic Birinapant, resulting in degradation of IAPs to enhance FADD-involving DR-induced apoptosis. In chronic lymphocytic leukemia, the HDAC inhibitor Romidepsin sensitizes tumor cells to TRAIL-induced apoptosis through enhancement of FADD recruitment to the DISC [164]. In ovarian cancer, the combination of the BH3-mimetic molecule AT-101 with cisplatin strongly sensitize cells towards apoptosis; they inhibit HDAC and DNA methyltransferase (DNMT) enzyme activities and they induce FADD expression among other apoptosis-related genes [163]. Carboplatin and Nortriptyline also favor FADD expression in tongue carcinoma [161] and bladder cancer cells [162], respectively.
5. Conclusions
The role of FADD in tumorigenesis is not fully understood. Its expression appears to be altered in many cancer types, but the underlying mechanisms are not always clear. Chromosomal alterations affecting FADD are more frequent than mutations or polymorphisms, but they do not always explain the changes in expression observed in tumor cells. Regulation of FADD expression by transcription factors and epigenetic mechanisms may also be altered in cancer, but limited evidence is available to confirm this. Furthermore, posttranslational modifications of FADD, especially phosphorylation and ubiquitination, may underlie the alterations of FADD functions in tumor cells. Besides its canonical role as an adaptor for cell death, FADD has been implicated in various signalosomes that would trigger tumorigenic pathways downstream of NF-κB-mediated gene expression, such as inflammation or proliferation. A role for FADD in directly regulating gene expression as a member of protein complexes in the nucleus has also been proposed, although the available evidence is still scarce. In addition, the implication of FADD in controlling energy metabolism is mainly based on recent evidence of protein‒protein interactions. In any case, many authors agree that FADD levels are a relevant factor to consider in the clinical management of cancer. However, discrepant results suggest that its role as a positive or negative prognostic factor would depend on the tumor type and cellular context. Future research will likely shed light on the molecular details of FADD functions, thus clarifying its clinical value for each cancer type.
Funding
This work was supported by the Spanish Ministry of Economy and Competitiveness (RTI2018-093330-B-I00 and BES-2013-065740 to J.F.-P.); Fundación Ramón Areces to J.F.-P.; the Autonomous Community of Madrid, Spain (B2017/BMD-3778 LINFOMAS-CM to J.F.-P.); the Scientific Foundation of the Spanish Association against Cancer (AECC 2018 PROYE18054PIRI to J.F.-P.); the Instituto de Salud Carlos III (ACCI-CIBERER-17 ISCIII to J.F.-P.), and the Universidad Autónoma de Madrid, Faculty of Sciences (grants for final Master’s dissertation to M.V.-M.). L.V.-M. is the recipient of a Master’s fellowship from the Universidad Autónoma de Madrid.
Acknowledgments
We would like to thank Tiaan Heunis for a critical reading of this manuscript. Institutional grants from Fundación Ramón Areces and Banco de Santander to CBMSO are also acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Yeh, W.C.; de la Pompa, J.L.; McCurrach, M.E.; Shu, H.B.; Elia, A.J.; Shahinian, A.; Ng, M.; Wakeham, A.; Khoo, W.; Mitchell, K.; et al. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 1998, 279, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Callegari, C.C.; Cavalli, I.J.; Lima, R.S.; Jucoski, T.S.; Torresan, C.; Urban, C.A.; Kuroda, F.; Anselmi, K.F.; Cavalli, L.R.; Ribeiro, E.M. Copy number and expression analysis of FOSL1, GSTP1, NTSR1, FADD and CCND1 genes in primary breast tumors with axillary lymph node metastasis. Cancer Genet. 2016, 209, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Bhojani, M.S.; Heaford, A.C.; Chang, D.C.; Laxman, B.; Thomas, D.G.; Griffin, L.B.; Yu, J.; Coppola, J.M.; Giordano, T.J.; et al. Phosphorylated FADD induces NF-kappaB, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc. Natl. Acad. Sci. USA 2005, 102, 12507–12512. [Google Scholar] [CrossRef] [PubMed]
- Chien, H.T.; Cheng, S.D.; Chuang, W.Y.; Liao, C.T.; Wang, H.M.; Huang, S.F. Clinical Implications of FADD Gene Amplification and Protein Overexpression in Taiwanese Oral Cavity Squamous Cell Carcinomas. PLoS ONE 2016, 11, e0164870. [Google Scholar] [CrossRef] [PubMed]
- Gibcus, J.H.; Menkema, L.; Mastik, M.F.; Hermsen, M.A.; de Bock, G.H.; van Velthuysen, M.L.; Takes, R.P.; Kok, K.; Alvarez Marcos, C.A.; van der Laan, B.F.; et al. Amplicon mapping and expression profiling identify the Fas-associated death domain gene as a new driver in the 11q13.3 amplicon in laryngeal/pharyngeal cancer. Clin. Cancer Res. 2007, 13, 6257–6266. [Google Scholar] [CrossRef]
