STAT3 and STAT5 Activation in Solid Cancers



Abstract
1. Introduction
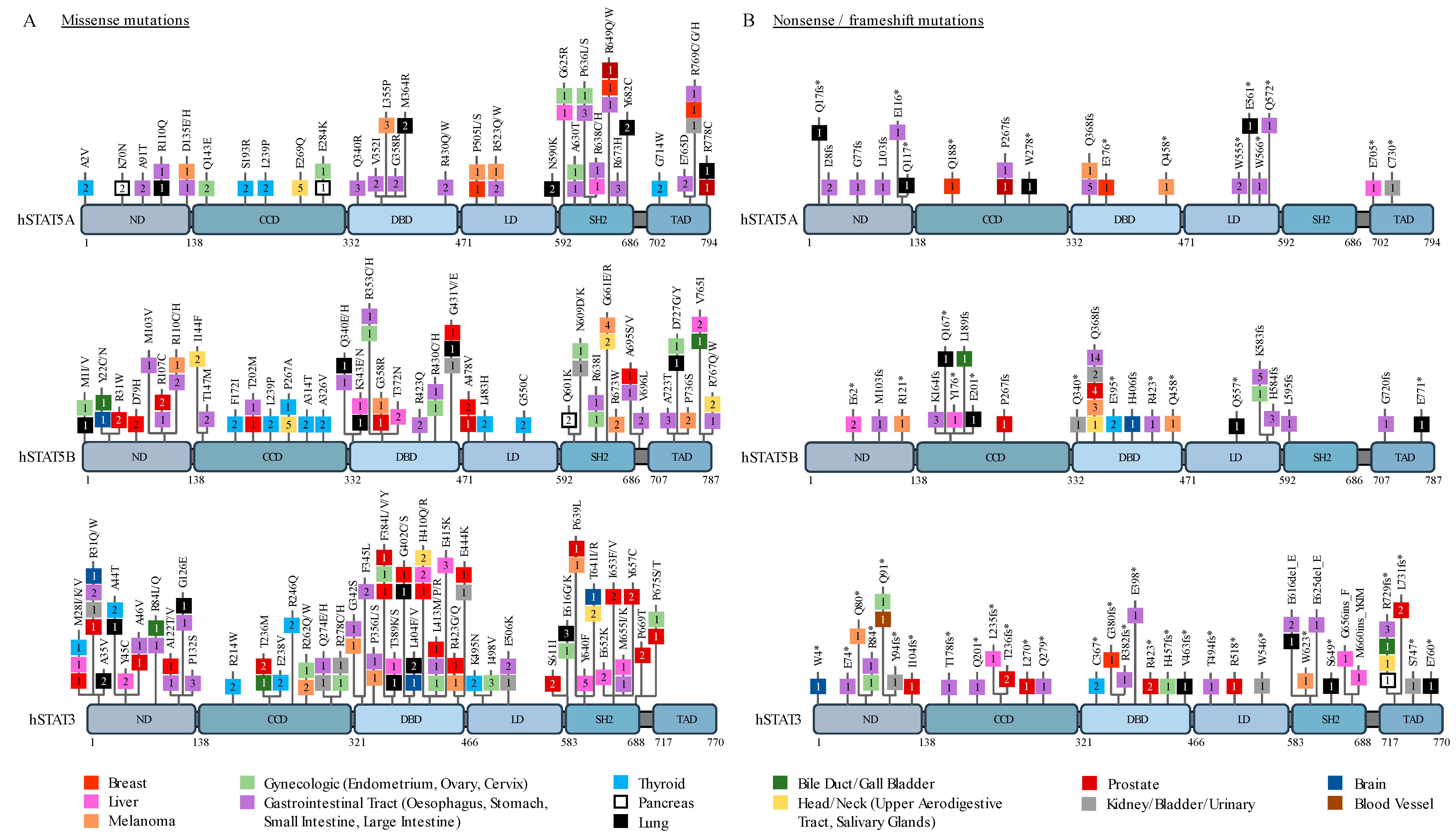
2. STAT3 and STAT5 in Solid Cancers
3. Mechanisms of Transformation by STAT3/5 Proteins in Solid Cancers
3.1. Cell Cycle and Apoptosis
3.2. Inflammation and Innate Immunity
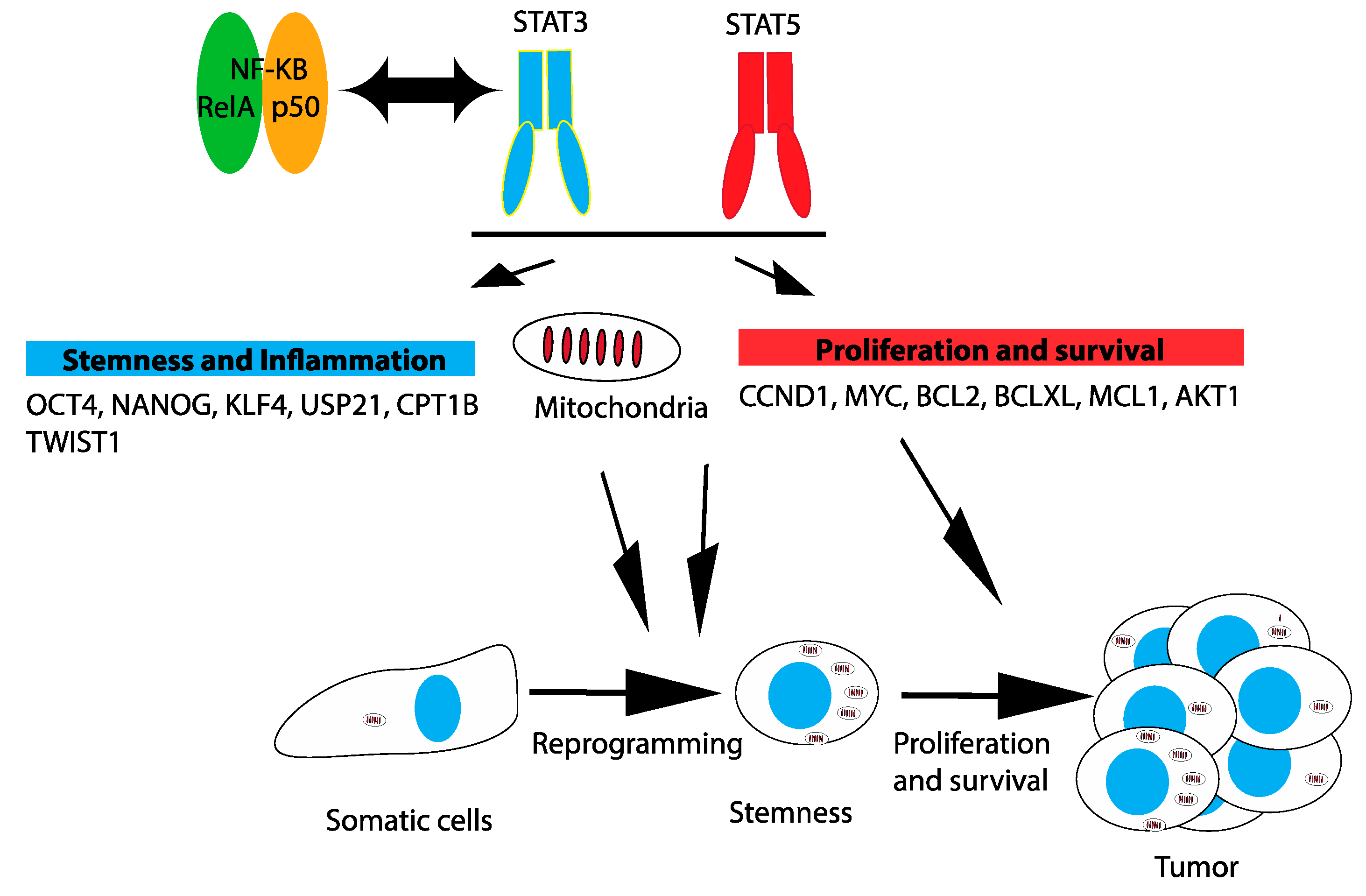
3.3. Mitochondria
3.4. Reprogramming and Stemness
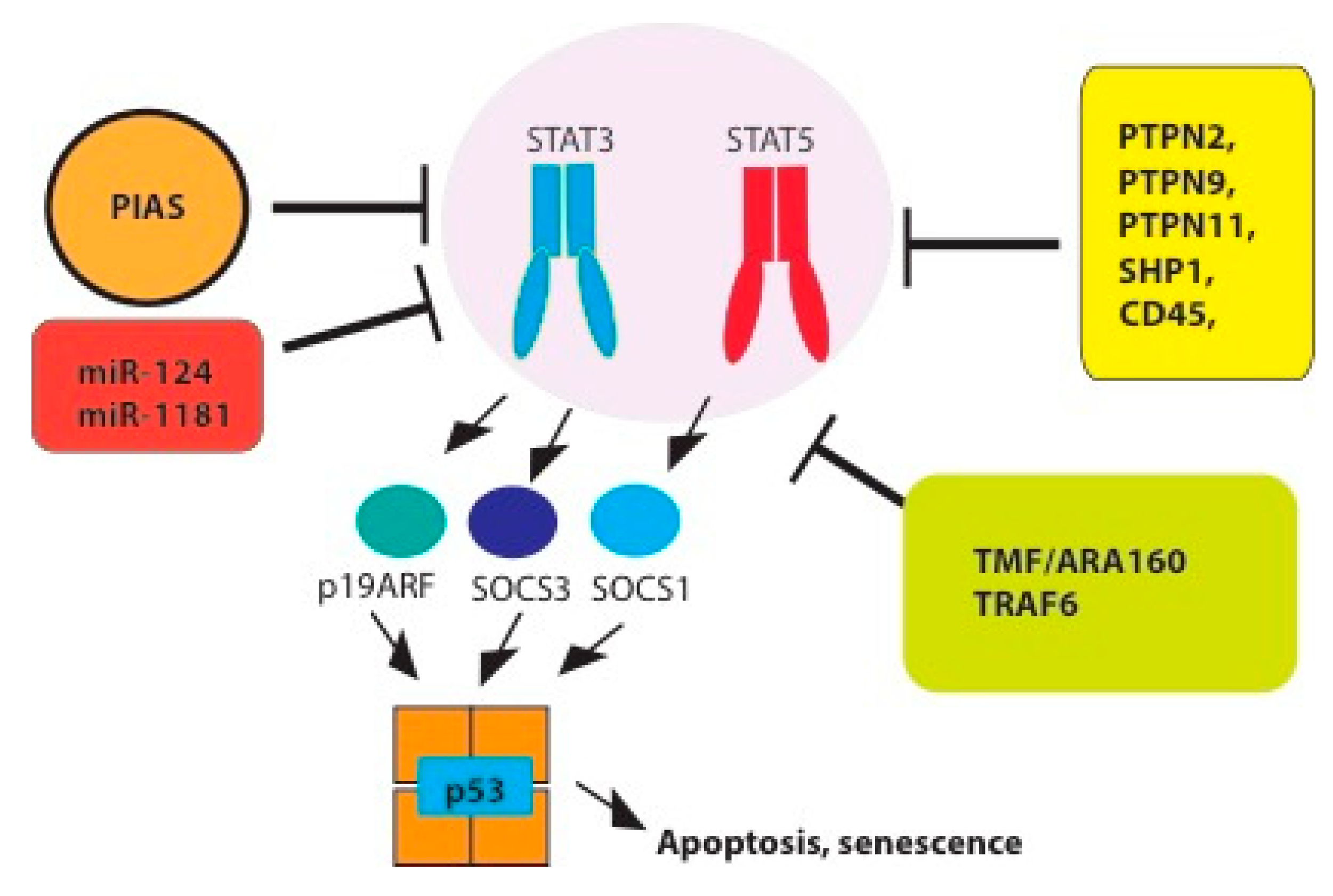
4. Tumor Suppressor Functions and Negative Regulation of STAT3/5 Signaling
4.1. Tyrosine Phosphatases
4.2. PIAS
4.3. E3 Ligases
4.4. MiRNAs
4.5. The Suppressor of Cytokine Signaling SOCS
4.6. P19ARF-p53 Pathway
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef] [PubMed]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagstrom, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. STAT3 Target Genes Relevant to Human Cancers. Cancers (Basel) 2014, 6, 897–925. [Google Scholar] [CrossRef]
- Basham, B.; Sathe, M.; Grein, J.; McClanahan, T.; D’Andrea, A.; Lees, E.; Rascle, A. In vivo identification of novel STAT5 target genes. Nucleic Acids Res. 2008, 36, 3802–3818. [Google Scholar] [CrossRef]
- Wu, P.; Wu, D.; Zhao, L.; Huang, L.; Shen, G.; Huang, J.; Chai, Y. Prognostic role of STAT3 in solid tumors: A systematic review and meta-analysis. Oncotarget 2016, 7, 19863–19883. [Google Scholar] [CrossRef]
- Mohanty, S.K.; Yagiz, K.; Pradhan, D.; Luthringer, D.J.; Amin, M.B.; Alkan, S.; Cinar, B. STAT3 and STAT5A are potential therapeutic targets in castration-resistant prostate cancer. Oncotarget 2017, 8, 85997–86010. [Google Scholar] [CrossRef]
- Mirtti, T.; Leiby, B.E.; Abdulghani, J.; Aaltonen, E.; Pavela, M.; Mamtani, A.; Alanen, K.; Egevad, L.; Granfors, T.; Josefsson, A.; et al. Nuclear Stat5a/b predicts early recurrence and prostate cancer-specific death in patients treated by radical prostatectomy. Hum. Pathol. 2013, 44, 310–319. [Google Scholar] [CrossRef]
- Klupp, F.; Diers, J.; Kahlert, C.; Neumann, L.; Halama, N.; Franz, C.; Schmidt, T.; Lasitschka, F.; Warth, A.; Weitz, J.; et al. Expressional STAT3/STAT5 Ratio is an Independent Prognostic Marker in Colon Carcinoma. Ann. Surg. Oncol. 2015, 22, S1548–S1555. [Google Scholar] [CrossRef]
- Peck, A.R.; Witkiewicz, A.K.; Liu, C.; Stringer, G.A.; Klimowicz, A.C.; Pequignot, E.; Freydin, B.; Tran, T.H.; Yang, N.; Rosenberg, A.L.; et al. Loss of nuclear localized and tyrosine phosphorylated Stat5 in breast cancer predicts poor clinical outcome and increased risk of antiestrogen therapy failure. J. Clin. Oncol. 2011, 29, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Kaltenecker, D.; Themanns, M.; Mueller, K.M.; Spirk, K.; Suske, T.; Merkel, O.; Kenner, L.; Luis, A.; Kozlov, A.; Haybaeck, J.; et al. Hepatic growth hormone-JAK2-STAT5 signalling: Metabolic function, non-alcoholic fatty liver disease and hepatocellular carcinoma progression. Cytokine 2018. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; Moriggl, R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim. Biophys. Acta 2011, 1815, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Lu, S. A meta-analysis of STAT3 and phospho-STAT3 expression and survival of patients with non-small-cell lung cancer. Eur. J. Surg. Oncol. 2014, 40, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.G.; Zhang, M.; Yang, F.; Gao, W.; Wang, Z.; Zhu, L.M. Clinical and prognostic significance of signal transducer and activator of transcription 3 and mucin 1 in patients with non-small cell lung cancer following surgery. Oncol. Lett. 2018, 15, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Wang, J.; Jiang, N.; Pan, H.; Li, D. Correlation between p-STAT3 overexpression and prognosis in lung cancer: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0182282. [Google Scholar] [CrossRef] [PubMed]
- Koperek, O.; Aumayr, K.; Schindl, M.; Werba, G.; Soleiman, A.; Schoppmann, S.; Sahora, K.; Birner, P. Phosphorylation of STAT3 correlates with HER2 status, but not with survival in pancreatic ductal adenocarcinoma. APMIS 2014, 122, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Xu, Y.; Ge, H.; Li, G.; Wu, J. Clinicopathological significance and prognostic role of p-STAT3 in patients with hepatocellular carcinoma. Onco-Targets Ther. 2018, 11, 1203–1214. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, J.; Li, W.; Chen, Y.; Liu, X.; Wang, J. Meta-analysis of STAT3 and phospho-STAT3 expression and survival of patients with breast cancer. Oncotarget 2018, 9, 13060–13067. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Salgado, R.; Brohee, S.; Zahavi, T.; Peretz, T.; Van den Eynden, G.; Rouas, G.; Salmon, A.; Francis, P.A.; Di Leo, A.; et al. p-STAT3 in luminal breast cancer: Integrated RNA-protein pooled analysis and results from the BIG 2-98 phase III trial. Int. J. Oncol. 2018, 52, 424–432. [Google Scholar] [CrossRef]
