Macrophage Migration Inhibitory Factor Is a Molecular Determinant of the Anti-EGFR Monoclonal Antibody Cetuximab Resistance in Human Colorectal Cancer Cells

 , , ,
, , , 


Abstract
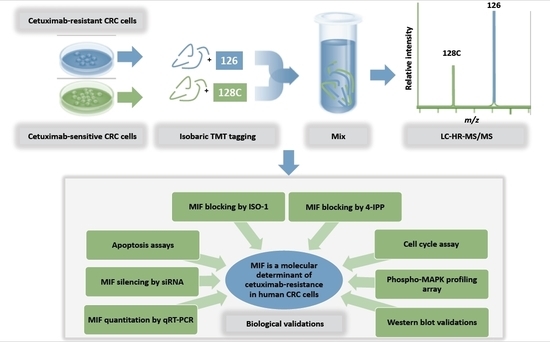
1. Introduction
2. Results
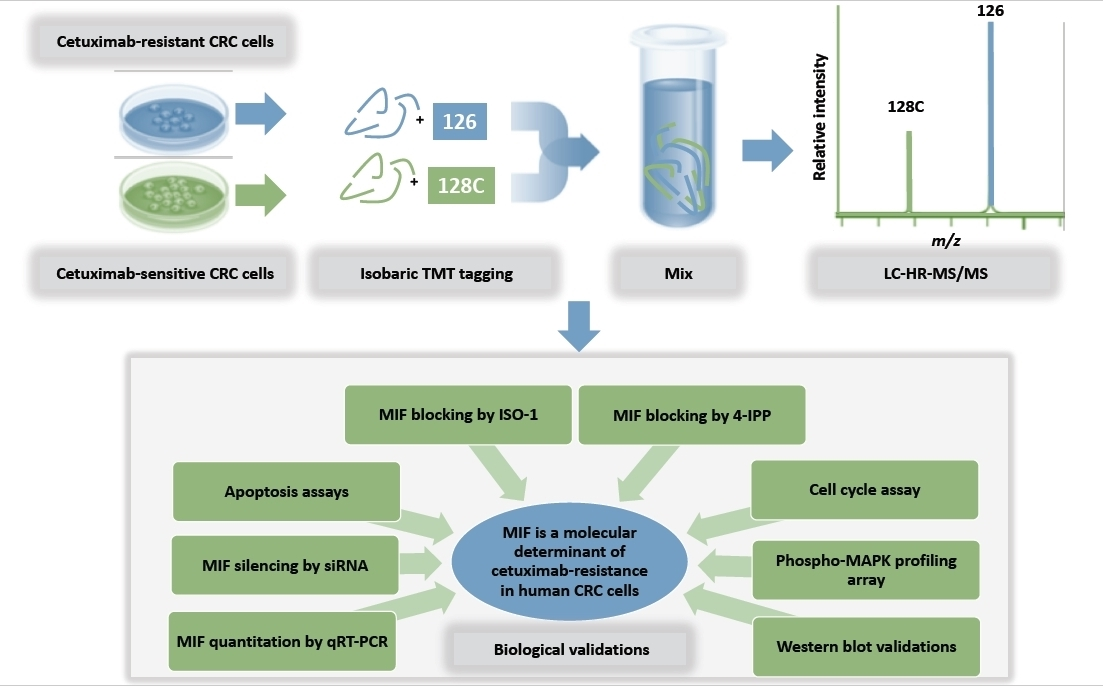
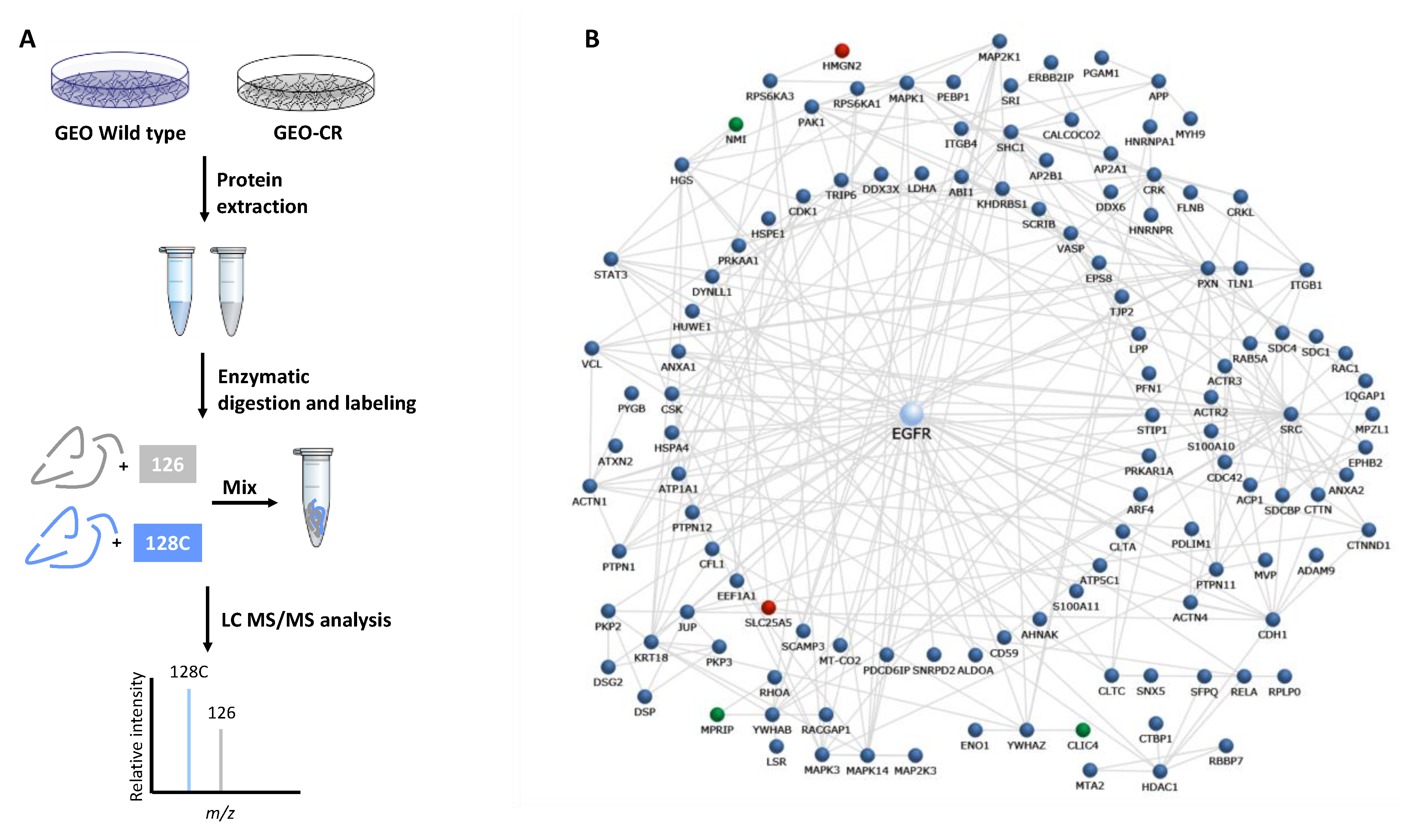
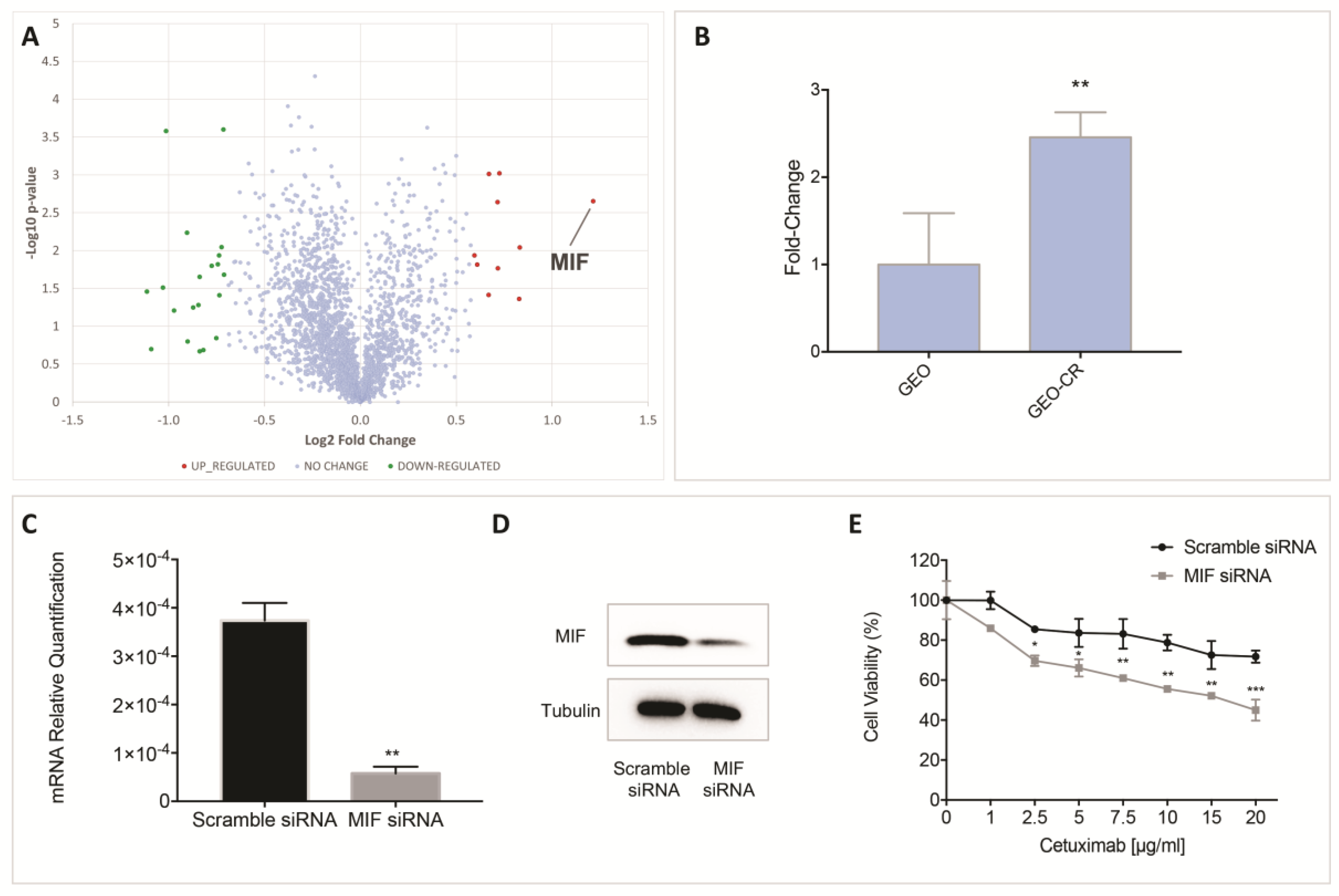
2.1. Proteomic Analysis by High Resolution nanoLC−MS/MS on an In Vitro Preclinical Model of Colon Cancer Cells with Acquired Resistance to Cetuximab
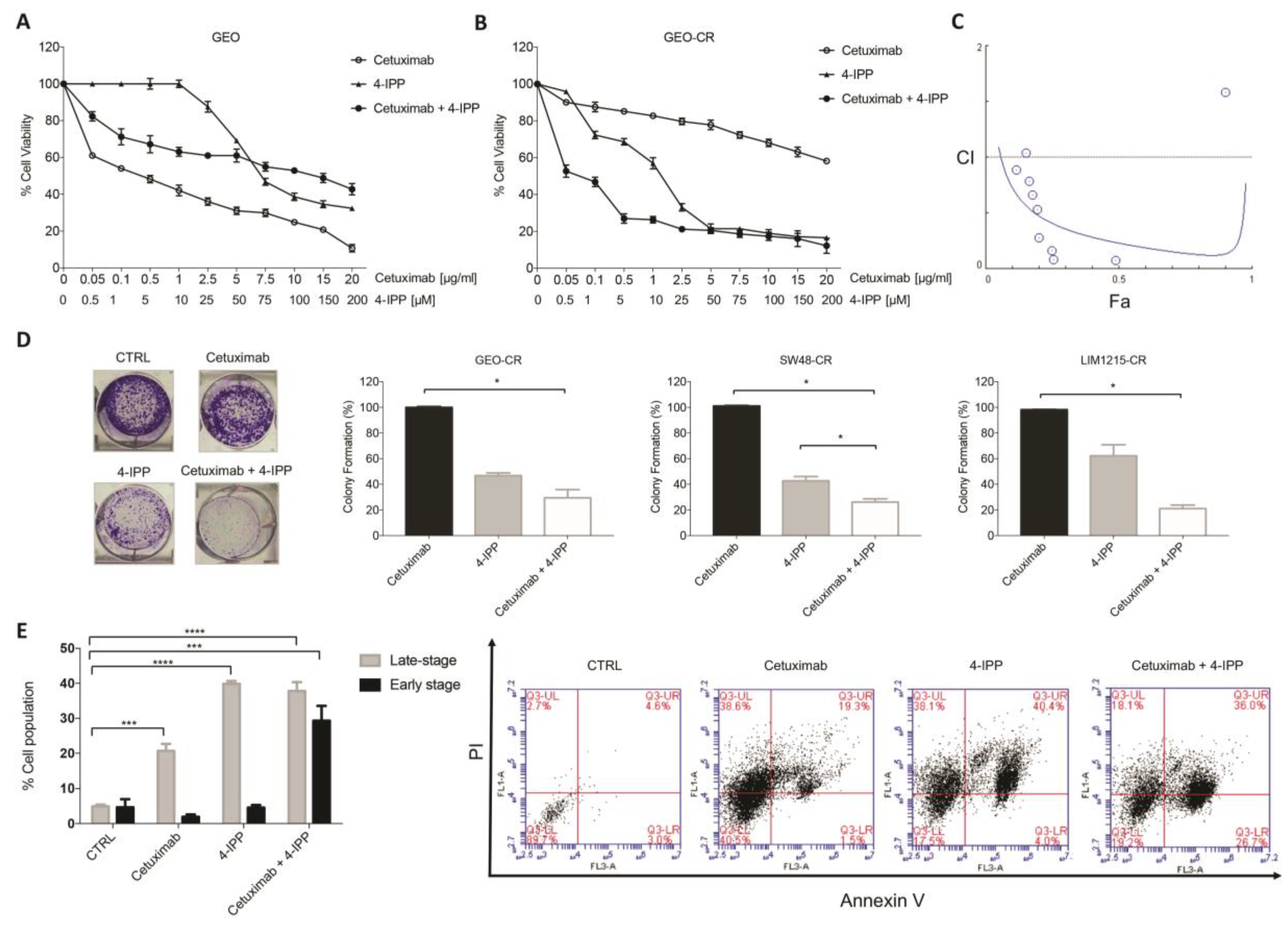
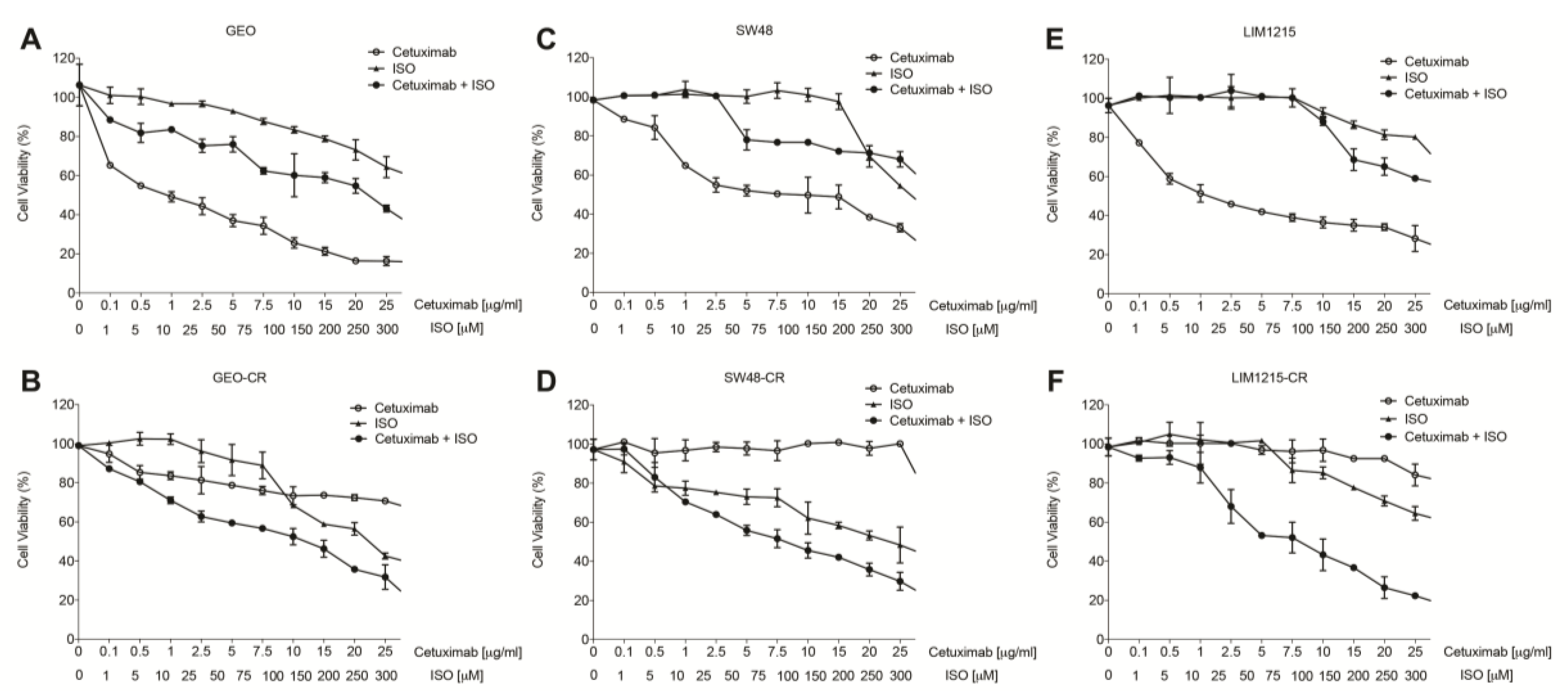
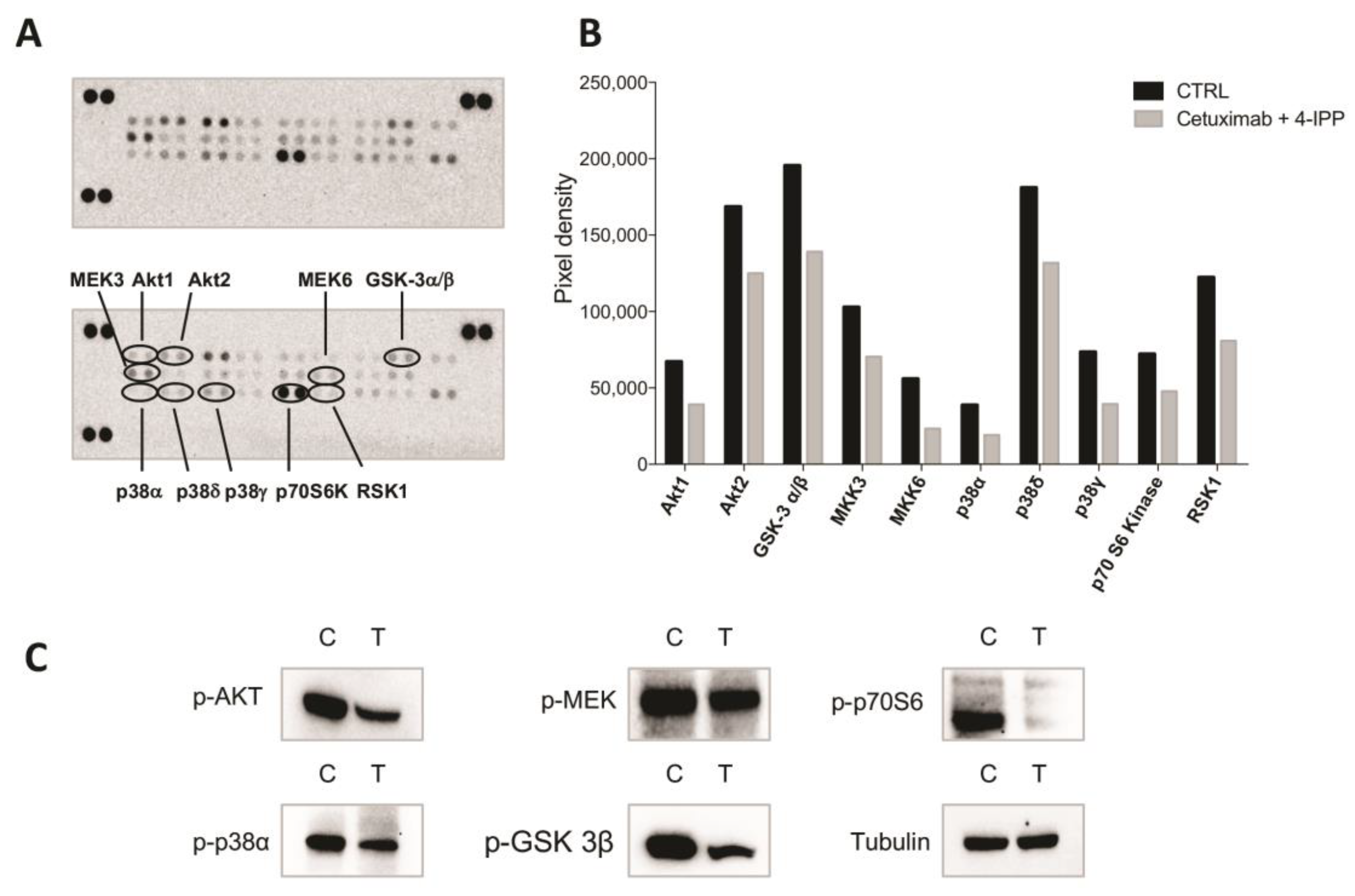
2.2. MIF Inhibition Affects Human Colon Cancer Cell Growth and Apoptosis
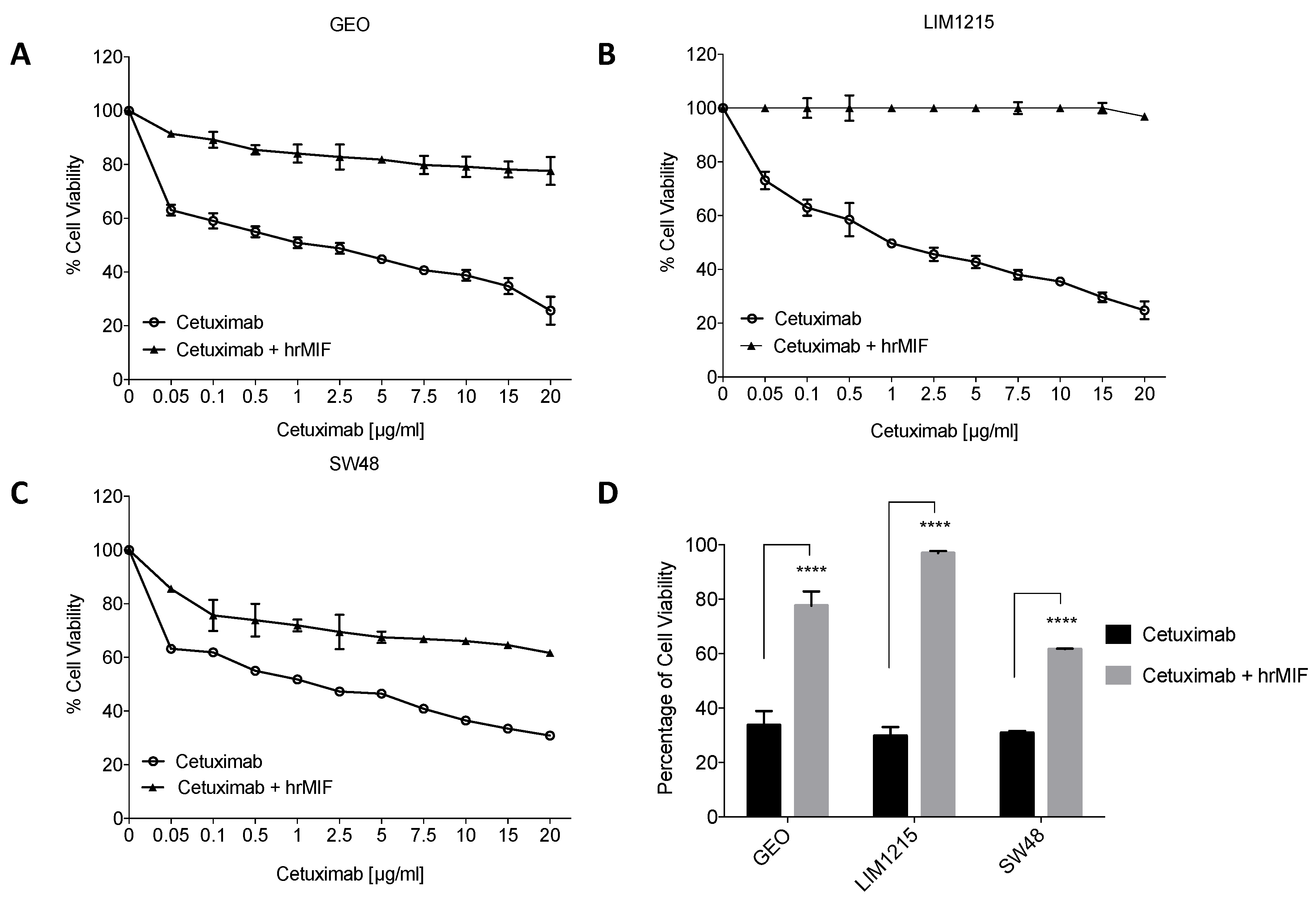
2.3. MIF Is Able to Trigger Cetuximab-Resistance in Sensitive Colon Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Human Cancer Cell Lines
4.3. Proliferation Assay
4.4. RNA Extraction and qRT-PCR
4.5. RNA Interference
4.6. Colony Formation Assay
4.7. Cell Cycle Assay
4.8. Apoptosis Assay
4.9. Phospho-MAPK Profiling Array
4.10. Western Blot Analysis
4.11. Sample Preparation for Proteomic Analyses
4.12. High Resolution nanoLC−Tandem Mass Spectrometry
4.13. Protein Identification and Quantitation
4.14. Bioinformatic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Chang, G.J.; Overman, M.J.; Eng, C.; Sargent, D.J.; Larson, D.W.; Grothey, A.; Vauthey, J.N.; Nagorney, D.M.; McWilliams, R.R. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J. Clin. Oncol. 2009, 27, 3677–3683. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Napolitano, S.; Della Corte, C.M.; Martini, G.; Martinelli, E.; Morgillo, F.; Ciardiello, F. Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of clinical evidence. ESMO Open 2016, 1, e000088. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G. Metastatic colorectal cancer: Current state and future directions. J. Clin. Oncol. 2015, 33, 1809–1824. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Morgillo, F.; Belli, G.; Cioffi, L.; Ciardiello, F. Molecular aspects of resistance to biological and non-biological drugs and strategies to overcome resistance in colorectal cancer. Curr. Med. Chem. 2014, 21, 1639–1653. [Google Scholar] [CrossRef]