- He, L.; Ren, Y.; Zheng, Q.; Wang, L.; Lai, Y.; Guan, S.; Zhang, X.; Zhang, R.; Wang, J.; Chen, D.; et al. Fas-associated protein with death domain (FADD) regulates autophagy through promoting the expression of Ras homolog enriched in brain (Rheb) in human breast adenocarcinoma cells. Oncotarget 2016, 7, 24572–24584. [Google Scholar] [CrossRef][Green Version]
- Pattje, W.J.; Melchers, L.J.; Slagter-Menkema, L.; Mastik, M.F.; Schrijvers, M.L.; Gibcus, J.H.; Kluin, P.M.; Hoegen-Chouvalova, O.; van der Laan, B.F.; Roodenburg, J.L.; et al. FADD expression is associated with regional and distant metastasis in squamous cell carcinoma of the head and neck. Histopathology 2013, 63, 263–270. [Google Scholar] [CrossRef]
- Schrijvers, M.L.; Pattje, W.J.; Slagter-Menkema, L.; Mastik, M.F.; Gibcus, J.H.; Langendijk, J.A.; van der Wal, J.E.; van der Laan, B.F.; Schuuring, E. FADD expression as a prognosticator in early-stage glottic squamous cell carcinoma of the larynx treated primarily with radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1220–1226. [Google Scholar] [CrossRef]
- Cimino, Y.; Costes, A.; Damotte, D.; Validire, P.; Mistou, S.; Cagnard, N.; Alifano, M.; Regnard, J.F.; Chiocchia, G.; Sautes-Fridman, C.; et al. FADD protein release mirrors the development and aggressiveness of human non-small cell lung cancer. Br. J. Cancer 2012, 106, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Marin-Rubio, J.L.; de Arriba, M.C.; Cobos-Fernandez, M.A.; Gonzalez-Sanchez, L.; Ors, I.; Sastre, I.; Fernandez-Piqueras, J.; Villa-Morales, M. Deregulated FADD expression and phosphorylation in T-cell lymphoblastic lymphoma. Oncotarget 2016. [Google Scholar] [CrossRef]
- Marin-Rubio, J.L.; Perez-Gomez, E.; Fernandez-Piqueras, J.; Villa-Morales, M. S194-P-FADD as a marker of aggressiveness and poor prognosis in human T-cell lymphoblastic lymphoma. Carcinogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Schuchmann, M.; Galle, P.R. Cell death and hepatocarcinogenesis: Dysregulation of apoptosis signaling pathways. J. Gastroenterol Hepatol. 2011, 26 (Suppl. 1), 213–219. [Google Scholar] [CrossRef]
- Tourneur, L.; Mistou, S.; Michiels, F.M.; Devauchelle, V.; Renia, L.; Feunteun, J.; Chiocchia, G. Loss of FADD protein expression results in a biased Fas-signaling pathway and correlates with the development of tumoral status in thyroid follicular cells. Oncogene 2003, 22, 2795–2804. [Google Scholar] [CrossRef][Green Version]
- Eytan, D.F.; Snow, G.E.; Carlson, S.; Derakhshan, A.; Saleh, A.; Schiltz, S.; Cheng, H.; Mohan, S.; Cornelius, S.; Coupar, J.; et al. SMAC Mimetic Birinapant plus Radiation Eradicates Human Head and Neck Cancers with Genomic Amplifications of Cell Death Genes FADD and BIRC2. Cancer Res. 2016, 76, 5442–5454. [Google Scholar] [CrossRef]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345. [Google Scholar] [CrossRef]
- Reddy, R.B.; Bhat, A.R.; James, B.L.; Govindan, S.V.; Mathew, R.; Ravindra, D.R.; Hedne, N.; Illiayaraja, J.; Kekatpure, V.; Khora, S.S.; et al. Meta-Analyses of Microarray Datasets Identifies ANO1 and FADD as Prognostic Markers of Head and Neck Cancer. PLoS ONE 2016, 11, e0147409. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Yun, J.A.; Jabeen, S.; Jeon, E.K.; Won, H.S.; Ko, Y.H.; Kim, S.Y. Prognostic significance of TMEM16A, PPFIA1, and FADD expression in invasive ductal carcinoma of the breast. World J. Surg. Oncol. 2014, 12, 137. [Google Scholar] [CrossRef]
- Lundgren, K.; Holm, K.; Nordenskjold, B.; Borg, A.; Landberg, G. Gene products of chromosome 11q and their association with CCND1 gene amplification and tamoxifen resistance in premenopausal breast cancer. Breast Cancer Res. 2008, 10, R81. [Google Scholar] [CrossRef]
- Norton, N.; Advani, P.P.; Serie, D.J.; Geiger, X.J.; Necela, B.M.; Axenfeld, B.C.; Kachergus, J.M.; Feathers, R.W.; Carr, J.M.; Crook, J.E.; et al. Assessment of Tumor Heterogeneity, as Evidenced by Gene Expression Profiles, Pathway Activation, and Gene Copy Number, in Patients with Multifocal Invasive Lobular Breast Tumors. PLoS ONE 2016, 11, e0153411. [Google Scholar] [CrossRef]
- Matsuda, R.; Enokida, H.; Chiyomaru, T.; Kikkawa, N.; Sugimoto, T.; Kawakami, K.; Tatarano, S.; Yoshino, H.; Toki, K.; Uchida, Y.; et al. LY6K is a novel molecular target in bladder cancer on basis of integrate genome-wide profiling. Br. J. Cancer 2011, 104, 376–386. [Google Scholar] [CrossRef]
- Proctor, A.J.; Coombs, L.M.; Cairns, J.P.; Knowles, M.A. Amplification at chromosome 11q13 in transitional cell tumours of the bladder. Oncogene 1991, 6, 789–795. [Google Scholar]
- Qin, S.L.; Chen, X.J.; Xu, X.; Shou, J.Z.; Bi, X.G.; Ji, L.; Han, Y.L.; Cai, Y.; Wei, F.; Ma, J.H.; et al. Detection of chromosomal alterations in bladder transitional cell carcinomas from Northern China by comparative genomic hybridization. Cancer Lett. 2006, 238, 230–239. [Google Scholar] [CrossRef]
- Zaharieva, B.M.; Simon, R.; Diener, P.A.; Ackermann, D.; Maurer, R.; Alund, G.; Knonagel, H.; Rist, M.; Wilber, K.; Hering, F.; et al. High-throughput tissue microarray analysis of 11q13 gene amplification (CCND1, FGF3, FGF4, EMS1) in urinary bladder cancer. J. Pathol. 2003, 201, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.; Aninat-Meyer, M.; Schluns, K.; Gellert, K.; Dietel, M.; Petersen, I. Chromosomal alterations in the clonal evolution to the metastatic stage of squamous cell carcinomas of the lung. Br. J. Cancer 2000, 82, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Irving, J.; Parker, R.; Kim, H.; Press, J.Z.; Longacre, T.A.; Chia, S.; Magliocco, A.; Makretsov, N.; Gilks, B.; et al. Amplification of EMSY, a novel oncogene on 11q13, in high grade ovarian surface epithelial carcinomas. Gynecol. Oncol. 2006, 100, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Kalloger, S.E.; Miller, M.A.; Shih Ie, M.; McKinney, S.E.; Santos, J.L.; Swenerton, K.; Spellman, P.T.; Gray, J.; Gilks, C.B.; et al. Amplification of 11q13 in ovarian carcinoma. Genes Chromosomes Cancer 2008, 47, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Schraml, P.; Schwerdtfeger, G.; Burkhalter, F.; Raggi, A.; Schmidt, D.; Ruffalo, T.; King, W.; Wilber, K.; Mihatsch, M.J.; Moch, H. Combined array comparative genomic hybridization and tissue microarray analysis suggest PAK1 at 11q13.5-q14 as a critical oncogene target in ovarian carcinoma. Am. J. Pathol. 2003, 163, 985–992. [Google Scholar] [CrossRef]
- Tourneur, L.; Delluc, S.; Levy, V.; Valensi, F.; Radford-Weiss, I.; Legrand, O.; Vargaftig, J.; Boix, C.; Macintyre, E.A.; Varet, B.; et al. Absence or low expression of fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome. Cancer Res. 2004, 64, 8101–8108. [Google Scholar] [CrossRef]
- Bhojani, M.S.; Chen, G.; Ross, B.D.; Beer, D.G.; Rehemtulla, A. Nuclear localized phosphorylated FADD induces cell proliferation and is associated with aggressive lung cancer. Cell Cycle 2005, 4, 1478–1481. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Srivatsan, E.S.; Wood, T.F.; Eubanks, P.J.; Ebrahimi, S.A.; Gatti, R.A.; Passaro, E., Jr.; Sawicki, M.P. Deletion mapping of endocrine tumors localizes a second tumor suppressor gene on chromosome band 11q13. Genes Chromosomes Cancer 1998, 22, 130–137. [Google Scholar] [CrossRef]
- Cheng, Y.; Chakrabarti, R.; Garcia-Barcelo, M.; Ha, T.J.; Srivatsan, E.S.; Stanbridge, E.J.; Lung, M.L. Mapping of nasopharyngeal carcinoma tumor-suppressive activity to a 1.8-megabase region of chromosome band 11q13. Genes Chromosomes Cancer 2002, 34, 97–103. [Google Scholar] [CrossRef]
- Srivatsan, E.S.; Ying, K.L.; Seeger, R.C. Deletion of chromosome 11 and of 14q sequences in neuroblastoma. Genes Chromosomes Cancer 1993, 7, 32–37. [Google Scholar] [CrossRef]
- Venugopalan, M.; Wood, T.F.; Wilczynski, S.P.; Sen, S.; Peters, J.; Ma, G.C.; Evans, G.A.; Srivatsan, E.S. Loss of heterozygosity in squamous cell carcinomas of the head and neck defines a tumor suppressor gene region on 11q13. Cancer Genet. Cytogenet 1998, 104, 124–132. [Google Scholar] [CrossRef]
- Zhuang, Z.; Merino, M.J.; Chuaqui, R.; Liotta, L.A.; Emmert-Buck, M.R. Identical allelic loss on chromosome 11q13 in microdissected in situ and invasive human breast cancer. Cancer Res. 1995, 55, 467–471. [Google Scholar] [PubMed]
- Soares, B.S.; Eguchi, K.; Frohman, L.A. Tumor deletion mapping on chromosome 11q13 in eight families with isolated familial somatotropinoma and in 15 sporadic somatotropinomas. J. Clin. Endocrinol. Metab. 2005, 90, 6580–6587. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, E.S.; Chakrabarti, R.; Zainabadi, K.; Pack, S.D.; Benyamini, P.; Mendonca, M.S.; Yang, P.K.; Kang, K.; Motamedi, D.; Sawicki, M.P.; et al. Localization of deletion to a 300 Kb interval of chromosome 11q13 in cervical cancer. Oncogene 2002, 21, 5631–5642. [Google Scholar] [CrossRef][Green Version]
- Chan, A.S.; Lam, W.K.; Wong, M.P.; Fu, K.H.; Lee, J.; Yew, W.W.; Chiu, S.W.; Lung, M.L. Chromosomal 11 alterations in non-small-cell lung carcinomas in Hong Kong. Lung Cancer 1996, 15, 51–65. [Google Scholar] [CrossRef]
- Gregory-Evans, C.Y.; Moosajee, M.; Hodges, M.D.; Mackay, D.S.; Game, L.; Vargesson, N.; Bloch-Zupan, A.; Ruschendorf, F.; Santos-Pinto, L.; Wackens, G.; et al. SNP genome scanning localizes oto-dental syndrome to chromosome 11q13 and microdeletions at this locus implicate FGF3 in dental and inner-ear disease and FADD in ocular coloboma. Hum. Mol. Genet. 2007, 16, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kim, G.H.; Byeon, J.H.; Eun, S.H.; Eun, B.L. Chromosome 11q13 deletion syndrome. Korean J. Pediatr. 2016, 59, S10–S13. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef]
- Shin, M.S.; Kim, H.S.; Lee, S.H.; Lee, J.W.; Song, Y.H.; Kim, Y.S.; Park, W.S.; Kim, S.Y.; Lee, S.N.; Park, J.Y.; et al. Alterations of Fas-pathway genes associated with nodal metastasis in non-small cell lung cancer. Oncogene 2002, 21, 4129–4136. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutation of FADD gene is rare in human colon and stomach cancers. APMIS 2004, 112, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld-Granot, G.; Toren, A.; Amariglio, N.; Brok-Simoni, F.; Rechavi, G. Mutation analysis of the FAS and TNFR apoptotic cascade genes in hematological malignancies. Exp. Hematol. 2001, 29, 228–233. [Google Scholar] [CrossRef]
- Dechant, M.J.; Fellenberg, J.; Scheuerpflug, C.G.; Ewerbeck, V.; Debatin, K.M. Mutation analysis of the apoptotic “death-receptors” and the adaptors TRADD and FADD/MORT-1 in osteosarcoma tumor samples and osteosarcoma cell lines. Int. J. Cancer 2004, 109, 661–667. [Google Scholar] [CrossRef]
- Bolze, A.; Byun, M.; McDonald, D.; Morgan, N.V.; Abhyankar, A.; Premkumar, L.; Puel, A.; Bacon, C.M.; Rieux-Laucat, F.; Pang, K.; et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am. J. Hum. Genet. 2010, 87, 873–881. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Eun, Y.G.; Chung, D.H.; Kim, S.W.; Lee, Y.C.; Kim, S.K.; Kwon, K.H. A Fas-associated via death domain promoter polymorphism (rs10898853, -16C/T) as a risk factor for papillary thyroid cancer. Eur. Surg. Res. 2014, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Villa-Morales, M.; Gonzalez-Gugel, E.; Shahbazi, M.N.; Santos, J.; Fernandez-Piqueras, J. Modulation of the Fas-apoptosis-signalling pathway by functional polymorphisms at Fas, FasL and Fadd and their implication in T-cell lymphoblastic lymphoma susceptibility. Carcinogenesis 2010, 31, 2165–2171. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Eskin, E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 2012, 8, e1002555. [Google Scholar] [CrossRef]
- Kim, P.K.; Dutra, A.S.; Chandrasekharappa, S.C.; Puck, J.M. Genomic structure and mapping of human FADD, an intracellular mediator of lymphocyte apoptosis. J. Immunol. 1996, 157, 5461–5466. [Google Scholar]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.; Whitfield, T.W.; Greven, M.C.; Pierce, B.G.; Dong, X.; Kundaje, A.; Cheng, Y.; et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012, 22, 1798–1812. [Google Scholar] [CrossRef]
- Hindryckx, P.; De Vos, M.; Jacques, P.; Ferdinande, L.; Peeters, H.; Olievier, K.; Bogaert, S.; Brinkman, B.; Vandenabeele, P.; Elewaut, D.; et al. Hydroxylase inhibition abrogates TNF-alpha-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. J. Immunol. 2010, 185, 6306–6316. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Nguyen, D.D.; Lee, D.G.; Kim, S.; Kang, K.; Rhee, J.K.; Chang, S. Integrative Bioinformatics and Functional Analyses of GEO, ENCODE, and TCGA Reveal FADD as a Direct Target of the Tumor Suppressor BRCA1. Int. J. Mol. Sci. 2018, 19, 1458. [Google Scholar] [CrossRef] [PubMed]
- Viringipurampeer, I.A.; Ferreira, T.; DeMaria, S.; Yoon, J.J.; Shan, X.; Moosajee, M.; Gregory-Evans, K.; Ngai, J.; Gregory-Evans, C.Y. Pax2 regulates a fadd-dependent molecular switch that drives tissue fusion during eye development. Hum. Mol. Genet. 2012, 21, 2357–2369. [Google Scholar] [CrossRef]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Llinas-Arias, P.; Esteller, M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: An update. Open Biol. 2017, 7, 170152. [Google Scholar] [CrossRef]
- Zhao, X.; Yin, H.; Li, N.; Zhu, Y.; Shen, W.; Qian, S.; He, G.; Li, J.; Wang, X. An Integrated Regulatory Network Based on Comprehensive Analysis of mRNA Expression, Gene Methylation and Expression of Long Non-coding RNAs (lncRNAs) in Myelodysplastic Syndromes. Front. Oncol. 2019, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Wichnieski, C.; Maheshwari, K.; Souza, L.C.; Nieves, F.; Tartari, T.; Garlet, G.P.; Carneiro, E.; Letra, A.; Silva, R.M. DNA methylation profiles of immune response-related genes in apical periodontitis. Int. Endod. J. 2019, 52, 5–12. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Chandrasoma, S.; Siegmund, K.D.; Weisenberger, D.J.; Cheng, J.C.; Toma, M.I.; Huland, H.; Jones, P.A.; Liang, G. Prognostic relevance of methylation markers in patients with non-muscle invasive bladder carcinoma. Eur. J. Cancer. 2005, 41, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Saberi, E.; Kordi-Tamandani, D.M.; Jamali, S.; Rigi-Ladiz, M.A. Analysis of methylation and mRNA expression status of FADD and FAS genes in patients with oral squamous cell carcinoma. Med. Oral Patol. Oral. Cir. Bucal. 2014, 19, e562–e568. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Kim, J.; Yu, J. Influence of oncogenic transcription factors on chromatin conformation and implications in prostate cancer. Appl. Clin. Genet. 2014, 7, 81–91. [Google Scholar] [CrossRef]
- Yin, Q.; Wu, M.; Liu, Q.; Lv, H.; Jiang, R. DeepHistone: A deep learning approach to predicting histone modifications. BMC Genom. 2019, 20, 193. [Google Scholar] [CrossRef] [PubMed]
- A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [CrossRef]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- Tamannai, M.; Farhangi, S.; Truss, M.; Sinn, B.; Wurm, R.; Bose, P.; Henze, G.; Riabowol, K.; von Deimling, A.; Tallen, G. The inhibitor of growth 1 (ING1) is involved in trichostatin A-induced apoptosis and caspase 3 signaling in p53-deficient glioblastoma cells. Oncol. Res. 2010, 18, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.; Sugita, F.; Nakamura, Y.; Sasaki, K.; Mitani, K. Fadd and Skp2 are possible downstream targets of RUNX1-EVI1. Int. J. Hematol. 2013, 97, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Tay, Y.M.; Tam, W.L.; Ang, Y.S.; Gaughwin, P.M.; Yang, H.; Wang, W.; Liu, R.; George, J.; Ng, H.H.; Perera, R.J.; et al. MicroRNA-134 modulates the differentiation of mouse embryonic stem cells, where it causes post-transcriptional attenuation of Nanog and LRH1. Stem cells 2008, 26, 17–29. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.F.; Yang, L.X.; Guo, R.W.; Liu, H.; Shi, Y.K.; Wang, H.; Ye, J.S.; Yang, Z.H.; Liang, X. miR-155 inhibits oxidized low-density lipoprotein-induced apoptosis of RAW264.7 cells. Mol. Cell Biochem. 2013, 382, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Yu, X.D.; Liu, Z.H.; Cheng, X.; Samartzis, D.; Jia, L.T.; Wu, S.X.; Huang, J.; Chen, J.; Luo, Z.J. Deregulated miR-155 promotes Fas-mediated apoptosis in human intervertebral disc degeneration by targeting FADD and caspase-3. J. Pathol. 2011, 225, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Liu, Z.; Li, B.S.; Tang, B.; Li, W.; Guo, G.; Shi, Y.; Wang, F.; Wu, Y.; Tong, W.D.; et al. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J. Infect. Dis. 2009, 200, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Noguchi, S.; Kumazaki, M.; Shinohara, H.; Miki, K.; Naoe, T.; Akao, Y. Epigenetic regulation of microRNA-128a expression contributes to the apoptosis-resistance of human T-cell leukaemia jurkat cells by modulating expression of fas-associated protein with death domain (FADD). Biochim. Biophys. Acta 2014, 1843, 590–602. [Google Scholar] [CrossRef]
- Tili, E.; Croce, C.M.; Michaille, J.J. miR-155: On the crosstalk between inflammation and cancer. Int. Rev. Immunol. 2009, 28, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Mouasni, S.; Tourneur, L. FADD at the Crossroads between Cancer and Inflammation. Trends Immunol. 2018, 39, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Tourneur, L.; Chiocchia, G. FADD: A regulator of life and death. Trends Immunol. 2010, 31, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, D.; Hua, Z. FADD and its phosphorylation. IUBMB Life 2004, 56, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.S.; Lee, S.J.; Kang, N.S.; Kim, E. Cooperative phosphorylation of FADD by Aur-A and Plk1 in response to taxol triggers both apoptotic and necrotic cell death. Cancer Res. 2011, 71, 7207–7215. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Volkland, J.; Blomberg, I.; Hoffmann, I.; Krammer, P.H.; Peter, M.E. Phosphorylation of FADD/ MORT1 at serine 194 and association with a 70-kDa cell cycle-regulated protein kinase. J. Immunol. 2000, 164, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Vilmont, V.; Filhol, O.; Hesse, A.M.; Coute, Y.; Hue, C.; Remy-Tourneur, L.; Mistou, S.; Cochet, C.; Chiocchia, G. Modulatory role of the anti-apoptotic protein kinase CK2 in the sub-cellular localization of Fas associated death domain protein (FADD). Biochim. Biophys. Acta 2015, 1853, 2885–2896. [Google Scholar] [CrossRef]
- Hua, Z.C.; Sohn, S.J.; Kang, C.; Cado, D.; Winoto, A. A function of Fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity 2003, 18, 513–521. [Google Scholar] [CrossRef]
- Yao, C.; Zhuang, H.; Cheng, W.; Lin, Y.; Du, P.; Yang, B.; Huang, X.; Chen, S.; Hu, Q.; Hua, Z.C. FADD phosphorylation impaired islet morphology and function. J. Cell Physiol. 2015, 230, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Foger, N.; Bulfone-Paus, S.; Chan, A.C.; Lee, K.H. Subcellular compartmentalization of FADD as a new level of regulation in death receptor signaling. FEBS J. 2009, 276, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.W.; Chandra, J.; Loegering, D.; Van Becelaere, K.; Kottke, T.J.; Gore, S.D.; Karp, J.E.; Sebolt-Leopold, J.; Kaufmann, S.H. Central role of Fas-associated death domain protein in apoptosis induction by the mitogen-activated protein kinase kinase inhibitor CI-1040 (PD184352) in acute lymphocytic leukemia cells in vitro. J. Biol. Chem. 2003, 278, 47326–47339. [Google Scholar] [CrossRef] [PubMed]
- Screaton, R.A.; Kiessling, S.; Sansom, O.J.; Millar, C.B.; Maddison, K.; Bird, A.; Clarke, A.R.; Frisch, S.M. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: A potential link between genome surveillance and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 5211–5216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Vallabhaneni, R.; Loughran, P.A.; Shapiro, R.; Yin, X.M.; Yuan, Y.; Billiar, T.R. Changes in FADD levels, distribution, and phosphorylation in TNFalpha-induced apoptosis in hepatocytes is caspase-3, caspase-8 and BID dependent. Apoptosis 2008, 13, 983–992. [Google Scholar] [CrossRef]
- Kim, H.; Lee, H.J.; Oh, Y.; Choi, S.G.; Hong, S.H.; Kim, H.J.; Lee, S.Y.; Choi, J.W.; Su Hwang, D.; Kim, K.S.; et al. The DUSP26 phosphatase activator adenylate kinase 2 regulates FADD phosphorylation and cell growth. Nat. Commun. 2014, 5, 3351. [Google Scholar] [CrossRef]
- Zhang, J.; Winoto, A. A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol. Cell Biol. 1996, 16, 2756–2763. [Google Scholar] [CrossRef]
- Drakos, E.; Leventaki, V.; Atsaves, V.; Schlette, E.J.; Lin, P.; Vega, F.; Miranda, R.N.; Claret, F.X.; Medeiros, L.J.; Rassidakis, G.Z. Expression of serine 194-phosphorylated Fas-associated death domain protein correlates with proliferation in B-cell non-Hodgkin lymphomas. Hum. Pathol. 2011, 42, 1117–1124. [Google Scholar] [CrossRef]
- Patel, S.; Murphy, D.; Haralambieva, E.; Abdulla, Z.A.; Wong, K.K.; Chen, H.; Gould, E.; Roncador, G.; Hatton, C.S.; Anderson, A.P.; et al. Increased Expression of Phosphorylated FADD in Anaplastic Large Cell and Other T-Cell Lymphomas. Biomark. insights 2014, 9, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.M.; Sebolt, K.A.; Hoff, B.A.; Boes, J.L.; Daniels, D.L.; Heist, K.A.; Galban, C.J.; Patel, R.M.; Zhang, J.; Beer, D.G.; et al. Phosphorylation of FADD by the kinase CK1alpha promotes KRASG12D-induced lung cancer. Sci. Signal. 2015, 8, ra9. [Google Scholar] [CrossRef] [PubMed]
- Schinske, K.A.; Nyati, S.; Khan, A.P.; Williams, T.M.; Johnson, T.D.; Ross, B.D.; Perez Tomas, R.; Rehemtulla, A. A Novel Kinase Inhibitor of FADD Phosphorylation Chemosensitizes through the Inhibition of NF-κB. Mol. Cancer Ther. 2011, 10, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.W.; Kim, J.H.; Ahn, Y.H.; Seo, J.; Ko, A.; Jeong, M.; Kim, S.J.; Ro, J.Y.; Park, K.M.; Lee, H.W.; et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat. Commun. 2012, 3, 978. [Google Scholar] [CrossRef] [PubMed]
- Feltham, R.; Silke, J. The small molecule that packs a punch: Ubiquitin-mediated regulation of RIPK1/FADD/caspase-8 complexes. Cell Death Differ. 2017, 24, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Lee, E.W.; Shin, J.; Seong, D.; Nam, Y.W.; Jeong, M.; Lee, S.H.; Lee, C.; Song, J. K6 linked polyubiquitylation of FADD by CHIP prevents death inducing signaling complex formation suppressing cell death. Oncogene 2018, 37, 4994–5006. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Tokunaga, F. Decreased linear ubiquitination of NEMO and FADD on apoptosis with caspase-mediated cleavage of HOIP. Biochem. Biophys. Res. Commun. 2017, 485, 152–159. [Google Scholar] [CrossRef]
- Tan, P.; Wang, A.; Chen, H.; Du, Y.; Qian, B.; Shi, H.; Zhang, Y.; Xia, X.; Fu, W. SPOP inhibits mice pancreatic stellate cell activation by promoting FADD degradation in cerulein-induced chronic pancreatitis. Exp. Cell Res. 2019, 111606. [Google Scholar] [CrossRef]
- Luo, J.; Chen, B.; Gao, C.X.; Xie, H.K.; Han, C.N.; Zhou, C.C. SPOP promotes FADD degradation and inhibits NF-kappaB activity in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 504, 289–294. [Google Scholar] [CrossRef]
- Chen, L.; Pei, H.; Lu, S.J.; Liu, Z.J.; Yan, L.; Zhao, X.M.; Hu, B.; Lu, H.G. SPOP suppresses osteosarcoma invasion via PI3K/AKT/NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 609–615. [Google Scholar] [CrossRef]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Oberst, A.; Pop, C.; Tremblay, A.G.; Blais, V.; Denault, J.B.; Salvesen, G.S.; Green, D.R. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J. Biol. Chem. 2010, 285, 16632–16642. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.P.; Nair, A.; Zhang, J. A novel function of RIP1 in postnatal development and immune homeostasis by protecting against RIP3-dependent necroptosis and FADD-mediated apoptosis. Front. Cell Dev. Biol. 2015, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Osborn, S.L.; Diehl, G.; Han, S.J.; Xue, L.; Kurd, N.; Hsieh, K.; Cado, D.; Robey, E.A.; Winoto, A. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13034–13039. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015, 22, 885–886. [Google Scholar] [CrossRef]
- Alappat, E.C.; Feig, C.; Boyerinas, B.; Volkland, J.; Samuels, M.; Murmann, A.E.; Thorburn, A.; Kidd, V.J.; Slaughter, C.A.; Osborn, S.L.; et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Mol. cell 2005, 19, 321–332. [Google Scholar] [CrossRef]
- Gomez-Angelats, M.; Cidlowski, J.A. Molecular evidence for the nuclear localization of FADD. Cell Death Differ. 2003, 10, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Beisner, D.R.; Chu, I.H.; Arechiga, A.F.; Hedrick, S.M.; Walsh, C.M. The requirements for Fas-associated death domain signaling in mature T cell activation and survival. J. Immunol. 2003, 171, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Osborn, S.L.; Sohn, S.J.; Winoto, A. Constitutive phosphorylation mutation in Fas-associated death domain (FADD) results in early cell cycle defects. J. Biol. Chem. 2007, 282, 22786–22792. [Google Scholar] [CrossRef] [PubMed]
- Papoff, G.; Trivieri, N.; Crielesi, R.; Ruberti, F.; Marsilio, S.; Ruberti, G. FADD-calmodulin interaction: A novel player in cell cycle regulation. Biochim. Biophys. Acta 2010, 1803, 898–911. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alappat, E.C.; Volkland, J.; Peter, M.E. Cell cycle effects by C-FADD depend on its C-terminal phosphorylation site. J. Biol. Chem. 2003, 278, 41585–41588. [Google Scholar] [CrossRef] [PubMed]
- Kabra, N.H.; Kang, C.; Hsing, L.C.; Zhang, J.; Winoto, A. T cell-specific FADD-deficient mice: FADD is required for early T cell development. Proc. Natl. Acad. Sci. USA 2001, 98, 6307–6312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kabra, N.H.; Cado, D.; Kang, C.; Winoto, A. FADD-deficient T cells exhibit a disaccord in regulation of the cell cycle machinery. J. Biol. Chem. 2001, 276, 29815–29818. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Thomas, E.; Barber, G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nature 2004, 432, 401–405. [Google Scholar] [CrossRef]
- Leulier, F.; Vidal, S.; Saigo, K.; Ueda, R.; Lemaitre, B. Inducible expression of double-stranded RNA reveals a role for dFADD in the regulation of the antibacterial response in Drosophila adults. Curr. Biol. 2002, 12, 996–1000. [Google Scholar] [CrossRef]
- Zhang, X.; Zang, S.; Li, C.; Wei, J.; Qin, Q. Molecular cloning and characterization of FADD from the orange-spotted grouper (Epinephelus coioides). Fish. Shellfish. Immunol. 2018, 74, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.P.; Henry, C.M.; Kearney, C.J.; Logue, S.E.; Feoktistova, M.; Tynan, G.A.; Lavelle, E.C.; Leverkus, M.; Martin, S.J. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 2013, 49, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Kawazu, M.; Ueno, T.; Fukumura, K.; Yamato, A.; Soda, M.; Yamashita, Y.; Choi, Y.L.; Yamasoba, T.; Mano, H. Cancer-associated missense mutations of caspase-8 activate nuclear factor-kappaB signaling. Cancer Sci. 2013, 104, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Shikama, Y.; Yamada, M.; Miyashita, T. Caspase-8 and caspase-10 activate NF-kappaB through RIP, NIK and IKKalpha kinases. Eur. J. Immunol. 2003, 33, 1998–2006. [Google Scholar] [CrossRef]
- Freer-Prokop, M.; O’Flaherty, J.; Ross, J.A.; Weyman, C.M. Non-canonical role for the TRAIL receptor DR5/FADD/caspase pathway in the regulation of MyoD expression and skeletal myoblast differentiation. Differentiation 2009, 78, 205–212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Dong, X.; Wang, H.; Li, J.; Yang, B.; Zhang, J.; Hua, Z.C. FADD regulates thymocyte development at the beta-selection checkpoint by modulating Notch signaling. Cell Death Dis. 2014, 5, e1273. [Google Scholar] [CrossRef]
- Pajerowski, A.G.; Nguyen, C.; Aghajanian, H.; Shapiro, M.J.; Shapiro, V.S. NKAP is a transcriptional repressor of notch signaling and is required for T cell development. Immunity 2009, 30, 696–707. [Google Scholar] [CrossRef]
- Song, T.; Liu, J.Y.; Yang, J.J. NKAP plays an oncogenic function partly through AKT signaling pathway in hepatocellular carcinoma. Neoplasma 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Gao, T.; Zhang, L.; Chen, X.; Pang, Q.; Wang, Y.; Wang, D.; Li, J.; Liu, Q. NKAP alters tumor immune microenvironment and promotes glioma growth via Notch1 signaling. J. Exp. Clin. Cancer Res. 2019, 38, 291. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, H.; Yin, Y.; Li, Q.; Zhang, M. NKAP functions as an oncogene and its expression is induced by CoCl2 treatment in breast cancer via AKT/mTOR signaling pathway. Cancer Manag. Res. 2018, 10, 5091–5100. [Google Scholar] [CrossRef] [PubMed]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Yang, B.Y.; Wang, J.Y.; Mo, X.; Zhang, J.; Hua, Z.C. FADD is essential for glucose uptake and survival of thymocytes. Biochem. Biophys. Res. Commun. 2014, 451, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Gan, Z.; Jiang, W.; Zhang, X.; Hua, Z.C. Comparative proteomics analysis reveals roles for FADD in the regulation of energy metabolism and proteolysis pathway in mouse embryonic fibroblast. Proteomics 2013, 13, 2398–2413. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Gan, Z.; Jiang, W.; Zhang, X.; Hua, Z.C. Functional specific roles of FADD: Comparative proteomic analyses from knockout cell lines. Mol. Biosyst. 2013, 9, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Wang, X.; Zha, D.; Gan, Z.; Cai, F.; Du, P.; Yang, Y.; Yang, B.; Zhang, X.; Yao, C.; et al. FADD is a key regulator of lipid metabolism. EMBO Mol. Med. 2016, 8, 895–918. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Muller, S.; Chen, Z.G.; Pan, L.; Tighiouart, M.; Shin, D.M.; Khuri, F.R.; Sun, S.Y. Prognostic impact of Fas-associated death domain, a key component in death receptor signaling, is dependent on the presence of lymph node metastasis in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2013, 14, 365–369. [Google Scholar] [CrossRef]
- Rasamny, J.J.; Allak, A.; Krook, K.A.; Jo, V.Y.; Policarpio-Nicolas, M.L.; Sumner, H.M.; Moskaluk, C.A.; Frierson, H.F., Jr.; Jameson, M.J. Cyclin D1 and FADD as biomarkers in head and neck squamous cell carcinoma. Otolaryngol. Head Neck Surg. 2012, 146, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Y.; Hammache, K.; He, L.; Zhu, B.; Cheng, W.; Hua, Z.C. The role of FADD in pancreatic cancer cell proliferation and drug resistance. Oncol. Lett. 2017, 13, 1899–1904. [Google Scholar] [CrossRef]
- Ramos-Miguel, A.; Garcia-Sevilla, J.A.; Barr, A.M.; Bayer, T.A.; Falkai, P.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A.; Honer, W.G.; Garcia-Fuster, M.J. Decreased cortical FADD protein is associated with clinical dementia and cognitive decline in an elderly community sample. Mol. Neurodegener. 2017, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Tanaka, N.; Shimada, K.; Matsumura, Y.; Miyake, M.; Anai, S.; Tomioka, A.; Okajima, E.; Hirayama, A.; Fujimoto, K.; et al. Phosphorylation status of Fas-associated death domain protein is associated with biochemical recurrence after radical prostatectomy. Urology 2013, 81, 607–610. [Google Scholar] [CrossRef]
- Matsuyoshi, S.; Shimada, K.; Nakamura, M.; Ishida, E.; Konishi, N. FADD phosphorylation is critical for cell cycle regulation in breast cancer cells. Br. J. Cancer 2006, 94, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Nakamura, M.; Ishida, E.; Konishi, N. Molecular roles of MAP kinases and FADD phosphorylation in prostate cancer. Histol. Histopathol. 2006, 21, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Wachters, J.E.; Schrijvers, M.L.; Slagter-Menkema, L.; Mastik, M.; Langendijk, J.A.; de Bock, G.H.; Roodenburg, J.L.; van der Laan, B.; van der Wal, J.E.; Schuuring, E. Phosphorylated FADD is not prognostic for local control in T1-T2 supraglottic laryngeal carcinoma treated with radiotherapy. Laryngoscope 2017, 127, E301–E307. [Google Scholar] [CrossRef]
- Yin, A.; Jiang, Y.; Zhang, X.; Luo, H. Overexpression of FADD enhances 5-fluorouracil-induced apoptosis in colorectal adenocarcinoma cells. Med. Oncol. 2010, 27, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.A.; Ng, W.H.; Lam, P.Y. FasL and FADD delivery by a glioma-specific and cell cycle-dependent HSV-1 amplicon virus enhanced apoptosis in primary human brain tumors. Mol. Cancer 2010, 9, 270. [Google Scholar] [CrossRef]
- Komata, T.; Koga, S.; Hirohata, S.; Takakura, M.; Germano, I.M.; Inoue, M.; Kyo, S.; Kondo, S.; Kondo, Y. A novel treatment of human malignant gliomas in vitro and in vivo: FADD gene transfer under the control of the human telomerase reverse transcriptase gene promoter. Int. J. Oncol. 2001, 19, 1015–1020. [Google Scholar] [CrossRef]
- Mishima, K.; Nariai, Y.; Yoshimura, Y. Carboplatin induces Fas (APO-1/CD95)-dependent apoptosis of human tongue carcinoma cells: Sensitization for apoptosis by upregulation of FADD expression. Int. J. Cancer 2003, 105, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Cheng, C.L.; Ho, H.C.; Wang, S.S.; Chiu, K.Y.; Su, C.K.; Ou, Y.C.; Lin, C.C. Nortriptyline induces mitochondria and death receptor-mediated apoptosis in bladder cancer cells and inhibits bladder tumor growth in vivo. Eur. J. Pharmacol. 2015, 761, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Karaca, B.; Atmaca, H.; Bozkurt, E.; Kisim, A.; Uzunoglu, S.; Karabulut, B.; Sezgin, C.; Sanli, U.A.; Uslu, R. Combination of AT-101/cisplatin overcomes chemoresistance by inducing apoptosis and modulating epigenetics in human ovarian cancer cells. Mol. Biol. Rep. 2013, 40, 3925–3933. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Harper, N.; Walewska, R.; Dyer, M.J.; Cohen, G.M. Enhanced Fas-associated death domain recruitment by histone deacetylase inhibitors is critical for the sensitization of chronic lymphocytic leukemia cells to TRAIL-induced apoptosis. Mol. Cancer Ther. 2009, 8, 3088–3097. [Google Scholar] [CrossRef]
- Shimada, K.; Matsuyoshi, S.; Nakamura, M.; Ishida, E.; Kishi, M.; Konishi, N. Phosphorylation of FADD is critical for sensitivity to anticancer drug-induced apoptosis. Carcinogenesis 2004, 25, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).