- Lin, G.S.; Chen, Y.P.; Lin, Z.X.; Wang, X.F.; Zheng, Z.Q.; Chen, L. STAT3 serine 727 phosphorylation influences clinical outcome in glioblastoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3141–3149. [Google Scholar]
- Vilardell, J.; Alcaraz, E.; Sarro, E.; Trilla, E.; Cuadros, T.; de Torres, I.; Plana, M.; Ramon, Y.C.S.; Pinna, L.A.; Ruzzene, M.; et al. Under-expression of CK2beta subunit in ccRCC represents a complementary biomarker of p-STAT3 Ser727 that correlates with patient survival. Oncotarget 2018, 9, 5736–5751. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Inoue, K.; Nagayasu, T.; Sekine, I. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol. Rep. 2006, 15, 1445–1451. [Google Scholar] [CrossRef]
- Morikawa, T.; Baba, Y.; Yamauchi, M.; Kuchiba, A.; Nosho, K.; Shima, K.; Tanaka, N.; Huttenhower, C.; Frank, D.A.; Fuchs, C.S.; et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin. Cancer Res. 2011, 17, 1452–1462. [Google Scholar] [CrossRef]
- Lee, J.L.; Wang, M.J.; Chen, J.Y. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J. Cell Biol. 2009, 185, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhu, F.; Zhang, M.; Li, Y.; Drennan, A.C.; Kimpara, S.; Rumball, I.; Selzer, C.; Cameron, H.; Kellicut, A.; et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2018, 115, E498–E505. [Google Scholar] [CrossRef] [PubMed]
- Creamer, B.A.; Sakamoto, K.; Schmidt, J.W.; Triplett, A.A.; Moriggl, R.; Wagner, K.U. Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol. Cell. Biol. 2010, 30, 2957–2970. [Google Scholar] [CrossRef]
- Tell, R.W.; Horvath, C.M. Bioinformatic analysis reveals a pattern of STAT3-associated gene expression specific to basal-like breast cancers in human tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 12787–12792. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef]
- Martincuks, A.; Andryka, K.; Kuster, A.; Schmitz-Van de Leur, H.; Komorowski, M.; Muller-Newen, G. Nuclear translocation of STAT3 and NF-kappaB are independent of each other but NF-kappaB supports expression and activation of STAT3. Cell. Signal. 2017, 32, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Paakinaho, V.; Baek, S.; Sung, M.H.; Hager, G.L. Synergistic gene expression during the acute phase response is characterized by transcription factor assisted loading. Nat. Commun. 2017, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Shin, H.J.; Gwak, H.S.; Park, S.; Chwae, Y.J. NF-kappaB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene 2012, 31, 3467–3481. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Singh, R.; Sabharwal, L.; Bando, H.; Meng, J.; Arima, Y.; Yamada, M.; Harada, M.; Jiang, J.J.; Kamimura, D.; et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 2014, 74, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Canino, C.; Luo, Y.; Marcato, P.; Blandino, G.; Pass, H.I.; Cioce, M. A STAT3-NFkB/DDIT3/CEBPbeta axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget 2015, 6, 12637–12653. [Google Scholar] [CrossRef] [PubMed]
- Thi, H.T.H.; Hong, S. Inflammasome as a Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Struhl, K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. USA 2013, 110, 972–977. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Parisotto, M.; Rowell, M.C.; Le Calve, B.; Igelmann, S.; Moineau-Vallee, K.; Saint-Germain, E.; Kalegari, P.; Bourdeau, V.; Kottakis, F.; et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 2019. [Google Scholar] [CrossRef]
- Luo, G.; Yu-Lee, L. Stat5b inhibits NFkappaB-mediated signaling. Mol. Endocrinol. 2000, 14, 114–123. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawashima, T.; Murata, K.; Akira, S.; Tonozuka, Y.; Minoshima, Y.; Feng, S.; Kumagai, H.; Tsuruga, H.; Ikeda, Y.; Asano, S.; et al. STAT5 induces macrophage differentiation of M1 leukemia cells through activation of IL-6 production mediated by NF-kappaB p65. J. Immunol. 2001, 167, 3652–3660. [Google Scholar] [CrossRef] [PubMed]
- Chueh, F.Y.; Leong, K.F.; Yu, C.L. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem. Biophys. Res. Commun. 2010, 402, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [PubMed]