- Valtorta, E.; Misale, S.; Sartore-Bianchi, A.; Nagtegaal, I.D.; Paraf, F.; Lauricella, C.; Dimartino, V.; Hobor, S.; Jacobs, B.; Ercolani, C.; et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cancer 2013, 133, 1259–1265. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, S.; Martini, G.; Rinaldi, B.; Martinelli, E.; Donniacuo, M.; Berrino, L.; Vitagliano, D.; Morgillo, F.; Barra, G.; De Palma, R.; et al. Primary and Acquired Resistance of Colorectal Cancer to Anti-EGFR Monoclonal Antibody Can Be Overcome by Combined Treatment of Regorafenib with Cetuximab. Clin. Cancer Res. 2015, 21, 2975–2983. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin. Cancer Res. 2013, 19, 6751–6765. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Napolitano, S.; Vitagliano, D.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; Ciardiello, D.; Ciardiello, F.; Martinelli, E. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin. Cancer Res. 2014, 20, 3775–3786. [Google Scholar] [CrossRef] [PubMed]
- Yonesaka, K.; Zejnullahu, K.; Okamoto, I.; Satoh, T.; Cappuzzo, F.; Souglakos, J.; Ercan, D.; Rogers, A.; Roncalli, M.; Takeda, M.; et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011, 3, 99ra86. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Malorni, L.; Cacace, G.; Cuccurullo, M.; Pocsfalvi, G.; Chambery, A.; Farina, A.; Di Maro, A.; Parente, A.; Malorni, A. Proteomic analysis of MCF-7 breast cancer cell line exposed to mitogenic concentration of 17beta-estradiol. Proteomics 2006, 6, 5973–5982. [Google Scholar] [CrossRef] [PubMed]
- Ou, K.; Kesuma, D.; Ganesan, K.; Yu, K.; Soon, S.Y.; Lee, S.Y.; Goh, X.P.; Hooi, M.; Chen, W.; Jikuya, H.; et al. Quantitative profiling of drug-associated proteomic alterations by combined 2-nitrobenzenesulfenyl chloride (NBS) isotope labeling and 2DE/MS identification. J. Proteome Res. 2006, 5, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, C.; Connor, M.; Kislinger, T.; Slingerland, J.; Emili, A. Global protein shotgun expression profiling of proliferating mcf-7 breast cancer cells. J. Proteome Res. 2005, 4, 674–689. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, S.; Lian, X.; Liu, L.; Zhao, S.; Xuan, Q.; Guo, L.; Liu, H.; Yang, Y.; Dong, T.; et al. Identification of proteins responsible for adriamycin resistance in breast cancer cells using proteomics analysis. Sci. Rep. 2015, 5, 9301. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Kang, H.; Timmermans, A.M.; Look, M.P.; Meijer-van Gelder, M.E.; den Bakker, M.A.; Jaitly, N.; Martens, J.W.; Luider, T.M.; Foekens, J.A.; et al. Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol. Cell. Proteom. 2009, 8, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Mundt, F.; Rajput, S.; Li, S.; Ruggles, K.V.; Mooradian, A.D.; Mertins, P.; Gillette, M.A.; Krug, K.; Guo, Z.; Hoog, J.; et al. Mass Spectrometry-Based Proteomics Reveals Potential Roles of NEK9 and MAP2K4 in Resistance to PI3K Inhibition in Triple-Negative Breast Cancers. Cancer Res. 2018, 78, 2732–2746. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.V.; Manning, H.C.; Coffey, R.J.; Liebler, D.C. Protein expression signatures for inhibition of epidermal growth factor receptor-mediated signaling. Mol. Cell. Proteom. 2012; 11, M111.015222. [Google Scholar] [CrossRef]
- Abe, Y.; Nagano, M.; Kuga, T.; Tada, A.; Isoyama, J.; Adachi, J.; Tomonaga, T. Deep Phospho- and Phosphotyrosine Proteomics Identified Active Kinases and Phosphorylation Networks in Colorectal Cancer Cell Lines Resistant to Cetuximab. Sci. Rep. 2017, 7, 10463. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Bianco, R.; Caputo, R.; Caputo, R.; Damiano, V.; Troiani, T.; Melisi, D.; De Vita, F.; De Placido, S.; Bianco, A.R.; et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin. Cancer Res. 2004, 10, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Troiani, T.; Morgillo, F.; Rodolico, G.; Vitagliano, D.; Morelli, M.P.; Tuccillo, C.; Vecchione, L.; Capasso, A.; Orditura, M.; et al. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin. Cancer Res. 2010, 16, 4990–5001. [Google Scholar] [CrossRef] [PubMed]
- Winner, M.; Meier, J.; Zierow, S.; Rendon, B.E.; Crichlow, G.V.; Riggs, R.; Bucala, R.; Leng, L.; Smith, N.; Lolis, E.; et al. A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 2008, 68, 7253–7257. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Arteaga, C.L. The epidermal growth factor receptor: From mutant oncogene in nonhuman cancers to therapeutic target in human neoplasia. J. Clin. Oncol. 2001, 19, 32s–40s. [Google Scholar] [PubMed]
- Galizia, G.; Lieto, E.; De Vita, F.; Orditura, M.; Castellano, P.; Troiani, T.; Imperatore, V.; Ciardiello, F. Cetuximab, a chimeric human mouse anti-epidermal growth factor receptor monoclonal antibody, in the treatment of human colorectal cancer. Oncogene 2007, 26, 3654–3660. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, F.; Sesboue, R.; Michel, P.; Sabourin, J.C.; Frebourg, T. Molecular determinants of anti-EGFR sensitivity and resistance in metastatic colorectal cancer. Br. J. Cancer 2010, 103, 1765–1772. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar]
- Mendelsohn, J. Blockade of receptors for growth factors: An anticancer therapy--the fourth annual Joseph H Burchenal American Association of Cancer Research Clinical Research Award Lecture. Clin. Cancer Res. 2000, 6, 747–753. [Google Scholar]
- Mendelsohn, J.; Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003, 21, 2787–2799. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef]
- Chekulayev, V.; Mado, K.; Shevchuk, I.; Koit, A.; Kaldma, A.; Klepinin, A.; Timohhina, N.; Tepp, K.; Kandashvili, M.; Ounpuu, L.; et al. Metabolic remodeling in human colorectal cancer and surrounding tissues: Alterations in regulation of mitochondrial respiration and metabolic fluxes. Biochem. Biophys. Rep. 2015, 4, 111–125. [Google Scholar] [CrossRef]
- Chevrollier, A.; Loiseau, D.; Reynier, P.; Stepien, G. Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim. Biophys. Acta 2011, 1807, 562–567. [Google Scholar] [CrossRef]
- Jang, J.Y.; Kim, Y.G.; Nam, S.J.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Kim, C.W. Targeting Adenine Nucleotide Translocase-2 (ANT2) to Overcome Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2016, 15, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Roger, T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Nobre, C.C.; de Araujo, J.M.; Fernandes, T.A.; Cobucci, R.N.; Lanza, D.C.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol. Oncol. Res. 2017, 23, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Weeks, A.N.; Lim, S.Y.; Yuzhalin, A.E.; Jones, K.; Muschel, R. Macrophage migration inhibitory factor: A key cytokine and therapeutic target in colon cancer. Cytokine Growth Factor Rev. 2015, 26, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef]
- Choudhary, S.; Hegde, P.; Pruitt, J.R.; Sielecki, T.M.; Choudhary, D.; Scarpato, K.; Degraff, D.J.; Pilbeam, C.C.; Taylor, J.A., 3rd. Macrophage migratory inhibitory factor promotes bladder cancer progression via increasing proliferation and angiogenesis. Carcinogenesis 2013, 34, 2891–2899. [Google Scholar] [CrossRef]
- Verjans, E.; Noetzel, E.; Bektas, N.; Schutz, A.K.; Lue, H.; Lennartz, B.; Hartmann, A.; Dahl, E.; Bernhagen, J. Dual role of macrophage migration inhibitory factor (MIF) in human breast cancer. BMC Cancer 2009, 9, 230. [Google Scholar] [CrossRef]
- He, X.X.; Chen, K.; Yang, J.; Li, X.Y.; Gan, H.Y.; Liu, C.Y.; Coleman, T.R.; Al-Abed, Y. Macrophage migration inhibitory factor promotes colorectal cancer. Mol. Med. 2009, 15, 1–10. [Google Scholar] [CrossRef]
- Dessein, A.F.; Stechly, L.; Jonckheere, N.; Dumont, P.; Monte, D.; Leteurtre, E.; Truant, S.; Pruvot, F.R.; Figeac, M.; Hebbar, M.; et al. Autocrine induction of invasive and metastatic phenotypes by the MIF-CXCR4 axis in drug-resistant human colon cancer cells. Cancer Res. 2010, 70, 4644–4654. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.K.; Kim, H.P.; Park, Y.L.; Jang, J.E.; Lim, Y.; Song, S.H.; Han, S.W.; Kim, T.Y. Macrophage migration inhibitory factor promotes resistance to MEK blockade in KRAS mutant colorectal cancer cells. Mol. Oncol. 2018, 12, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Varney, M.L.; Singh, R.K. Expression of interleukin 8 and its receptors in human colon carcinoma cells with different metastatic potentials. Clin. Cancer Res. 2001, 7, 3298–3304. [Google Scholar] [PubMed]
- Napolitano, S.; Martini, G.; Martinelli, E.; Belli, V.; Parascandolo, A.; Laukkanen, M.O.; Sforza, V.; Morgillo, F.; Ciardiello, D.; Ciardiello, F.; et al. Therapeutic efficacy of SYM004, a mixture of two anti-EGFR antibodies in human colorectal cancer with acquired resistance to cetuximab and MET activation. Oncotarget 2017, 8, 67592–67604. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Russo, R.; D’Ambrosio, S.; Dello Iacono, I.; Rega, C.; Carlino, E.; Argenzio, O.; Russo, L.; D’Abrosca, B.; Chambery, A.; et al. Physiological characterization and quantitative proteomic analyses of metabolically engineered E. coli K4 strains with improved pathways for capsular polysaccharide biosynthesis. Biotechnol. Bioeng. 2018, 115, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Nebbioso, A.; Cuomo, F.; Rotili, D.; Cobellis, G.; Bontempo, P.; Baldi, A.; Spugnini, E.P.; Citro, G.; Chambery, A.; et al. RIP1-HAT1-SIRT Complex Identification and Targeting in Treatment and Prevention of Cancer. Clin. Cancer Res. 2018, 24, 2886–2900. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Description | Gene | GEO_CR/GEO | Coverage | # PSMs | # Peptides | MW [kDa] |
|---|---|---|---|---|---|---|---|
| P14174 | Macrophage Migration inhibitory factor | MIF | 2.3 | 26.1 | 10 | 2 | 12.5 |
| Q99439 | Calponin-2 | CNN2 | 1.8 | 40.5 | 30 | 7 | 33.7 |
| Q7LBR1 | Charged multivesicular body protein 1b | CHMP1B | 1.8 | 7.5 | 4 | 2 | 22.1 |
| Q9UEE9-1 | Craniofacial development protein 1 | CFDP1 | 1.7 | 6.7 | 4 | 2 | 33.6 |
| P84101-1 | Small EDRK-rich factor 2 | SERF2 | 1.6 | 40.7 | 6 | 3 | 6.9 |
| P05204 | Non-histone chromosomal protein HMG-17 | HMGN2 | 1.6 | 30.0 | 23 | 3 | 9.4 |
| P05141 | ADP/ATP translocase 2 | SLC25A5 | 1.6 | 42.3 | 184 | 5 | 32.8 |
| P56381 | ATP synthase subunit epsilon, mitochondrial | ATPSF1E | 1.6 | 29.4 | 5 | 2 | 5.8 |
| P09234 | U1 small nuclear ribonucleoprotein C | SNRPC | 1.5 | 13.2 | 5 | 2 | 17.4 |
| Q01844-5 | Isoform 5 of RNA-binding protein EWS | EWSR1 | 1.5 | 11.5 | 22 | 6 | 68.9 |
| Q5VW32 | BRO1 domain-containing protein BROX | BROX | 0.6 | 11.7 | 10 | 4 | 46.4 |
| O14493 | Claudin-4 | CLDN4 | 0.6 | 16.7 | 10 | 2 | 22.1 |
| Q99795 | Cell surface A33 antigen | GPA33 | 0.6 | 33.9 | 59 | 10 | 35.6 |
| O94901-9 | Isoform 9 of SUN domain-containing protein 1 | SUN1 | 0.6 | 4.7 | 5 | 3 | 101.9 |
| P21912 | Succinate dehydrogenase [ubiquinone] iron-sulfur subunit | SDHB | 0.6 | 14.3 | 6 | 4 | 31.6 |
| Q13287 | N-myc-interactor | NMI | 0.6 | 9.4 | 4 | 3 | 35.0 |
| Q9UBC2-2 | Isoform 2 of Epidermal growth factor receptor substrate 15-like 1 | EPSISL1 | 0.6 | 2.0 | 3 | 2 | 99.5 |
| Q9BX40 | protein LSM14 homolog B | LSM14B | 0.6 | 8.1 | 2 | 2 | 42.0 |
| Q16222-1 | UDP-N-acetylhexosamine pyrophosphorylase | UAP1 | 0.6 | 6.9 | 5 | 3 | 58.7 |
| Q6WCQ1-2 | Isoform 2 of Myosin phosphatase Rho-interacting protein | MPRIP | 0.6 | 5.1 | 4 | 3 | 118.0 |
| Q13753-1 | Laminin subunit gamma-2 | LAMC2 | 0.6 | 3.5 | 7 | 4 | 130.9 |
| P29034 | protein S100-A2 | S100A2 | 0.6 | 27.6 | 7 | 5 | 11.1 |
| O00461 | Golgi integral membrane protein 4 | GOLIM4 | 0.5 | 5.2 | 5 | 3 | 81.8 |
| Q9Y696 | Chloride intracellular channel protein 4 | CLIC4 | 0.5 | 13.4 | 5 | 2 | 28.8 |
| P19957 | Elafin | PI3 | 0.5 | 20.5 | 6 | 2 | 12.3 |
| O75110 | Probable phospholipid-transporting ATPase IIA | ATP9A | 0.5 | 1.6 | 5 | 2 | 118.5 |
| P80188 | Neutrophil gelatinase-associated lipocalin | LCN2 | 0.5 | 43.9 | 46 | 7 | 22.6 |
| Q9P265 | Disco-interacting protein 2 homolog B | DIP2B | 0.5 | 1.7 | 3 | 2 | 171.4 |
| Q9BPX3 | Condensin complex subunit 3 | NCAPG | 0.5 | 5.5 | 6 | 4 | 114.3 |
| P50440-1 | glycine amidinotransferase | GATM | 0.5 | 7.8 | 5 | 3 | 48.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, R.; Matrone, N.; Belli, V.; Ciardiello, D.; Valletta, M.; Esposito, S.; Pedone, P.V.; Ciardiello, F.; Troiani, T.; Chambery, A. Macrophage Migration Inhibitory Factor Is a Molecular Determinant of the Anti-EGFR Monoclonal Antibody Cetuximab Resistance in Human Colorectal Cancer Cells. Cancers 2019, 11, 1430. https://doi.org/10.3390/cancers11101430
Russo R, Matrone N, Belli V, Ciardiello D, Valletta M, Esposito S, Pedone PV, Ciardiello F, Troiani T, Chambery A. Macrophage Migration Inhibitory Factor Is a Molecular Determinant of the Anti-EGFR Monoclonal Antibody Cetuximab Resistance in Human Colorectal Cancer Cells. Cancers. 2019; 11(10):1430. https://doi.org/10.3390/cancers11101430
Chicago/Turabian StyleRusso, Rosita, Nunzia Matrone, Valentina Belli, Davide Ciardiello, Mariangela Valletta, Sabrina Esposito, Paolo Vincenzo Pedone, Fortunato Ciardiello, Teresa Troiani, and Angela Chambery. 2019. "Macrophage Migration Inhibitory Factor Is a Molecular Determinant of the Anti-EGFR Monoclonal Antibody Cetuximab Resistance in Human Colorectal Cancer Cells" Cancers 11, no. 10: 1430. https://doi.org/10.3390/cancers11101430
APA StyleRusso, R., Matrone, N., Belli, V., Ciardiello, D., Valletta, M., Esposito, S., Pedone, P. V., Ciardiello, F., Troiani, T., & Chambery, A. (2019). Macrophage Migration Inhibitory Factor Is a Molecular Determinant of the Anti-EGFR Monoclonal Antibody Cetuximab Resistance in Human Colorectal Cancer Cells. Cancers, 11(10), 1430. https://doi.org/10.3390/cancers11101430