Experimental Models to Define the Genetic Predisposition to Liver Cancer

 ,
, 

Abstract
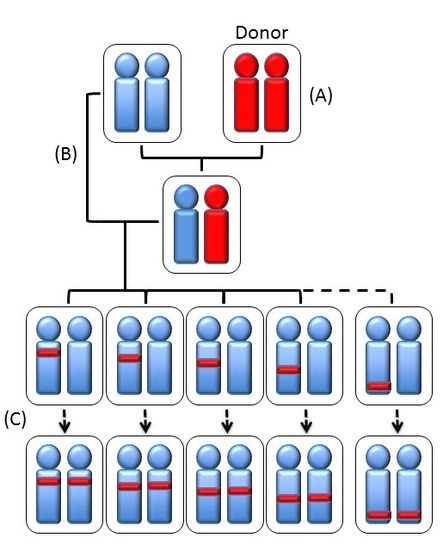
1. Introduction
2. The Individual Susceptibility to Liver Cancer
3. The Genetic Model to Study the Rodent Susceptibility to Liver Carcinogenesis
4. The Loci Controlling the Susceptibility to HCC
5. The Genes Involved
6. Phenotypic Effects of the Variation of the Genetic Susceptibility to Liver Cancer
6.1. The Role of c-Myc
6.2. The Ras-Family Genes and Related Pathways
6.3. Mybl2 and AKT and ERK1/2 Signaling
6.4. The Methionine Cycle
7. Human Hepatocarcinogenesis
7.1. Cell Cycle Deregulation
7.2. ERK Signaling
7.3. PI3K/AKT/mTOR Signaling
7.4. The Role of MYBL2
7.5. The Role of Cell Cycle
7.6. The Role of DNA Repair
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bosch, F.X.; Ribes, J.; Diaz, M.; Cleries, R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004, 127, S5–S16. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Feo, F.; Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D.F. Genetic alterations in liver carcinogenesis. Crit. Rev. Oncog. 2000, 11, 19–62. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; London, V.T. Epidemiology and natural history of hepatocellular carcinoma. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 3–23. [Google Scholar] [CrossRef] [PubMed]
- El Serag, H.B.; Mason, A.C. Risk factors for the rising rates of primary liver cancer in the United States. Arch. Intern. Med. 2000, 160, 3227–3230. [Google Scholar] [CrossRef] [PubMed]
- Ogimoto, I.; Shibata, A.; Kurozawa, Y.; Nose, T.; Yoshimura, T.; Suzuki, H.; Iwai, N.; Sakata, R.; Fujita, Y.; Ichikawa, S.; et al. Risk of death due to hepatocellular carcinoma among drinkers and ex-drinkers. Univariate analysis of JACC study data. Kurume Med. J. 2004, 51, 59–70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V.; WHO Intenational Agency for Research on cancer Monograph Working Group. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293. [Google Scholar] [CrossRef]
- Kuper, H.; Tzonou, A.; Kaklamani, E.; Hsieh, C.C.; Lagiou, P.; Adami, H.O. Tobacco smoking, alcohol consumption and their interaction in the causation of hepatocellular carcinoma. Int. J. Cancer 2000, 85, 498–502. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco Smoke and Involuntary Smoking; IARC: Lyon, France, 2004. [Google Scholar]
- Dragani, T.A. Risk of HCC: Genetic heterogeneity and complex genetics. J. Hepatol. 2010, 5, 252–257. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Frau, M.; Tomasi, M.L.; Feo, F.; Pascale, R.M. Deregulation of signalling pathways in prognostic subtypes of hepatocellular carcinoma: Novel insights from interspecies comparison. Biochim. Biophys. Acta 2012, 1826, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Degawa, M. Induction of cytochrome P450 isoforms by carcinogenic aromatic amines and carcinogenic susceptibility of rodent animals. Pharmacogenetics 1995, 5, S80–S83. [Google Scholar] [CrossRef] [PubMed]
- Raunio, H.; Rautio, A.; Gullsten, H.; Pelkonen, O. Polymorphisms of CYP2A6 and its practical consequences. Br. J. Clin. Pharmacol. 2001, 52, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Higashi, T.; Kano, S.; Tateishi, N.; Kido, Y.; Mori, T.; Higashi, K.; Sakamoto, Y. Decreased activation of carcinogens in the liver of carcinogen-resistant rats. Cancer Res. 1989, 49, 5352–5357. [Google Scholar] [PubMed]
- Fujita, K.; Kamataki, T. Predicting the mutagenicity of tobacco-related Nnitrosamines in humans using 11 strains of Salmonella typhimurium YG7108, each coexpressing a form of human cytochrome P450 along with NADPH-cytochrome P450 reductase. Environ. Mol. Mutagen. 2001, 38, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.L.; Gallagher, E.P.; Bammler, T.K.; Kunze, K.L. Role of cytochrome P4501A2 in chemical carcinogenesis: Implications for human variability in expression and enzyme activity. Pharmacogenetics 1995, 5, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Quattrochi, L.C.; Vu, T.; Tukey, R.H. The human CYP1A2 gene and induction of 3-methylcholanthrene. A region of DNA that supports Ahreceptor binding and promoter-specific induction. J. Biol. Chem. 1994, 269, 6949–6954. [Google Scholar]
- Dragani, T.A.; Manenti, G.; Della Porta, G. Genetic susceptibility to murine hepatocarcinogenesis is associated with high growth rate of NDEA-initiated hepatocytes. J. Cancer Res. Clin. Oncol. 1987, 113, 223–229. [Google Scholar] [CrossRef]
- Manenti, G.; Binelli, G.; Gariboldi, M.; Canzian, F.; De Gregorio, L.; Falvella, F.S.; Dragani, T.A.; Pierotti, M.A. Multiple loci affect genetic predisposition to hepatocarcinogenesis in mice. Genomics 1994, 23, 118–124. [Google Scholar] [CrossRef]
- Dragani, T.A. Manenti, G.; Della Porta, G. Quantitative analysis of genetic susceptibility to liver and lung carcinogenesis in mice. Cancer Res. 1991, 51, 6299–6303. [Google Scholar]
- Drinkwater, N.R.; Ginsler, J.J. Genetic control of hepatocarcinogenesis in C57BL/6J and C3H/HeJ inbred mice. Carcinogenesis 1986, 7, 1701–1707. [Google Scholar] [CrossRef]
- Bilger, A.; Bennett, L.M.; Carabeo, R.A.; Chiaverotti, T.A.; Dvorak, C.; Liss, K.M.; Schadewald, S.A.; Pitot, H.C.; Drinkwater, N.R. A potent modifier of liver cancer risk on distal mouse chromosome 1: Linkage analysis and characterization of congenic lines. Genetics 2004, 167, 859–866. [Google Scholar] [CrossRef]
- Asamoto, M.; Tsuda, H.; Kato, T.; Ito, N.; Masuki, T.; Hashimoto, Y.; Nagase, S. Strain differences in susceptibility to 2-acetylaminofluorene and phenobarbital promotion of rat hepatocarcinogenesis: Immunohistochemical analysis of cytochrome P450 isozyme induction by 2-acetylaminofluorene and phenobarbital. Jpn. J. Cancer Res. 1989, 80, 1041–1046. [Google Scholar] [CrossRef]
- Yoshimoto, F.; Masuda, S.; Higashi, T.; Nishii, T.; Takamisawa, I.; Tateishi, N.; Sakamoto, Y. Comparison of drug-metabolizing activities in the livers of carcinogen-sensitive parent rats and carcinogen-resistant descendants. Cancer Res. 1985, 45, 6155–6159. [Google Scholar]
- Denda, A.; Kitayama, W.; Konishi, Y.; Yan, Y.; Fukamachi, Y.; Miura, M.; Gotoh, S.; Ikemura, K.; Abe, T.; Higashi, T.; et al. Genetic properties for the suppression of development of putative preneoplastic glutathione S-transferase placental form-positive foci in the liver of carcinogenresistant DRH strain rats. Cancer Lett. 1999, 140, 59–67. [Google Scholar] [CrossRef]
- Zeng, Z.-Z.; Higashi, S.; Kitayama, S.; Denda, A.; Yan, Y.; Matsuo, K.; Konishi, Y.; Hiai, H.; Higashi, K. Genetic resistance to chemical carcinogen-induced preneoplastic hepatic lesions in DRH strain rats. Cancer Res. 2000, 60, 2876–2881. [Google Scholar]
- Yan, Y.; Zeng, Z.Z.; Higashi, S.; Denda, A.; Konishi, Y.; Onishi, S.; Ueno, H.; Higashi, K.; Hiai, H. Resistance of DRH strain rats to chemical carcinogenesis of liver: Genetic analysis of later progression stage. Carcinogenesis 2002, 23, 189–196. [Google Scholar] [CrossRef][Green Version]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Gaspa, L.; Dragani, T.A.; Feo, F. The BN rat strain carries dominant hepatocarcinogen resistance loci. Carcinogenesis 1996, 17, 1765–1768. [Google Scholar] [CrossRef][Green Version]
- Wood, G.A.; Korkola, J.E.; Lee, V.M.; Sarma, D.S.R.; Archer, M.C. Resistance of Copenhagen rats to chemical induction of glutathione S-transferase 7-7-positive liver foci. Carcinogenesis 1997, 18, 1745–1750. [Google Scholar] [CrossRef][Green Version]
- Wood, G.A.; Sarma, D.S.R.; Archer, M.C. Resistance to the promotion of glutathione S-transferase 7-7-positive liver lesions in Copenhagen rats. Carcinogenesis 1999, 20, 1169–1175. [Google Scholar] [CrossRef][Green Version]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogeninduced hyperplastic lesions in a new model of sequential analysis of liver carcinogenesis. Am. J. Pathol. 1997, 88, 595–618. [Google Scholar]
- Mott, R.; Flint, J. Simultaneous detection and fine mapping of quantitative trait loci in mouse using heterogeneous stocks. Genetics 2002, 160, 1609–1618. [Google Scholar]
- Snell, G.D. Methods for study of histocompatibility genes. J. Genet. 1948, 49, 87–108. [Google Scholar] [CrossRef]
- Cowley, A.W., Jr.; Liang, M.; Roman, R.J.; Greene, A.S.; Jacob, H.J. Consomic rat model system for physiological genomics. Acta Physiol. Scand. 2004, 181, 585–592. [Google Scholar] [CrossRef]
- Flin, J.; Waldar, W.; Shifman, S.; Mott, R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat. Rev. Genet. 2005, 6, 271–286. [Google Scholar] [CrossRef]
- Darvasi, A. Experimental strategies for the genetic dissection of complex traits in animal models. Nat. Genet. 1998, 18, 19–24. [Google Scholar] [CrossRef]
- Wang, D.; Lemon, W.J.; You, M. Linkage disequilibrium mapping of novel lung tumor susceptibility quantitative trait loci in mice. Oncogene 2002, 21, 6858–6865. [Google Scholar] [CrossRef][Green Version]
- Manenti, A.; Stafford, L.; De Gregorio, M.; Gariboldi, F.S.; Favella, P.; Avner, T.A.; Dragani, T.A. Linkage disequilibrium and physical mapping of Pas1 in mice. Genome Res. 1999, 9, 639–646. [Google Scholar]
- Poole, T.M.; Drinkwater, N.R. Two genes abrogate the inhibition of murine hepatocarcinogenesis by ovarian hormones. Proc. Natl. Acad. Sci. USA 1996, 93, 5848–5853. [Google Scholar] [CrossRef]
- Lee, G.H.; Bennett, L.M.; Carabeo, R.A.; Drinkwater, N.R. Identification of hepatocarcinogenesis-resistance genes in DBA/2 mice. Genetics 1995, 139, 387–395. [Google Scholar]
- Smith, G.S.; Walfgord, R.L.; Weith, A. Lifespan and incidence of cancer and other diseases in selected long-lived inbred mice and their F1 hybrids. J. Natl. Cancer Inst. 1973, 50, 1195–1213. [Google Scholar] [CrossRef]
- Diwan, B.A.; Rice, J.M.; Ohshima, M.M.; Ward, J.M. Interstrain differences in susceptibility to liver carcinogenesis initiated by N-nitrosodiethylamine and its promotion by phenobarbital in C57BL/6NCr, C3H/HeNCrMTVand DBA/2NCr mice. Carcinogenesis 1986, 7, 215–220. [Google Scholar] [CrossRef]
- Lee, G.H.; Drinkwater, N.R. The Hcr (hepatocarcinogen resistance) loci of DBA/2J mice partially suppress phenotypic expression of the Hcs (hepatocarcinogen sensitivity) loci of C3H/HeJ mice. Carcinogenesis 1995, 16, 1993–1996. [Google Scholar] [CrossRef]
- Tatematsu, M.; Nagamine, Y.; Farber, E. Redifferentiation as a basis for remodeling of carcinogen-induced hepatocyte nodules to normal appearing liver. Cancer Res. 1983, 43, 5049–5058. [Google Scholar]
- Melhem, M.F.; Kunz, H.W.; Gill, T.J., III. A major histocompatibility complex-linked locus in the rat critically influences resistance to diethylnitrosamine carcinogenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 1967–1971. [Google Scholar] [CrossRef]
- DeMiglio, M.R.; Canzian, F.; Pascale, R.M.; Simile, M.M.; Muroni, M.R.; Calvisi, D.F.; Romeo, G.; Feo, F. Identification of genetic loci controlling hepatocarcinogenesis on rat chromosomes 7 and 10. Cancer Res. 1999, 59, 4651–4657. [Google Scholar]
- De Miglio, M.R.; Pascale, R.M.; Simile, M.M.; Muroni, M.R.; Calvisi, D.F.; Virdis, P.; Bosinco, G.M.; Frau, M.; Seddaiu, M.A.; Ladu, S.; et al. Chromosome mapping of multiple loci affecting the genetic predisposition to rat liver carcinogenesis. Cancer Res. 2002, 62, 4459–4463. [Google Scholar]
- De Miglio, M.R.; Pascale, R.M.; Simile, M.M.; Muroni, M.R.; Virdis, P.; Kwong, K.M.-T.; Wong, L.K.L.; Bosinco, G.M.; Pulina, F.R.; Calvisi, D.F.; et al. Polygenic control of hepatocarcinogenesis in Copenhagen x F344 rats. Int. J. Cancer 2004, 111, 9–16. [Google Scholar] [CrossRef]
- De Miglio, M.R.; Simile, M.M.; Muroni, M.R.; Calvisi, D.F.; Virdis, P.; Asara, G.; Frau, M.; Bosinco, G.M.; Seddaiu, M.A.; Daino, L.; et al. Phenotypic reversion of rat neoplastic liver nodules is under genetic control. Int. J. Cancer 2003, 105, 70–75. [Google Scholar] [CrossRef]
- De Miglio, M.R.; Virdis, P.; Calvisi, D.F.; Frau, M.; Muroni, M.R.; Simile, M.M.; Daino, L.; Careddu, G.M.; Sanna-Passino, E.; Pascale, R.M.; et al. Mapping a sex hormone-sensitive gene determining female resistance to liver carcinogenesis in a congenic F344.BN-Hcs4 rat. Cancer Res. 2006, 66, 10384–10390. [Google Scholar] [CrossRef]
- Canzian, F.; Ushijima, T.; Pascale, R.M.; Sugimura, T.; Dragani, T.A.; Nagao, M. Construction of a phylogenetic tree for inbred strains of rat by arbitrarily primed polymerase chain reaction (AP-PCR). Mamm. Genome 1995, 6, 231–235. [Google Scholar] [CrossRef]
- Sargent, L.; Dragan, Y.; Xu, Y.H.; Sattler, G.; Wiley, J.; Pitot, H.C. Specific chromosomal changes in albumin simian virus 40 T antigen transgenic rat liver neoplasms. Cancer Res. 1996, 56, 2985–2991. [Google Scholar]
- De Miglio, M.R.; Muroni, M.R.; Simile, M.M.; Virdis, P.; Asara, G.; Frau, M.; Calvisi, D.F.; Seddaiu, M.A.; Pascale, R.M.; Feo, F. Frequent loss of heterozygosity at the Hcr1 (hepatocarcinogenesis resistance) locus on chromosome 10 in primary hepatocellular carcinomas from LFF1 rat strain. Hepatology 2001, 33, 1110–1117. [Google Scholar] [CrossRef]
- Pascale, R.M.; De Miglio, M.R.; Muroni, M.R.; Simile, M.M.; Daino, L.; Seddaiu, M.A.; Nufris, A.; Gaspa, L.; Deiana, L.; Feo, F. c-Myc amplification in pre-malignant and malignant lesions induced in rat liver by the resistant hepatocyte model. Int. J. Cancer. 1996, 68, 136–142. [Google Scholar] [CrossRef]
- De Miglio, M.R.; Simile, M.M.; Muroni, M.R.; Pusceddu, S.; Calvisi, D.; Carru, A.; Seddaiu, M.A.; Daino, L.; Deiana, L.; Pascale, R.M.; et al. Correlation of c-myc overexpression and amplification with progression of preneoplastic liver lesions to malignancy in the poorly susceptible Wistar rat strain. Mol. Carcinog. 1999, 25, 21–29. [Google Scholar] [CrossRef]
- Falvella, F.S.; Pascale, R.M.; Gariboldi, M.; Manenti, G.; De Miglio, M.R.; Simile, M.M.; Dragani, T.A.; Feo, F. Stearoyl-CoA desaturase 1 (Scd1) gene overexpression is associated with genetic predisposition to hepatocarcinogenesis in mice and rats. Carcinogenesis 2002, 23, 1933–1936. [Google Scholar] [CrossRef]
- Manenti, G.; Galvan, A.; Falvella, F.S.; Pascale, R.M.; Spada, E.; Milani, S.; Gonzalez Neira, A.; Feo, F.; Dragani, T.A. Genetic control of resistance to hepatocarcinogenesis by the mouse Hpcr3 locus. Hepatology 2008, 48, 617–623. [Google Scholar] [CrossRef]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, H.G.; Lee, Y.H.; Sears, N.C.; Coulouarn, C.; Conner, E.A.; Factor, V.M.; Roskams, M.T.; Thorgeirsson, S.S. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachman, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Cigliano, A.; Pilo, M.G.; Li, L.; Latte, G.; Szydlowska, M.; Simile, M.M.; Paliogiannis, P.; Che, L.; Pes, G.M.; Palmieri, G.; et al. Deregulated c-Myc requires a functional HSF1 for experimental and human hepatocarcinogenesis. Oncotarget 2017, 8, 90638–90650. [Google Scholar] [CrossRef]
- Xu, Z.; Xu, M.; Liu, P.; Zhang, S.; Shang, R.; Qiao, Y.; Che, L.; Ribback, S.; Cigliano, A.; Evert, K.; et al. The mTORC2-Akt cascade is crucial for c-Myc to promote hepatocarcinogenesis in mice and men. Hepatology 2019. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pinna, F.; Pellegrino, R.; Sanna, V.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Frau, M.; Tomasi, M.L.; et al. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int. J. Cancer 2008, 123, 2057–2064. [Google Scholar] [CrossRef]
- Wierstra, I.; Alves, J. FOXM1, a typical proliferation-associated transcription factor. Biol. Chem. 2007, 388, 1257–1274. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pinna, F.; Ladu, S.; Pellegrino, R.; Simile, M.M.; Frau, M.; De Miglio, M.R.; Tomasi, L.; Sanna, V.; Muroni, M.R.; et al. Forkhead box M1B is a determinant of rat susceptibility to hepatocarcinogenesis and sustains ERK activity in human HC. Gut 2009, 58, 679–687. [Google Scholar] [CrossRef]
- Shin, E.; Kim, S.H.; Jeong, H.Y.; Jang, J.J.; Lee, K. Nuclear expression of S-phase kinase-associated protein 2 predicts poor prognosis of hepatocellular carcinoma. APMIS 2012, 120, 349–357. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pinna, F.; Ladu, S.; Muroni, M.R.; Frau, M.; Demartis, I.; Tomasi, M.L.; Sini, M.; Simile, M.M.; Seddaiu, M.A.; et al. The degradation of cell cycle regulators by SKP2/CKS1 ubiquitin ligase is genetically controlled in rodent liver cancer and contributes to determine the susceptibility to the disease. Int. J. Cancer 2010, 126, 1275–1281. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Pinna, F.; Frau, M.; Tomasi, M.L.; Sini, M.; Simile, M.M.; Bonelli, P.; Muroni, M.R.; Seddaiu, M.A.; et al. SKP2 and CKS1 promote degradation of cell cycle regulators and are associated with hepatocellular carcinoma prognosis. Gastroenterology 2009, 137, 1816–1826. [Google Scholar] [CrossRef]
- Xu, H.; Choe, C.; Shin, S.H.; Park, S.W.; Kim, H.S.; Jung, S.H.; Yim, S.H.; Kim, T.M.; Chung, Y.J. Silencing of KIF14 interferes with cell cycle progression and cytokinesis by blocking the p27(Kip1) ubiquitination pathway in hepatocellular carcinoma. Exp. Mol. Med. 2014, 46, e97. [Google Scholar] [CrossRef]
- Huang, Y.; Tong, S.; Tai, A.W.; Hussain, M.; Lok, A.S. Hepatitis B virus core promoter mutations contribute to hepatocarcinogenesis by deregulating SKP2 and its target p21. Gastroenterology 2011, 141, 1412–1421. [Google Scholar] [CrossRef]
- Delogu, S.; Wang, C.; Cigliano, A.; Utpatel, K.; Sini, M.; Longerich, T.; Waldburger, N.; Breuhahn, K.; Jiang, L.; Ribback, S.; et al. SKP2 cooperates with N-Ras or AKT to induce liver tumor development in mice. Oncotarget 2015, 6, 2222–2234. [Google Scholar] [CrossRef]
- Parekh, P.; Rao, K.B.V. Overexpression of cyclin D1 is associated with elevated levels of MAP kinases, Akt and Pak1 during diethylnitrosamine-induced progressive liver carcinogenesis. Cell Biol. Int. 2007, 31, 35–43. [Google Scholar] [CrossRef]
- Factor, V.; Oliver, A.L.; Panta, G.R.; Thorgeirsson, S.S.; Sonenshein, G.E.; Arsura, M. Roles of Akt/PKB and IKK complex in constitutive induction of NF-kappaB in hepatocellular carcinomas of transforming growth factor alpha/c-myc transgenic mice. Hepatology 2001, 34, 32–41. [Google Scholar] [CrossRef]
- Datta, J.; Majumder, S.; Kutay, H.; Motiwala, T.; Frankel, W.; Costa, R.; Cha, H.C.; MacDougald, O.A.; Jacob, S.T.; Ghoshal, K. Metallothionein expression is suppressed in primary human hepatocellular carcinomas and is mediated through inactivation of CCAAT/enhancer binding protein alpha by phosphatidylinositol 3-kinase signaling cascade. Cancer Res. 2007, 67, 2736–2746. [Google Scholar]
- Ho, C.; Wang, C.; Mattu, S.; Destefanis, G.; Ladu, S.; Delogu, S.; Armbruster, J.; Fan, L.; Lee, S.A.; Jiang, L.; et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012, 55, 833–845. [Google Scholar]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6, 20484. [Google Scholar] [CrossRef]
- Frau, M.; Ladu, S.; Calvisi, D.F.; Simile, M.M.; Bonelli, P.; Daino, L.; Tomasi, M.L.; Seddaiu, M.A.; Feo, F.; Pascale, R.M. Mybl2 expression is under genetic control and contributes to determine a hepatocellular carcinoma susceptible phenotype. J. Hepatol. 2011, 55, 111–119. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Simile, M.M.; Ladu, S.; Frau, M.; Tomasi, M.-L.; Demartis, M.I.; Daino, L.; Seddaiu, M.A.; Brozzetti, S.; Feo, F.; et al. Activation of v-Myb avian myeloblastosis viral oncogene homolog-like2 (MYBL2)–LIN9 complex contributes to human hepatocarcinogenesis and identifies a subset of hepatocellular carcinoma with mutant p53. Hepatology 2011, 53, 1226–1236. [Google Scholar] [CrossRef]
- Xu, Y.; Qu, X.; Zhang, X.; Luo, Y.; Zhang, Y.; Luo, Y.; Hou, K.; Liu, Y. Midkine positively regulates the proliferation of human gastric cancer cells. Cancer Lett. 2009, 279, 137–144. [Google Scholar] [CrossRef]
- Xiao, L.; Gong, L.L.; Yuan, D.; Deng, M.; Zeng, X.M.; Chen, L.L.; Zhang, L.; Yan, Q.; Liu, J.P.; Hu, X.H.; et al. Protein phosphatase-1 regulates Akt1 signal transduction pathway to control gene expression, cell survival and differentiation. Cell Death Differ. 2010, 17, 1448–1462. [Google Scholar] [CrossRef]
- Li, X.; Tao, J.; Cigliano, A.; Sini, M.; Calderaro, J.; Azoulay, D.; Wang, C.; Liu, Y.; Jiang, L.; Evert, K.; et al. Co-activation of PIK3CA and Yap promotes development of hepatocellular and cholangiocellular tumors in mouse and human liver. Oncotarget 2015, 6, 10102–10115. [Google Scholar] [CrossRef]
- Bremer, J.; Greenberg, D.M. Biosynthesis of choline in vitro. Biochim. Biophys. Acta 1960, 37, 173–175. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Ramani, K.; Mato, J.M.; Lu, S.C. Role of methionine adenosyltransferase genes in hepatocarcinogenesis. Cancers 2011, 3, 1480–1497. [Google Scholar] [CrossRef]
- Garcea, R.; Pascale, R.; Daino, L.; Frassetto, S.; Cozzolino, P.; Ruggio, M.E.; Vannini, M.G.; Gaspa, L.; Feo, F. Variations in ornithine decarboxylase activity and S-adenosyl-methionine and S-methylthioadenosine contents during the development of diethylnitrosamine-induced liver hyperplastic nodules and hepatocellular carcinomas. Carcinogenesis 1987, 8, 653–658. [Google Scholar] [CrossRef]
- Mato, M.; Corrales, F.J.; Lu, S.C.; Avila, M.A. S-Adenosylmethionine: A control switch that regulates liver function. FASEB J. 2002, 16, 15–26. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Simile, M.M.; Ladu, S.; Pellegrino, R.; De Murtas, V.; Pinna, F.; Tomasi, M.L.; Frau, M.; Virdis, P.; De Miglio, M.R.; et al. Altered methionine metabolism and global DNA methylation in liver cancer: Relationship with genomic instability and prognosis. Int. J. Cancer 2007, 121, 2410–2420. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Metabolic regulatory properties of S-adenosylmethionine and S-adenosylhomocysteine. Clin. Chem. Lab. Med. 2007, 45, 1694–1699. [Google Scholar] [CrossRef]
- Garcea, R.; Daino, L.; Pascale, R.M.; Simile, M.M.; Puddu, M.; Frassetto, S.; Cozzolino, P.; Seddaiu, M.A.; Gaspa, L.; Feo, F. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: Role of remodeling and apoptosis. Cancer Res. 1989, 49, 1850–1856. [Google Scholar]
- Pascale, R.M.; Marras, V.; Simile, M.M.; Daino, L.; Pinna, G.; Bennati, S.; Carta, M.; Seddaiu, M.A.; Massarelli, G.; Feo, F. Chemoprevention of rat liver carcinogenesis by S-adenosyl-l-methionine: A long-term study. Cancer Res. 1992, 52, 4979–4986. [Google Scholar]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Nufris, A.; Daino, L.; Seddaiu, M.A.; Rao, P.M. Chemoprevention by S-adenosylmethionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis 1995, 16, 427–430. [Google Scholar] [CrossRef]
- Simile, M.M.; Saviozzi, M.; De Miglio, M.R.; Muroni, M.R.; Nufris, A.; Pascale, R.M.; Malvaldi, G.; Feo, F. Persistent chemopreventive effect of S-adenosyl-L-methionine on the development of liver putative preneoplastic lesions induced by thiobenzamide in diethylnitrosamine-initiated rats. Carcinogenesis 1996, 17, 1533–1537. [Google Scholar] [CrossRef]
- Lu, S.C.; Ramani, K.; Ou, X.; Lin, M.; Yu, V.; Ko, K.; Park, R.; Bottiglieri, T.; Tsukamoto, H.; Kanel, G.; et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology 2009, 50, 462–471. [Google Scholar] [CrossRef]
- Cai, J.; Mao, Z.; Hwang, J.J.; Lu, S.C. Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res. 1998, 58, 1444–1450. [Google Scholar]
- Frau, M.L.; Tomasi, M.M.; Simile, M.I.; Demartis, F.; Salis, G.; Latte, D.F.; Calvisi, M.A.; Seddaiu, L.; Daino, L.; Feo, C.F.; et al. Role of transcriptional and post-transcriptional regulation of methionineadenosyltransferases in liver cancer progression. Hepatology 2012, 56, 165–175. [Google Scholar] [CrossRef]
- Lu, S.C.; Alvarez, L.; Huang, Z.Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 5560–5565. [Google Scholar] [CrossRef]
- Torres, L.; Avila, M.A.; Carretero, M.V.; Latasa, M.U.; Caballería, J.; López-Rodas, G.; Boukaba, A.; Lu, S.C.; Franco, L.; Mato, J.M. Liver-specific methionine adenosyltransferase MAT1A gene expression is associated with a specific pattern of promoter methylation and histone acetylation: Implications for MAT1A silencing during transformation. FASEB J. 2000, 14, 95–102. [Google Scholar] [CrossRef]
- Tomasi, M.L.; Li, T.W.; Li, M.; Mato, J.M.; Lu, S.C. Inhibition of human Methionine Adsenosyltransferase 1A transcription by coding region methylation. J. Cell. Physiol. 2012, 227, 1583–1591. [Google Scholar] [CrossRef]
- Yang, H.; Huang, Z.Z.; Zeng, Z.; Chen, C.; Selby, R.R.; Lu, S.C. Role of promoter methylation in increased methionine adenosyltransferase 2A expression in human liver cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, 184–190. [Google Scholar] [CrossRef]
- Vázquez-Chantada, M.; Fernández-Ramos, D.; Embade, N.; Martínez-Lopez, N.; Varela-Rey, M.; Woodhoo, A.; Luka, Z.; Wagner, C.; Anglim, P.P.; Finnell, R.H.; et al. HuR/-methyl-HuR and AUF1 regulate the MAT expressed during liver proliferation, differentiation, and carcinogenesis. Gastroenterology 2010, 138, 1943–1953. [Google Scholar] [CrossRef]
- Brennan, M.C.M.; Steitz, J.A. HuR and mRNA stability. Cell. Mol. Life Sci. 2001, 58, 266–277. [Google Scholar] [CrossRef]
- Lal, A.; Mazan-Mamczarz, K.; Kawai, T.; Yang, X.; Martindale, J.L.; Gorospe, M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004, 23, 3092–3102. [Google Scholar] [CrossRef]
- Eden, F.; Gaudet, A.; Waghmare, R.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Mann, C.D.; Neal, C.P.; Garcea, G.; Manson, M.M.; Dennison, A.R.; Berry, D.P. Prognostic molecular markers in hepatocellular carcinoma: A systematic review. Eur. J. Cancer 2007, 43, 979–992. [Google Scholar] [CrossRef]
- Martinez-Chantar, M.L.; Corrales, F.J.; Martinez-Cruz, A.; Garcia-Trevijano, E.R.; Huang, Z.Z.; Chen, L.; Kanel, G.; Avila, M.A.; Mato, J.M.; Lu, S.C. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase1A. FASEB J. 2002, 16, 1292–1294. [Google Scholar] [CrossRef]
- Tomasi, M.L.; Iglesias-Ara, A.; Yang, H.; Ramani, K.; Feo, F.; Pascale, M.R.; Martinez-Chantar, M.L.; Mato, J.M.; Lu, S.C. S-adenosylmethionine regulates apurinic/apyrimidinic endonuclease 1 stability: Implication in hepatocarcinogenesis. Gastroenterology 2009, 136, 1025–1036. [Google Scholar] [CrossRef]
- Mol, C.D.; Hosfield, J.D.; Tainer, J.A. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: The 3′ ends justify the means. Mutat. Res. 2000, 460, 211–229. [Google Scholar] [CrossRef]
- Yang, H.; Cho, M.E.; Li, T.W.; Peng, H.; Ko, K.S.; Mato, J.M.; Lu, S.C. MicroRNAs regulate methionine adenosyltransferase 1A expression in hepatocellular carcinoma. J. Clin. Investig. 2013, 123, 285–298. [Google Scholar] [CrossRef]
- Lo, T.; Tsai, W.C.; Chen, S.T. MicroRNA-21-3p, a berberine-induced miRNA, directly down-regulates human methionine adenosyltransferases 2A and 2B and inhibits hepatoma cell growth. PLoS ONE 2013, 8, e75628. [Google Scholar] [CrossRef]
- Simile, M.M.; Peitta, G.; Tomasi, K.L.; Brozzetti, S.; Feo, C.F.; Porcu, A.; Cigliano, A.; Calvisi, D.F.; Feo, F.; Pascale, R.M. MicroRNA-203 impacts on the growth, aggressiveness and prognosis of hepatocellular carcinoma by targeting MAT2A and MAT2B genes. Oncotarget 2019, 10, 2835–2854. [Google Scholar] [CrossRef]
- Cai, R.L.; Meng, W.; Lu, H.Y.; Lin, W.Y.; Jiang, F.; Shen, F.M. Segregation analysis of hepatocellular carcinoma in a moderately high-incidence area of East China. World J. Gastroenterol. 2003, 9, 2428–2432. [Google Scholar] [CrossRef]
- Fernandez, E.; La Vecchia, C.; D’Avanzo, B.; Negri, E.; Franceschi, S. Family history and the risk of liver, gallbladder, and pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 1994, 31, 209–212. [Google Scholar]
- Hemminki, K.; Li, X. Familial risks of cancer as a guide to gene identification and mode of inheritance. Int. J. Cancer 2004, 110, 291–294. [Google Scholar] [CrossRef]
- Feo, F.; De Miglio, M.R.; Simile, M.M.; Muroni, M.R.; Calvisi, D.F.; Frau, M.; Pascale, R.M. Hepatocellular carcinoma as a complex polygenic disease. Interpretative analysis of recent developments on genetic predisposition. BBA Cancer Rev. 2006, 1765, 126–147. [Google Scholar]
- Nahon, P.; Zucman-Rossi, J. Single nucleotide polymorphisms and risk of hepatocellular carcinoma in cirrhosis. J. Hepatol. 2012, 57, 663–674. [Google Scholar] [CrossRef]
- Ezzikouri, S.; Benjelloun, S.; Pineau, P. Human genetic variation and the risk of hepatocellular carcinoma development. Hepatol. Int. 2013, 7, 820–831. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, J.; Shen, X. Genome-wide association study: New genetic insights into HBV/HCV-related hepatocellular carcinoma genomes. Scand. J. Gastroenterol. 2017, 52, 209–215. [Google Scholar] [CrossRef]
- Mohamadkhani, A.; Pourasgari, M.; Poustchi, H. Significant SNPs Related to Telomere Length and Hepatocellular Carcinoma Risk in Chronic Hepatitis B Carriers. Asian Pac. J. Cancer Prev. 2018, 19, 585–590. [Google Scholar]
- Zhang, Y.J.; Jiang, W.; Chen, C.J.; Lee, C.S.; Kahn, S.M.; Santella, R.M.; Weinstein, I.B. Amplification and overexpression of cyclin D1 in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 1993, 196, 1010–1016. [Google Scholar] [CrossRef]
- Ohashi, R.; Gao, C.; Miyazaki, M.; Hamazaki, K.; Tsuji, T.; Inoue, Y.; Uemura, T.; Hirai, R.; Shimizu, N.; Namba, M. Enhanced expression of cyclin E and cyclin A in human hepatocellular carcinoma. Anticancer Res. 2001, 21, 657–662. [Google Scholar]
- Qin, L.X.; Tang, Z.Y. The prognostic molecular markers in hepatocellular carcinoma. World J. Gastroenterol. 2002, 8, 385–392. [Google Scholar] [CrossRef]
- Stepanova, L.; Leng, X.; Parker, S.B.; Harper, J.W. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996, 10, 1491–1502. [Google Scholar] [CrossRef]
- Gaubatz, S.; Lees, J.A.; Lindeman, G.J.; Livingston, D.M. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol. Cell. Biol. 2001, 21, 1384–1392. [Google Scholar] [CrossRef]
- Crosby, M.E.; Almasan, A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol. Ther. 2004, 3, 1208–1211. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Frau, M.; Muroni, M.R.; Seddaiu, M.A.; Daino, L.; Muntoni, M.D.; De Miglio, M.R.; Thorgeirsson, S.S.; et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology 2005, 42, 1310–1319. [Google Scholar] [CrossRef]
- Matsuda, Y. Molecular mechanism underlying the functional loss of cyclin-dependent kinase inhibitors p16 and p27 in hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 1734–1740. [Google Scholar] [CrossRef]
- Li, Q.; Deng, C.; Zhang, T.; Li, X. Association of GSTP1 and P16 promoter methylation with the risk of HBV-related hepatocellular carcinoma: A meta-analysis. OncoTargets Ther. 2018, 11, 5789–5796. [Google Scholar] [CrossRef]
- Wang, I.C.; Chen, Y.J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, G.; Costa, R.H. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2–Cks1) ubiquitin ligase. Mol. Cell. Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pinna, F.; Meloni, F.; Ladu, S.; Pellegrino, R.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Virdis, P.; et al. Dual-Specificity phosphatase 1 ubiquitination in extracellular signal-regulated-kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 2008, 68, 4192–4200. [Google Scholar] [CrossRef]
- Lin, Y.W.; Yang, J.L. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J. Biol. Chem. 2006, 281, 915–926. [Google Scholar] [CrossRef]
- Peyrou, M.; Bourgoin, L.; Foti, M. PTEN in liver diseases and cancer. World J. Gastroenterol. 2010, 16, 4627–4633. [Google Scholar] [CrossRef]
- Le, J.W.; Soung, Y.H.; Kim, S.Y.; Lee, H.W.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 2005, 24, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Alexia, J.C.; Fallot, G.; Lasfer, M.; Schweizer-Groyer, G.; Groyer, A. An evaluation of the role of insulin-like growth factors (IGF) and of type-I IGF receptor signaling in hepatocarcinogenesis and in the resistance of hepatocarcinoma cells againstdrug-induced apoptosis. Biochem. Pharmacol. 2004, 68, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Carlin, C.R.; Simon, D.; Mattison, B.; Knowles, B.B. Expression and biosynthetic variation of the epidermal growth factor receptor in human hepatocellular schmiyzcarcinoma-derived cell lines. Mol. Cell. Biol. 1988, 8, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Wohlschlaeger, J.; Lang, H.; Sotiropoulos, G.C.; Malago, M.; Steveling, K.; Reis, H.; Cicinnati, V.R.; Schmid, K.W.; Baba, H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J. Hepatol. 2008, 48, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.H.; Huang, C.C.; Lin, P.R.; Chang, H.W.; Ger, L.P.; Lin, Y.W.; Tai, M.H. Expression and prognostic role of tumour suppressor gene PTEN/MMAC1/TEP1 in hepatocellularcarcinoma. Cancer 2003, 97, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Sakamoto, M.; Yamasaki, S.; Todo, S.; Hirohashi, S. AKT phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer 2005, 103, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signalling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Sakamoto, M.; Yasuda, J.; Takamura, M.; Fujita, N.; Tsuruo, T.; Todo, S.; Hirohashi, S. Critical involvement of the phosphatidylinositol 3-kinase/Akt pathway in anchorage-independent growth and hematogeneous intrahepatic metastasis of liver cancer. Cancer Res. 2002, 62, 2971–2975. [Google Scholar]
- Sahin, F.; Kannangai, R.; Adegbola, O.; Wang, J.; Su, G.; Torbenson, M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 2004, 10, 8421–8425. [Google Scholar] [CrossRef]
- Rizell, M.; Andersson, M.; Cahlin, C.; Hafström, L.; Olausson, M.; Lindnér, P. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int. J. Clin. Oncol. 2008, 13, 66–70. [Google Scholar] [CrossRef]
- Cao, H.; Xu, Z.; Wang, J.; Cigliano, A.; Pilo, M.G.; Ribback, S.; Zhang, S.; Qiao, Y.; Che, L.; Pascale, R.M.; et al. Functional role of SGK3 in PI3K/Pten driven liver tumor development. BMC Cancer 2019, 19, 343. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hu, J.; Cao, H.; Pilo, M.G.; Cigliano, A.; Shao, Z.; Xu, M.; Ribback, S.; Dombrowski, F.; Calvisi, D.F.; et al. Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Exp. Mol. Med. 2018, 50, e417. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Chen, X.; Latzko, P.; Malz, M.; Gaida, M.M.; Felix, K.; Ladu, S.; Singer, S.; Pinna, F.; Gretz, N.; et al. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology 2013, 144, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Evert, M.; Frau, M.; Tomasi, M.L.; Latte, G.; Simile, M.M.; Seddaiu, M.A.; Zimmermann, A.; Ladu, S.; Staniscia, T.; Brozzetti, S.; et al. Deregulation of DNA-dependent protein kinase catalytic subunit contributes to human hepatocarcinogenesis development and has a putative prognostic value. Br. J. Cancer 2013, 109, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Joseph, C.; Latte, G.; Evert, M.; Feo, F.; Calvisi, D.F. DNA-PKcs: A promising therapeutic target in human hepatocellular carcinoma? DNA Repair 2016, 47, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Loi, R.; Perra, A.; Factor, V.M.; Ledda-Columbano, G.M.; Columbano, A.; Thorgeirsson, S.S. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology 2010, 51, 1401–1409. [Google Scholar] [CrossRef]
- Frau, M.; Simile, M.M.; Tomasi, M.L.; Demartis, M.I.; Daino, L.; Seddaiu, M.A.; Brozzetti, S.; Feo, C.F.; Massarelli, G.; Solinas, G.; et al. An expression signature of phenotypic resistance to hepatocellular carcinoma identified by cross-species gene expression analysis. Cell. Oncol. 2012, 35, 163–173. [Google Scholar] [CrossRef]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Chr | QTL |
|---|---|---|
| C3H/He x A/J | 7 | Hcs1 |
| 8 | Hcs2 | |
| 12 | Hcs3 | |
| (C3H/He x Mus spretus) x C57BL/6J | 2 | Hcs4 |
| 5 | Hcs5 | |
| 19 | Hcs6 | |
| RCS | 1 | Hcs7 |
| Br x B6 | 17 | Hcf1 |
| 1 | Hcf2 | |
| DBA/2J | 4 | Hcr1 |
| 10 | Hcr2 | |
| (C3H/He x BALB/c) F2 | 15 | Hpcr3 |
| Strain | Chr | QTL |
|---|---|---|
| MHC-recombinant | 20 | RCC |
| BN × BFF1 | 7 | Hcs1 |
| 1 | Hcs2 | |
| BFF2, CFF2, DHR | 1 | Hcs3/Drh1 |
| CFF2 | 16 | Hcs4 |
| 1 | Hcs5 | |
| 14 | Hcs6 | |
| 15 | Hcs7 | |
| (F344 × DRH) F2 | 4 | Drh2a/b |
| BN × BFF1 | 10 | Hcr1 |
| 4 | Hcr2 | |
| 8 | Hcr3 | |
| 4 | Hcr9 | |
| BFF2 | 8 | Hcr10 |
| CFF2 | 6 | Hcr11 |
| 3 | Hcr12 | |
| CFF2 | 4 | Hcr13 |
| 18 | Hcr14 | |
| BFF1 | 7 | Lnnr1 |
| 1 | Lnnr2 | |
| 2 | Lnnr3 | |
| 13 | Lnnr4 | |
| 13 | Lnnr5 | |
| 13 | Lnnr6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascale, R.M.; Simile, M.M.; Peitta, G.; Seddaiu, M.A.; Feo, F.; Calvisi, D.F. Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers 2019, 11, 1450. https://doi.org/10.3390/cancers11101450
Pascale RM, Simile MM, Peitta G, Seddaiu MA, Feo F, Calvisi DF. Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers. 2019; 11(10):1450. https://doi.org/10.3390/cancers11101450
Chicago/Turabian StylePascale, Rosa M., Maria M. Simile, Graziella Peitta, Maria A. Seddaiu, Francesco Feo, and Diego F. Calvisi. 2019. "Experimental Models to Define the Genetic Predisposition to Liver Cancer" Cancers 11, no. 10: 1450. https://doi.org/10.3390/cancers11101450
APA StylePascale, R. M., Simile, M. M., Peitta, G., Seddaiu, M. A., Feo, F., & Calvisi, D. F. (2019). Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers, 11(10), 1450. https://doi.org/10.3390/cancers11101450