- Macias, E.; Rao, D.; Carbajal, S.; Kiguchi, K.; DiGiovanni, J. Stat3 binds to mtDNA and regulates mitochondrial gene expression in keratinocytes. J. Investig. Dermatol. 2014, 134, 1971–1980. [Google Scholar] [CrossRef]
- Rincon, M.; Pereira, F.V. A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function. Int. J. Mol. Sci. 2018, 19, 1656. [Google Scholar] [CrossRef]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 a La Carte. Int. J. Mol. Sci. 2018, 19, 2820. [Google Scholar] [CrossRef]
- Luo, D.; Fraga-Lauhirat, M.; Millings, J.; Ho, C.; Villarreal, E.M.; Fletchinger, T.C.; Bonfiglio, J.V.; Mata, L.; Nemesure, M.D.; Bartels, L.E.; et al. Phospho-valproic acid (MDC-1112) suppresses glioblastoma growth in preclinical models through the inhibition of STAT3 phosphorylation. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Sala, D.; Cunningham, T.J.; Stec, M.J.; Etxaniz, U.; Nicoletti, C.; Dall’Agnese, A.; Puri, P.L.; Duester, G.; Latella, L.; Sacco, A. The Stat3-Fam3a axis promotes muscle stem cell myogenic lineage progression by inducing mitochondrial respiration. Nat. Commun. 2019, 10, 1796. [Google Scholar] [CrossRef]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef]
- Qiu, H.; Lizano, P.; Laure, L.; Sui, X.; Rashed, E.; Park, J.Y.; Hong, C.; Gao, S.; Holle, E.; Morin, D.; et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 2011, 124, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Gadir, N.; Haim-Vilmovsky, L.; Kraut-Cohen, J.; Gerst, J.E. Localization of mRNAs coding for mitochondrial proteins in the yeast Saccharomyces cerevisiae. RNA 2011, 17, 1551–1565. [Google Scholar] [CrossRef]
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Gucek, M.; Xu, H. The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J. 2016, 35, 1045–1057. [Google Scholar] [CrossRef]
- Hansen, K.G.; Herrmann, J.M. Transport of Proteins into Mitochondria. Protein J. 2019. [Google Scholar] [CrossRef]
- Richard, A.J.; Hang, H.; Stephens, J.M. Pyruvate dehydrogenase complex (PDC) subunits moonlight as interaction partners of phosphorylated STAT5 in adipocytes and adipose tissue. J. Biol. Chem. 2017, 292, 19733–19742. [Google Scholar] [CrossRef] [PubMed]
- Kidder, B.L.; Yang, J.; Palmer, S. Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS ONE 2008, 3, e3932. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Guo, G.; Wray, J.; Eyres, I.; Nichols, J.; Grotewold, L.; Morfopoulou, S.; Humphreys, P.; Mansfield, W.; Walker, R.; et al. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 2009, 5, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Li, P.; Tong, C.; Ying, Q.L. Embryonic stem cell self-renewal pathways converge on the transcription factor Tfcp2l1. EMBO J. 2013, 32, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Liu, J.; Chen, C.; Liu, Z.; Jiang, C.; Chu, H.; Pan, W.; Wang, X.; Zhang, L.; Li, B.; et al. The deubiquitinase USP21 maintains the stemness of mouse embryonic stem cells via stabilization of Nanog. Nat. Commun. 2016, 7, 13594. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; van Oosten, A.L.; Theunissen, T.W.; Guo, G.; Silva, J.C.; Smith, A. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell Stem Cell 2010, 7, 319–328. [Google Scholar] [CrossRef]
- Tang, Y.; Luo, Y.; Jiang, Z.; Ma, Y.; Lin, C.J.; Kim, C.; Carter, M.G.; Amano, T.; Park, J.; Kish, S.; et al. Jak/Stat3 signaling promotes somatic cell reprogramming by epigenetic regulation. Stem Cells 2012, 30, 2645–2656. [Google Scholar] [CrossRef]
- Carbognin, E.; Betto, R.M.; Soriano, M.E.; Smith, A.G.; Martello, G. Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J. 2016, 35, 618–634. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Gong, A.H.; Wei, P.; Zhang, S.; Yao, J.; Yuan, Y.; Zhou, A.D.; Lang, F.F.; Heimberger, A.B.; Rao, G.; Huang, S. FoxM1 Drives a Feed-Forward STAT3-Activation Signaling Loop That Promotes the Self-Renewal and Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Jiang, D. Resveratrol eliminates cancer stem cells of osteosarcoma by STAT3 pathway inhibition. PLoS ONE 2018, 13, e0205918. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Yu, X.; Huang, H.; Zhou, W.; Xiang, C.; Huang, H.; Miele, L.; Liu, Z.; Bebek, G.; Bao, S.; et al. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. Cell Stem Cell 2018, 22, 104–118.e6. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Zhang, R.; Li, Z.; Yin, Y.; Fang, X.; Ding, X.; Cai, Y.; Yang, S.; Mu, H.; Zong, D.; et al. Nuclear Smad6 promotes gliomagenesis by negatively regulating PIAS3-mediated STAT3 inhibition. Nat. Commun. 2018, 9, 2504. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wu, J.; Shi, J.; Huo, Y.M.; Dai, W.; Geng, J.; Lu, P.; Yang, M.W.; Fang, Y.; Wang, W.; et al. IL22RA1/STAT3 Signaling Promotes Stemness and Tumorigenicity in Pancreatic Cancer. Cancer Res. 2018, 78, 3293–3305. [Google Scholar] [CrossRef]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150. [Google Scholar] [CrossRef]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef]
- Chen, M.W.; Yang, S.T.; Chien, M.H.; Hua, K.T.; Wu, C.J.; Hsiao, S.M.; Lin, H.; Hsiao, M.; Su, J.L.; Wei, L.H. The STAT3-miRNA-92-Wnt Signaling Pathway Regulates Spheroid Formation and Malignant Progression in Ovarian Cancer. Cancer Res. 2017, 77, 1955–1967. [Google Scholar] [CrossRef]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, P.S.; Zheng, Q.; Bhagrath, M.; Mulkearns-Hubert, E.E.; Myers, M.G.; Lathia, J.D.; Reizes, O. STAT3 activation by leptin receptor is essential for TNBC stem cell maintenance. Endocr. Relat. Cancer 2017, 24, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.J.; Lai, H.M.; Chang, Y.W.; Chen, G.Y.; Lee, J.L. Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 2011, 30, 3186–3199. [Google Scholar] [CrossRef] [PubMed]
- Schepers, H.; van Gosliga, D.; Wierenga, A.T.; Eggen, B.J.; Schuringa, J.J.; Vellenga, E. STAT5 is required for long-term maintenance of normal and leukemic human stem/progenitor cells. Blood 2007, 110, 2880–2888. [Google Scholar] [CrossRef] [PubMed]
- Talati, P.G.; Gu, L.; Ellsworth, E.M.; Girondo, M.A.; Trerotola, M.; Hoang, D.T.; Leiby, B.; Dagvadorj, A.; McCue, P.A.; Lallas, C.D.; et al. Jak2-Stat5a/b Signaling Induces Epithelial-to-Mesenchymal Transition and Stem-Like Cell Properties in Prostate Cancer. Am. J. Pathol. 2015, 185, 2505–2522. [Google Scholar] [CrossRef] [PubMed]
- Boutillon, F.; Pigat, N.; Sala, L.S.; Reyes-Gomez, E.; Moriggl, R.; Guidotti, J.E.; Goffin, V. STAT5a/b Deficiency Delays, but does not Prevent, Prolactin-Driven Prostate Tumorigenesis in Mice. Cancers (Basel) 2019, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.R.; Naylor, M.J.; Asselin-Labat, M.L.; Blazek, K.D.; Gardiner-Garden, M.; Hilton, H.N.; Kazlauskas, M.; Pritchard, M.A.; Chodosh, L.A.; Pfeffer, P.L.; et al. The ETS transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev. 2008, 22, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Metser, G.; Shin, H.Y.; Wang, C.; Yoo, K.H.; Oh, S.; Villarino, A.V.; O’Shea, J.J.; Kang, K.; Hennighausen, L. An autoregulatory enhancer controls mammary-specific STAT5 functions. Nucleic Acids Res. 2016, 44, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Stute, P.; Sielker, S.; Wood, C.E.; Register, T.C.; Lees, C.J.; Dewi, F.N.; Williams, J.K.; Wagner, J.D.; Stefenelli, U.; Cline, J.M. Life stage differences in mammary gland gene expression profile in non-human primates. Breast Cancer Res. Treat. 2012, 133, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Morales, L.D.; Jang, I.S.; Cho, Y.Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.; Cao, F.; Jiang, H.; Li, A.; Li, J.; Qiu, L.; Shen, H.; Chang, W.; Zhou, C.; et al. SHP2 associates with nuclear localization of STAT3: Significance in progression and prognosis of colorectal cancer. Sci. Rep. 2017, 7, 17597. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.C.; Shiau, C.W.; Tai, W.T.; Hung, M.H.; Chu, P.Y.; Hsieh, F.S.; Lin, H.; Yu, H.C.; Chen, K.F. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene 2015, 34, 5252–5263. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wen, R.; Yang, S.; Schuman, J.; Zhang, E.E.; Yi, T.; Feng, G.S.; Wang, D. Identification of Shp-2 as a Stat5A phosphatase. J. Biol. Chem. 2003, 278, 16520–16527. [Google Scholar] [CrossRef]
- Xiao, W.; Ando, T.; Wang, H.Y.; Kawakami, Y.; Kawakami, T. Lyn- and PLC-beta3-dependent regulation of SHP-1 phosphorylation controls Stat5 activity and myelomonocytic leukemia-like disease. Blood 2010, 116, 6003–6013. [Google Scholar] [CrossRef]
- Brantley, E.C.; Nabors, L.B.; Gillespie, G.Y.; Choi, Y.H.; Palmer, C.A.; Harrison, K.; Roarty, K.; Benveniste, E.N. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: Implications for STAT-3 activation and gene expression. Clin. Cancer Res. 2008, 14, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Sundvall, M.; Korhonen, A.; Vaparanta, K.; Anckar, J.; Halkilahti, K.; Salah, Z.; Aqeilan, R.I.; Palvimo, J.J.; Sistonen, L.; Elenius, K. Protein inhibitor of activated STAT3 (PIAS3) protein promotes SUMOylation and nuclear sequestration of the intracellular domain of ErbB4 protein. J. Biol. Chem. 2012, 287, 23216–23226. [Google Scholar] [CrossRef]
- Jang, H.D.; Yoon, K.; Shin, Y.J.; Kim, J.; Lee, S.Y. PIAS3 suppresses NF-kappaB-mediated transcription by interacting with the p65/RelA subunit. J. Biol. Chem. 2004, 279, 24873–24880. [Google Scholar] [CrossRef]
- Liu, Y.; Bridges, R.; Wortham, A.; Kulesz-Martin, M. NF-kappaB repression by PIAS3 mediated RelA SUMOylation. PLoS ONE 2012, 7, e37636. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, L.; Shi, H.; Wu, C. p53 Nterminal binding and stabilisation by PIAS3 inhibits MDM2induced p53 ubiquitination and regulates cell growth. Mol. Med. Rep. 2014, 9, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.; Tsruya, R.; Levitsky, P.; Pomp, O.; Taller, M.; Weisberg, S.; Parris, W.; Kulkarni, S.; Malovani, H.; Pawson, T.; et al. TMF/ARA160 is a BC-box-containing protein that mediates the degradation of Stat3. Oncogene 2004, 23, 8908–8919. [Google Scholar] [CrossRef] [PubMed]
- Abrham, G.; Volpe, M.; Shpungin, S.; Nir, U. TMF/ARA160 downregulates proangiogenic genes and attenuates the progression of PC3 xenografts. Int. J. Cancer 2009, 125, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yuan, Y.; Jin, C.; Chen, H.; Leng, L.; He, F.; Wang, J. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS ONE 2012, 7, e49567. [Google Scholar] [CrossRef] [PubMed]
- Deschenes-Simard, X.; Gaumont-Leclerc, M.F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013, 27, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Du, P.; Cui, P.; Qin, Y.; Hu, C.; Wu, J.; Zhou, Z.; Zhang, W.; Qin, L.; Huang, G. LncRNA PVT1 promotes angiogenesis via activating the STAT3/VEGFA axis in gastric cancer. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Chang, D.; Du, H.Z.; Zhao, Y.P. Genome-wide screen identifies PVT1 as a regulator of Gemcitabine sensitivity in human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2011, 407, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chen, X.; Li, J.; Guo, Y.; Li, H.; Pan, X.; Jiang, J.; Liu, H.; Wu, B. Salivary HOTAIR and PVT1 as novel biomarkers for early pancreatic cancer. Oncotarget 2016, 7, 25408–25419. [Google Scholar] [CrossRef]
- Pan, X.; Li, B.; Fan, N.; Li, J.; Cai, F.; Zhao, G.; Zheng, G.; Gao, C. Long Noncoding RNA PVT1 as a Potent Predictor of Prognosis in Cancers: A Meta-Analysis. Clin. Lab. 2017, 63, 1657–1666. [Google Scholar] [CrossRef]
- Hatziapostolou, M.; Polytarchou, C.; Aggelidou, E.; Drakaki, A.; Poultsides, G.A.; Jaeger, S.A.; Ogata, H.; Karin, M.; Struhl, K.; Hadzopoulou-Cladaras, M.; et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011, 147, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Li, J.; Wang, J.; Luo, X.; Ai, J.; Liu, Y.; Wang, N.; Liang, H.; Zhang, M.; Chen, N.; et al. microRNA-124 regulates cardiomyocyte differentiation of bone marrow-derived mesenchymal stem cells via targeting STAT3 signaling. Stem Cells 2012, 30, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, Y.; Yue, X.; Li, H.; Luo, X.; Wang, Y.; Wang, K.; Wan, J. MiR-124 suppresses growth of human colorectal cancer by inhibiting STAT3. PLoS ONE 2013, 8, e70300. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, F.; Kong, L.Y.; Xu, S.; Doucette, T.; Ferguson, S.D.; Yang, Y.; McEnery, K.; Jethwa, K.; Gjyshi, O.; et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. 2013, 73, 3913–3926. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Shi, Y.; Zhang, Y.; Sun, J. CircRNA_100782 regulates pancreatic carcinoma proliferation through the IL6-STAT3 pathway. Onco-Targets Ther. 2017, 10, 5783–5794. [Google Scholar] [CrossRef]
- Jiang, J.; Li, Z.; Yu, C.; Chen, M.; Tian, S.; Sun, C. MiR-1181 inhibits stem cell-like phenotypes and suppresses SOX2 and STAT3 in human pancreatic cancer. Cancer Lett. 2015, 356, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, W.W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Oskooei, V.K.; Azari, I.; Taheri, M. Suppressor of cytokine signaling (SOCS) genes are downregulated in breast cancer. World J. Surg. Oncol. 2018, 16, 226. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. JAKSTAT 2013, 2, e24053. [Google Scholar] [CrossRef]
- Suzuki, A.; Hanada, T.; Mitsuyama, K.; Yoshida, T.; Kamizono, S.; Hoshino, T.; Kubo, M.; Yamashita, A.; Okabe, M.; Takeda, K.; et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 2001, 193, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kanda, H.; Shikauchi, Y.; Saiura, A.; Matsubara, K.; Kitagawa, T.; Yamamoto, J.; Kubo, T.; Yoshikawa, H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005, 24, 6406–6417. [Google Scholar] [CrossRef] [PubMed]
- Rigby, R.J.; Simmons, J.G.; Greenhalgh, C.J.; Alexander, W.S.; Lund, P.K. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene 2007, 26, 4833–4841. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Yeganeh, M.; Ramanathan, S.; Leblanc, C.; Pomerleau, V.; Ferbeyre, G.; Saucier, C.; Ilangumaran, S. SOCS1 controls liver regeneration by regulating HGF signaling in hepatocytes. J. Hepatol. 2011, 55, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, M.; Gui, Y.; Kandhi, R.; Bobbala, D.; Tobelaim, W.S.; Saucier, C.; Yoshimura, A.; Ferbeyre, G.; Ramanathan, S.; Ilangumaran, S. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21(CIP1/WAF1) in the liver. Oncogene 2016, 35, 4200–4211. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.C.; Zhou, J.; He, J.Y.; Wei, Y.G.; Qin, Y.; Li, B. Aberrant promoter methylation of SOCS-1 gene may contribute to the pathogenesis of hepatocellular carcinoma: A meta-analysis. J. BUON 2016, 21, 142–151. [Google Scholar] [PubMed]
- Suzuki, M.; Shigematsu, H.; Shivapurkar, N.; Reddy, J.; Miyajima, K.; Takahashi, T.; Gazdar, A.F.; Frenkel, E.P. Methylation of apoptosis related genes in the pathogenesis and prognosis of prostate cancer. Cancer Lett. 2006, 242, 222–230. [Google Scholar] [CrossRef]
- Kobayashi, N.; Uemura, H.; Nagahama, K.; Okudela, K.; Furuya, M.; Ino, Y.; Ito, Y.; Hirano, H.; Inayama, Y.; Aoki, I.; et al. Identification of miR-30d as a novel prognostic maker of prostate cancer. Oncotarget 2012, 3, 1455–1471. [Google Scholar] [CrossRef]
- Chevrier, M.; Bobbala, D.; Villalobos-Hernandez, A.; Khan, M.G.; Ramanathan, S.; Saucier, C.; Ferbeyre, G.; Geha, S.; Ilangumaran, S. Expression of SOCS1 and the downstream targets of its putative tumor suppressor functions in prostate cancer. BMC Cancer 2017, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Hernandez, A.; Bobbala, D.; Kandhi, R.; Khan, M.G.; Mayhue, M.; Dubois, C.M.; Ferbeyre, G.; Saucier, C.; Ramanathan, S.; Ilangumaran, S. SOCS1 inhibits migration and invasion of prostate cancer cells, attenuates tumor growth and modulates the tumor stroma. Prostate Cancer Prostatic Dis. 2017, 20, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Mallette, F.A.; Deschenes-Simard, X.; Ramanathan, S.; Gagnon, J.; Moores, A.; Ilangumaran, S.; Ferbeyre, G. SOCS1 links cytokine signaling to p53 and senescence. Mol. Cell 2009, 36, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Calabrese, V.; Ilangumaran, S.; Ferbeyre, G. SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging 2010, 2, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Saint-Germain, E.; Mignacca, L.; Vernier, M.; Bobbala, D.; Ilangumaran, S.; Ferbeyre, G. SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging 2017, 9, 2137–2162. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Bouamar, H.; Jiang, D.; Wang, L.; Lin, A.P.; Ortega, M.; Aguiar, R.C. MicroRNA 155 control of p53 activity is context dependent and mediated by Aicda and Socs1. Mol. Cell. Biol. 2015, 35, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Shan, X.; Qian, J.; Ji, Q.; Wang, L.; Wang, X.; Li, M.; Ding, H.; Liu, Q.; Chen, L.; et al. The suppressor of cytokine signaling SOCS1 promotes apoptosis of intestinal epithelial cells via p53 signaling in Crohn’s disease. Exper. Mol. Pathol. 2016, 101, 1–11. [Google Scholar] [CrossRef]
- Mallette, F.A.; Gaumont-Leclerc, M.F.; Ferbeyre, G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007, 21, 43–48. [Google Scholar] [CrossRef]
- Mallette, F.A.; Gaumont-Leclerc, M.F.; Huot, G.; Ferbeyre, G. Myc Down-regulation as a Mechanism to Activate the Rb Pathway in STAT5A-induced Senescence. J. Biol. Chem. 2007, 282, 34938–34944. [Google Scholar] [CrossRef]
- Mallette, F.A.; Moiseeva, O.; Calabrese, V.; Mao, B.; Gaumont-Leclerc, M.F.; Ferbeyre, G. Transcriptome analysis and tumor suppressor requirements of STAT5-induced senescence. Ann. N. Y. Acad. Sci. 2010, 1197, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Barash, I. Stat5 in the mammary gland: Controlling normal development and cancer. J. Cell. Physiol. 2006, 209, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, M.T.; Xie, J.; Torhorst, J.; Bubendorf, L.; Haas, P.; Kononen, J.; Sauter, G.; Rui, H. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J. Clin. Oncol. 2004, 22, 2053–2060. [Google Scholar] [CrossRef]
- Peck, A.R.; Witkiewicz, A.K.; Liu, C.; Klimowicz, A.C.; Stringer, G.A.; Pequignot, E.; Freydin, B.; Yang, N.; Ertel, A.; Tran, T.H.; et al. Low levels of Stat5a protein in breast cancer are associated with tumor progression and unfavorable clinical outcomes. Breast Cancer Res. 2012, 14, R130. [Google Scholar] [CrossRef] [PubMed]
- Castro, P.; Giri, D.; Lamb, D.; Ittmann, M. Cellular senescence in the pathogenesis of benign prostatic hyperplasia. Prostate 2003, 55, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, J.L.; Timmerman, L.; Fridlyand, J.; Bastian, B.C. Mechanisms of cell-cycle arrest in Spitz nevi with constitutive activation of the MAP-kinase pathway. Am. J. Pathol. 2004, 164, 1783–1787. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Mooi, W.J.; Peeper, D.S. BRAF(E600) in benign and malignant human tumours. Oncogene 2008, 27, 877–895. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Vernier, M.; Bourdeau, V.; Gaumont-Leclerc, M.F.; Moiseeva, O.; Begin, V.; Saad, F.; Mes-Masson, A.M.; Ferbeyre, G. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011, 25, 41–50. [Google Scholar] [CrossRef]
- Dorr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Dabritz, J.H.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Kase, J.; Dorr, J.R.; Milanovic, M.; Lenze, D.; Grau, M.; Beuster, G.; Ji, S.; Reimann, M.; Lenz, P.; et al. Opposing roles of NF-kappaB in anti-cancer treatment outcome unveiled by cross-species investigations. Genes Dev. 2011, 25, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Zardo, G.; Tiirikainen, M.I.; Hong, C.; Misra, A.; Feuerstein, B.G.; Volik, S.; Collins, C.C.; Lamborn, K.R.; Bollen, A.; Pinkel, D.; et al. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nat. Genet. 2002, 32, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Lin, S.F.; Chang, J.G.; Yang, M.Y.; Hung, S.Y.; Chang, C.S. Epigenetic alteration of the SOCS1 gene in chronic myeloid leukaemia. Br. J. Haematol. 2003, 123, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Wong, K.Y.; Loong, F.; Srivastava, G. SOCS1 and SHP1 hypermethylation in mantle cell lymphoma and follicular lymphoma: Implications for epigenetic activation of the Jak/STAT pathway. Leukemia 2004, 18, 356–358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ekmekci, C.G.; Gutierrez, M.I.; Siraj, A.K.; Ozbek, U.; Bhatia, K. Aberrant methylation of multiple tumor suppressor genes in acute myeloid leukemia. Am. J. Hematol. 2004, 77, 233–240. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Lindeman, G.J.; Choong, D.Y.; Wittlin, S.; Brentzell, L.; Phillips, W.; Campbell, I.G.; Visvader, J.E. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene 2004, 23, 7726–7733. [Google Scholar] [CrossRef]
- Hatirnaz, O.; Ure, U.; Ar, C.; Akyerli, C.; Soysal, T.; Ferhanoglu, B.; Ozcelik, T.; Ozbek, U. The SOCS-1 gene methylation in chronic myeloid leukemia patients. Am. J. Hematol. 2007, 82, 729–730. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, H.W.; Lu, M.H.; He, X.H.; Li, Y.; Gu, H.; Liu, M.F.; Wang, E.D. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010, 70, 3119–3127. [Google Scholar] [CrossRef]
- Merkel, O.; Hamacher, F.; Griessl, R.; Grabner, L.; Schiefer, A.I.; Prutsch, N.; Baer, C.; Egger, G.; Schlederer, M.; Krenn, P.W.; et al. Oncogenic role of miR-155 in anaplastic large cell lymphoma lacking the t(2;5) translocation. J. Pathol. 2015, 236, 445–456. [Google Scholar] [CrossRef]
- Zhao, X.D.; Zhang, W.; Liang, H.J.; Ji, W.Y. Overexpression of miR -155 promotes proliferation and invasion of human laryngeal squamous cell carcinoma via targeting SOCS1 and STAT3. PLoS ONE 2013, 8, e56395. [Google Scholar] [CrossRef]
- Saint-Germain, E.; Mignacca, L.; Huot, G.; Acevedo, M.; Moineau-Vallee, K.; Calabrese, V.; Bourdeau, V.; Rowell, M.C.; Ilangumaran, S.; Lessard, F.; et al. Phosphorylation of SOCS1 inhibits the SOCS1-p53 tumor suppressor axis. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Xu, Z.; Zhang, Y.; Jiang, P.; Huang, G.; Chen, S.; Lyu, X.; Zheng, P.; Zhao, X.; Zeng, Y.; et al. FXR induces SOCS3 and suppresses hepatocellular carcinoma. Oncotarget 2015, 6, 34606–34616. [Google Scholar] [CrossRef]
- Attia, Y.M.; Tawfiq, R.A.; Ali, A.A.; Elmazar, M.M. The FXR Agonist, Obeticholic Acid, Suppresses HCC Proliferation & Metastasis: Role of IL-6/STAT3 Signalling Pathway. Sci. Rep. 2017, 7, 12502. [Google Scholar] [CrossRef] [PubMed]
- Sugase, T.; Takahashi, T.; Serada, S.; Fujimoto, M.; Hiramatsu, K.; Ohkawara, T.; Tanaka, K.; Miyazaki, Y.; Makino, T.; Kurokawa, Y.; et al. SOCS1 Gene Therapy Improves Radiosensitivity and Enhances Irradiation-Induced DNA Damage in Esophageal Squamous Cell Carcinoma. Cancer Res. 2017, 77, 6975–6986. [Google Scholar] [CrossRef] [PubMed]
- Sugase, T.; Takahashi, T.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Hiramatsu, K.; Nishida, T.; Hirota, S.; Saito, Y.; Tanaka, K.; et al. SOCS1 gene therapy has antitumor effects in imatinib-resistant gastrointestinal stromal tumor cells through FAK/PI3 K signaling. Gastric Cancer 2018, 21, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Kunimura, N.; Kitagawa, K.; Fukui, Y.; Saito, H.; Narikiyo, K.; Ishiko, M.; Otsuki, N.; Nibu, K.I.; Fujisawa, M.; et al. Overexpression of SOCS3 mediated by adenovirus vector in mouse and human castration-resistant prostate cancer cells increases the sensitivity to NK cells in vitro and in vivo. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- de la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef]
- Pencik, J.; Schlederer, M.; Gruber, W.; Unger, C.; Walker, S.M.; Chalaris, A.; Marie, I.J.; Hassler, M.R.; Javaheri, T.; Aksoy, O.; et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat. Commun. 2015, 6, 7736. [Google Scholar] [CrossRef]
- Lessard, F.; Morin, F.; Ivanchuk, S.; Langlois, F.; Stefanovsky, V.; Rutka, J.; Moss, T. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol. Cell 2010, 38, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001, 2, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Han, S.Y.; Song, J. Regulatory Network of ARF in Cancer Development. Mol. Cell 2018, 41, 381–389. [Google Scholar] [CrossRef]
- Ferbeyre, G.; de Stanchina, E.; Lin, A.W.; Querido, E.; McCurrach, M.E.; Hannon, G.J.; Lowe, S.W. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell. Biol. 2002, 22, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Wiebringhaus, R.; Susani, M.; Culig, Z.; Kenner, L. IL-6/STAT3/ARF: The guardians of senescence, cancer progression and metastasis in prostate cancer. Swiss Med. Wkly. 2015, 145, w14215. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.E.; Kitagawa, M.; Su, W.C.; You, Z.H.; Iwamoto, Y.; Fu, X.Y. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 1996, 272, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; O’Brien, C.A.; Roberson, P.K.; Manolagas, S.C. Transcriptional activation of the p21(WAF1,CIP1,SDI1) gene by interleukin-6 type cytokines. A prerequisite for their pro-differentiating and anti-apoptotic effects on human osteoblastic cells. J. Biol. Chem. 1998, 273, 21137–21144. [Google Scholar] [CrossRef] [PubMed]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K.; et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef] [PubMed]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 2010, 138, 1003–1011. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.C.; Lee, S.E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. STAT3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lafdil, F.; Wang, L.; Park, O.; Yin, S.; Niu, J.; Miller, A.M.; Sun, Z.; Gao, B. Hepatoprotective versus oncogenic functions of STAT3 in liver tumorigenesis. Am. J. Pathol. 2011, 179, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Schneller, D.; Machat, G.; Sousek, A.; Proell, V.; van Zijl, F.; Zulehner, G.; Huber, H.; Mair, M.; Muellner, M.K.; Nijman, S.M.; et al. p19(ARF)/p14(ARF) controls oncogenic functions of signal transducer and activator of transcription 3 in hepatocellular carcinoma. Hepatology 2011, 54, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Chen, Y.; Wu, C.; Wu, Z.Y.; Tweardy, D.J.; Alshareef, A.; Liao, L.D.; Xue, Y.J.; Wu, J.Y.; Chen, B.; et al. The Opposing Function of STAT3 as an Oncoprotein and Tumor Suppressor Is Dictated by the Expression Status of STAT3beta in Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. 2016, 22, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, E.; Egloff, A.M.; Sasaki, C.; Kountourakis, P.; Burtness, B.; Fountzilas, G.; Dafni, U.; Zaramboukas, T.; Rampias, T.; Rimm, D.; et al. Nuclear localization of signal transducer and activator of transcription 3 in head and neck squamous cell carcinoma is associated with a better prognosis. Clin. Cancer Res. 2010, 16, 2427–2434. [Google Scholar] [CrossRef]
- Gordziel, C.; Bratsch, J.; Moriggl, R.; Knosel, T.; Friedrich, K. Both STAT1 and STAT3 are favourable prognostic determinants in colorectal carcinoma. Br. J. Cancer 2013, 109, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Ettl, T.; Stiegler, C.; Zeitler, K.; Agaimy, A.; Zenk, J.; Reichert, T.E.; Gosau, M.; Kuhnel, T.; Brockhoff, G.; Schwarz, S. EGFR, HER2, survivin, and loss of pSTAT3 characterize high-grade malignancy in salivary gland cancer with impact on prognosis. