Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation




Abstract
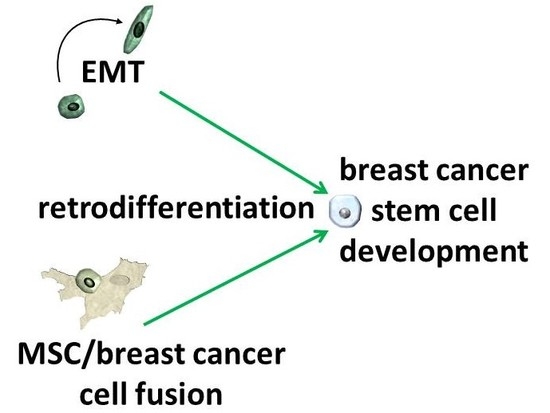
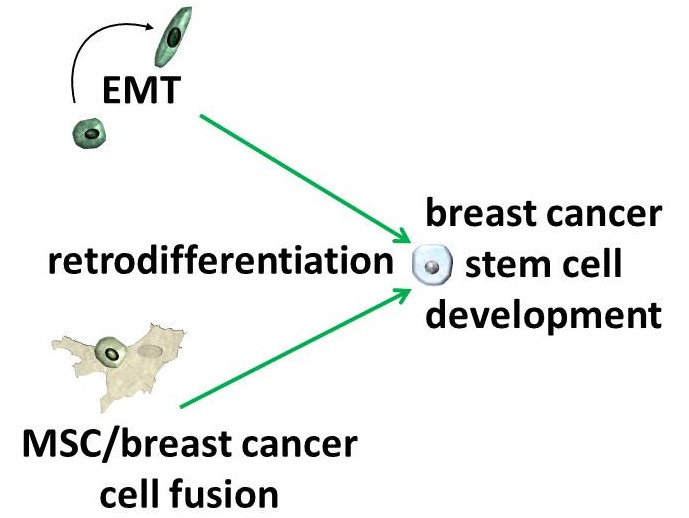{kind=link}
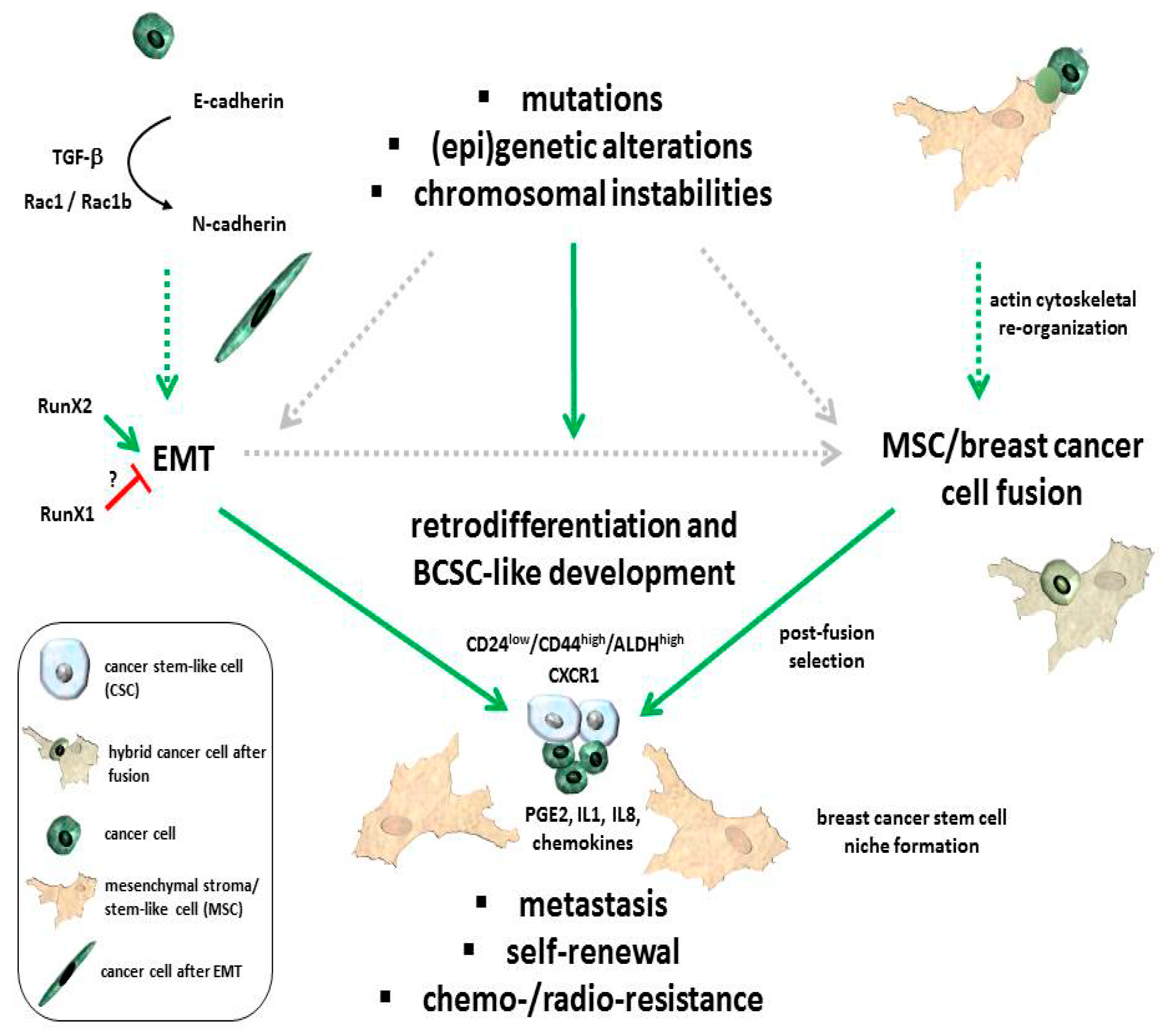{kind=link}
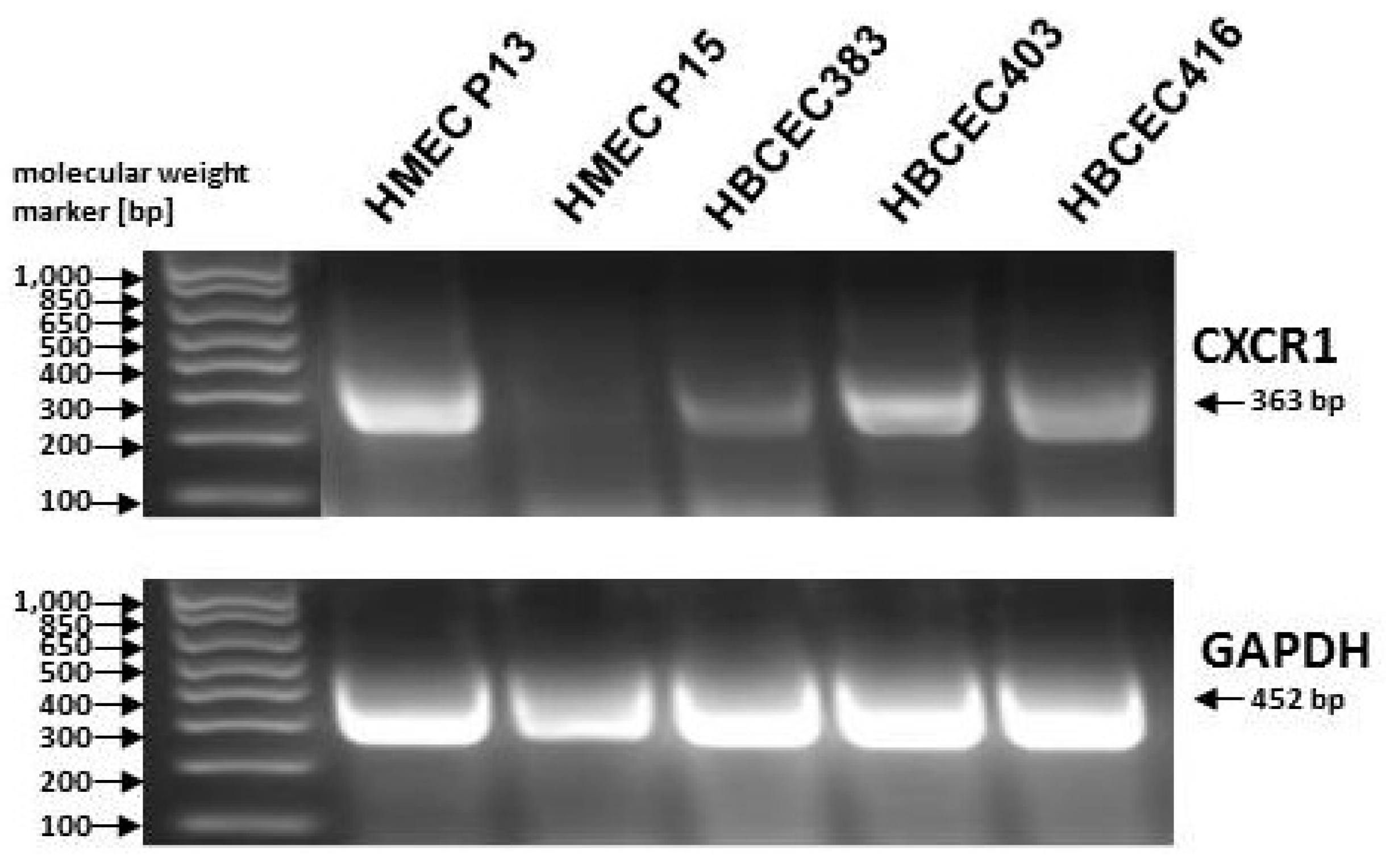{kind=link}
1. Introduction
2. Interaction and Cell Fusion of MSC with Breast Cancer Cells
3. EMT and the Formation of Breast Cancer Stem Cells (BCSCs)
4. Breast Cancer Stem Cell Properties and Corresponding Carcinoma Stem Cell Niche
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALDH-1 | aldehyde dehydrogenase-1 |
| α-SMA | α-smooth muscle actin |
| (B)CSCs | (breast) cancer stem cells |
| CA+-MSC | cancer-associated and -supporting MSC |
| CA--MSC | cancer-associated and -suppressing MSC |
| CCL5 | CC-chemokine ligand 5 |
| CSCN | cancer stem cell niche |
| CXCR1 | CXC-motif chemokine receptor 1 |
| DKK-1 | Dickkopf-related protein 1 |
| EMT | epithelial-mesenchymal transition |
| EGF | epidermal growth factor |
| Fsp 1 | fibroblast-specific protein 1 |
| GAPDH | glyceraldehyde-3-phosphate-dehydrogenase |
| HBCEC | human breast cancer-derived epithelial cells |
| HGF | hepatocyte growth factor |
| HMEC | human mammary epithelial cells |
| IFNβ | interferon β |
| IL1 | interleukin 1 |
| IL8 | interleukin 8 |
| MSC | mesenchymal stroma/stem-like cells |
| MET | mesenchymal-epithelial transition |
| microRNA | miRNA |
| PDAC | pancreatic ductal adenocarcinoma |
| PGE2 | prostaglandin E2 |
| RUNX1, 2 | runt-related transcription factor 1, 2 |
| TAMs | tumor-associated macrophages |
| TGFβ | transforming growth factor-beta |
| TNFα | tumor necrosis factor alpha |
| Zeb | Zinc-finger E-box homeobox |
References
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H. Blockade of TGF-beta signaling: A potential target for cancer immunotherapy? Expert Opin. Ther. Targets 2019, 23, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Boregowda, S.V.; Booker, C.N.; Phinney, D.G. Mesenchymal Stem Cells: The Moniker Fits the Science. Stem Cells 2018, 36, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P. “Mesenchymal” stem cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Bohm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Phinney, D.G. Building a consensus regarding the nature and origin of mesenchymal stem cells. J. Cell Biochem. 2002, 85, 7–12. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef]
- Friedenstein, A.; Kuralesova, A.I. Osteogenic precursor cells of bone marrow in radiation chimeras. Transplantation 1971, 12, 99–108. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. New MSC: MSCs as pericytes are Sentinels and gatekeepers. J. Orthop. Res. 2017, 35, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Yianni, V.; Sharpe, P.T. Perivascular-Derived Mesenchymal Stem Cells. J. Dent. Res. 2019, 98, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes-Camboa, N.; Cattaneo, P.; Sun, Y.; Moore-Morris, T.; Gu, Y.; Dalton, N.D.; Rockenstein, E.; Masliah, E.; Peterson, K.L.; Stallcup, W.B.; et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell 2017, 20, 345–359.e5. [Google Scholar] [CrossRef] [PubMed]
- Carrancio, S.; Blanco, B.; Romo, C.; Muntion, S.; Lopez-Holgado, N.; Blanco, J.F.; Brinon, J.G.; San Miguel, J.F.; Sanchez-Guijo, F.M.; del Canizo, M.C. Bone marrow mesenchymal stem cells for improving hematopoietic function: An in vitro and in vivo model. Part 2: Effect on bone marrow microenvironment. PLoS ONE 2011, 6, e26241. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Abe, R.; Fujita, Y.; Ando, S.; Inokuma, D.; Shimizu, H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J. Immunol. 2008, 180, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Kachgal, S.; Kniazeva, E.; Mori, H.; Costes, S.V.; George, S.C.; Putnam, A.J. Mesenchymal cells stimulate capillary morphogenesis via distinct proteolytic mechanisms. Exp. Cell Res. 2010, 316, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Prasanna, S.J. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci. Rep. 2016, 6, 38308. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.; Yang, Y.; von der Ohe, J.; Melzer, C.; Hillemanns, P.; Feuerhake, F.; Hass, R. SCCOHT tumors acquire chemoresistance and protection by interacting mesenchymal stroma/stem cells within the tumor microenvironment. Int. J. Oncol. 2016, 49, 2453–2463. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.H.; Chang, M.C.; Tsai, K.S.; Hung, M.C.; Chen, H.L.; Hung, S.C. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene 2013, 32, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Fritz, A.J.; Zaidi, S.K.; van Wijnen, A.J.; Nickerson, J.A.; Imbalzano, A.N.; Lian, J.B.; Stein, J.L.; Stein, G.S. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J. Cell. Physiol. 2018, 233, 9136–9144. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.; Boxberger, S.; Jorgensen, B.; Feldmann, S.; Ehninger, G.; Bornhauser, M.; Werner, C. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells 2004, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Batlle, R.; Andres, E.; Gonzalez, L.; Llonch, E.; Igea, A.; Gutierrez-Prat, N.; Berenguer-Llergo, A.; Nebreda, A.R. Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38alpha through TGF-beta and JNK signaling. Nat. Commun. 2019, 10, 3071. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, M.P.; Fuentes-Julian, S.; Blazquez-Martinez, A.; Pascual, C.Y.; Aller, M.A.; Arias, J.; Arnalich-Montiel, F. Immunosuppressive properties of mesenchymal stem cells: Advances and applications. Curr. Mol. Med. 2012, 12, 574–591. [Google Scholar] [CrossRef]
- Yang, Y.; Otte, A.; Hass, R. Human mesenchymal stroma/stem cells exchange membrane proteins and alter functionality during interaction with different tumor cell lines. Stem Cells Dev. 2015, 24, 1205–1222. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Otte, A. Mesenchymal stem cells as all-round supporters in a normal and neoplastic microenvironment. Cell Commun. Signal. 2012, 10, 26. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal. 2018, 16, 2. [Google Scholar] [CrossRef]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of MSC with tumor cells. Cell Commun. Signal. 2016, 14, 20. [Google Scholar] [CrossRef]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef]
- Melzer, C.; Rehn, V.; Yang, Y.; Bahre, H.; von der Ohe, J.; Hass, R. Taxol-Loaded MSC-Derived Exosomes Provide a Therapeutic Vehicle to Target Metastatic Breast Cancer and Other Carcinoma Cells. Cancers (Basel) 2019, 11, 798. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bucan, V.; Baehre, H.; von der Ohe, J.; Otte, A.; Hass, R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int. J. Oncol. 2015, 47, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Coffman, L.G.; Pearson, A.T.; Frisbie, L.G.; Freeman, Z.; Christie, E.; Bowtell, D.D.; Buckanovich, R.J. Ovarian Carcinoma-Associated Mesenchymal Stem Cells Arise from Tissue-Specific Normal Stroma. Stem Cells 2019, 37, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Hass, R. Concise Review: Crosstalk of Mesenchymal Stroma/Stem-Like Cells with Cancer Cells Provides Therapeutic Potential. Stem Cells 2018, 36, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Von Der Ohe, J.; Hass, R. In Vitro Fusion of Normal and Neoplastic Breast Epithelial Cells with Human Mesenchymal Stroma/Stem Cells Partially Involves Tumor Necrosis Factor Receptor Signaling. Stem Cells 2018, 36, 12. [Google Scholar] [CrossRef] [PubMed]
- Weiler, J.; Dittmar, T. Minocycline impairs TNF-alpha-induced cell fusion of M13SV1-Cre cells with MDA-MB-435-pFDR1 cells by suppressing NF-kappaB transcriptional activity and its induction of target-gene expression of fusion-relevant factors. Cell Commun. Signal. 2019, 17, 71. [Google Scholar] [CrossRef]
- Ogle, B.M.; Cascalho, M.; Platt, J.L. Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 2005, 6, 567–575. [Google Scholar] [CrossRef]
- Martin, F.T.; Dwyer, R.M.; Kelly, J.; Khan, S.; Murphy, J.M.; Curran, C.; Miller, N.; Hennessy, E.; Dockery, P.; Barry, F.P.; et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: Stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat. 2010, 124, 317–326. [Google Scholar] [CrossRef]
- Xue, J.; Zhu, Y.; Sun, Z.; Ji, R.; Zhang, X.; Xu, W.; Yuan, X.; Zhang, B.; Yan, Y.; Yin, L.; et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 2015, 15, 793. [Google Scholar] [CrossRef]
- Wei, H.J.; Nickoloff, J.A.; Chen, W.H.; Liu, H.Y.; Lo, W.C.; Chang, Y.T.; Yang, P.C.; Wu, C.W.; Williams, D.F.; Gelovani, J.G.; et al. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget 2014, 5, 9514–9529. [Google Scholar] [CrossRef] [PubMed]
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Mandel, K.; Yang, Y.; Schambach, A.; Glage, S.; Otte, A.; Hass, R. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013, 22, 3114–3127. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Hass, R. MSC stimulate ovarian tumor growth during intercellular communication but reduce tumorigenicity after fusion with ovarian cancer cells. Cell Commun. Signal. 2018, 16, 67. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K. The cancer cell—leukocyte fusion theory of metastasis. Adv. Cancer Res. 2008, 101, 397–444. [Google Scholar] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Bartosh, T.J.; Ullah, M.; Zeitouni, S.; Beaver, J.; Prockop, D.J. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc. Natl. Acad. Sci. USA 2016, 113, E6447–E6456. [Google Scholar] [CrossRef] [PubMed]
- Chitwood, C.A.; Dietzsch, C.; Jacobs, G.; McArdle, T.; Freeman, B.T.; Banga, A.; Noubissi, F.K.; Ogle, B.M. Breast tumor cell hybrids form spontaneously in vivo and contribute to breast tumor metastases. APL Bioeng. 2018, 2, 031907. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Von Der Ohe, J.; Hass, R. In vivo cell fusion between mesenchymal stroma/stem-like cells and breast cancer cells. Cancers 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Hass, R. Involvement of Actin Cytoskeletal Components in Breast Cancer Cell Fusion with Human Mesenchymal Stroma/Stem-Like Cells. Int. J. Mol. Sci. 2019, 20, 876. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Mallini, P.; Lennard, T.; Kirby, J.; Meeson, A. Epithelial-to-mesenchymal transition: What is the impact on breast cancer stem cells and drug resistance. Cancer Treat. Rev. 2014, 40, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach-Carson, M.C.; Levine, H. Tumor Budding: The Name is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed]
- Ewald, A.J.; Huebner, R.J.; Palsdottir, H.; Lee, J.K.; Perez, M.J.; Jorgens, D.M.; Tauscher, A.N.; Cheung, K.J.; Werb, Z.; Auer, M. Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J. Cell Sci. 2012, 125 Pt 11, 2638–2654. [Google Scholar] [CrossRef]
- Foubert, E.; De Craene, B.; Berx, G. Key signalling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res. 2010, 12, 206. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Hwang, J.; Andres Blanco, M.; Wei, Y.; Lukacisin, M.; Romano, R.A.; Smalley, K.; Liu, S.; Yang, Q.; Ibrahim, T.; et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell Biol. 2012, 14, 1212–1222. [Google Scholar] [CrossRef]
- Watanabe, K.; Villarreal-Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Dev. Cell 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, H.J.; Jang, M.H.; Gwak, J.M.; Lee, K.S.; Kim, E.J.; Kim, H.J.; Lee, H.E.; Park, S.Y. Epithelial-mesenchymal transition increases during the progression of in situ to invasive basal-like breast cancer. Hum. Pathol. 2013, 44, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [PubMed]
- Spaderna, S.; Schmalhofer, O.; Wahlbuhl, M.; Dimmler, A.; Bauer, K.; Sultan, A.; Hlubek, F.; Jung, A.; Strand, D.; Eger, A.; et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008, 68, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Chimge, N.O.; Baniwal, S.K.; Little, G.H.; Chen, Y.B.; Kahn, M.; Tripathy, D.; Borok, Z.; Frenkel, B. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: The role of SNAI2. Breast Cancer Res. 2011, 13, R127. [Google Scholar] [CrossRef] [PubMed]
- Van Bragt, M.P.; Hu, X.; Xie, Y.; Li, Z. RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. eLife 2014, 3, e03881. [Google Scholar] [CrossRef] [PubMed]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Liu, C.; Gao, F.; Mitchel, R.E.; Zhao, L.; Yang, Y.; Lei, J.; Cai, J. miR-200c enhances radiosensitivity of human breast cancer cells. J. Cell. Biochem. 2013, 114, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Corominas-Faja, B.; Cufi, S.; Vazquez-Martin, A.; Martin-Castillo, B.; Iglesias, J.M.; Lopez-Bonet, E.; Martin, A.G.; Menendez, J.A. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle 2012, 11, 4020–4032. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-beta-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664.e8. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011, 21, 275–289. [Google Scholar] [CrossRef]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.; Li, X.Y.; Lu, N.; An, T.; Liu, Z.P.; Fu, R.; Lv, W.C.; Zhang, Y.W.; Xu, X.J.; Grant Rowe, R.; et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nat. Cell Biol. 2016, 18, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Perdigao-Henriques, R.; Petrocca, F.; Altschuler, G.; Thomas, M.P.; Le, M.T.; Tan, S.M.; Hide, W.; Lieberman, J. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene 2016, 35, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. To differentiate or not—Routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef]
- Dey, P.; Rathod, M.; De, A. Targeting stem cells in the realm of drug-resistant breast cancer. Breast Cancer Targets Ther. 2019, 11, 115–135. [Google Scholar] [CrossRef]
- Chen, W.; Qin, Y.; Liu, S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin. Transl. Med. 2018, 7, 27. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Oktay, Y.; Vougas, K.; Louka, M.; Vorgias, C.E.; Georgakilas, A.G. Determinants of resistance to chemotherapy and ionizing radiation in breast cancer stem cells. Cancer Lett. 2016, 380, 485–493. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Honeth, G.; Bendahl, P.O.; Ringner, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lovgren, K.; Grabau, D.; Ferno, M.; Borg, A.; Hegardt, C. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Sivridis, E.; Fiska, A.; Koukourakis, M.I. The CD44+/CD24- phenotype relates to ‘triple-negative’ state and unfavorable prognosis in breast cancer patients. Med. Oncol. 2011, 28, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Fekir, K.; Dubois-Pot-Schneider, H.; Desert, R.; Daniel, Y.; Glaise, D.; Rauch, C.; Morel, F.; Fromenty, B.; Musso, O.; Cabillic, F.; et al. Retrodifferentiation of Human Tumor Hepatocytes to Stem Cells Leads to Metabolic Reprogramming and Chemoresistance. Cancer Res. 2019, 79, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Hass, R. Retrodifferentiation—A mechanism for cellular regeneration? Biol. Chem. 2009, 390, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.; Kuperwasser, C. Human breast cancer stem cell markers CD44 and CD24: Enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007, 9, 303. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Lehnert, H.; Ungefroren, H.; Hass, R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol. Cancer 2017, 16, 28. [Google Scholar] [CrossRef]
- Li, H.J.; Reinhardt, F.; Herschman, H.R.; Weinberg, R.A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012, 2, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Bertram, C. Characterization of human breast cancer epithelial cells (HBCEC) derived from long term cultured biopsies. J. Exp. Clin. Cancer Res. 2009, 28, 127. [Google Scholar] [CrossRef] [PubMed]
- Bertram, C.; Hass, R. MMP-7 is involved in the aging of primary human mammary epithelial cells (HMEC). Exp. Gerontol. 2008, 43, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Arfuso, F.; Sethi, G.; Dharmarajan, A.; Warrier, S. Encapsulated human mesenchymal stem cells (eMSCs) as a novel anti-cancer agent targeting breast cancer stem cells: Development of 3D primed therapeutic MSCs. Int. J. Biochem. Cell Biol. 2019, 110, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.L.; Kuo, Y.C.; Ho, Y.S.; Huang, Y.H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers (Basel) 2019, 11, 1334. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Han, X.; Liu, S.; Wang, L. CSCs in Breast Cancer-One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers (Basel) 2019, 11, 1128. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Gentile, P.; Fabbri, G.; Cervelli, V.; Orlandi, A. The Role of Breast Cancer Stem Cells as a Prognostic Marker and a Target to Improve the Efficacy of Breast Cancer Therapy. Cancers (Basel) 2019, 11, 1021. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, J.H.; Nam, J.S. Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers (Basel) 2019, 11, 965. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hass, R.; von der Ohe, J.; Ungefroren, H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers 2019, 11, 1432. https://doi.org/10.3390/cancers11101432
Hass R, von der Ohe J, Ungefroren H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers. 2019; 11(10):1432. https://doi.org/10.3390/cancers11101432
Chicago/Turabian StyleHass, Ralf, Juliane von der Ohe, and Hendrik Ungefroren. 2019. "Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation" Cancers 11, no. 10: 1432. https://doi.org/10.3390/cancers11101432
APA StyleHass, R., von der Ohe, J., & Ungefroren, H. (2019). Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers, 11(10), 1432. https://doi.org/10.3390/cancers11101432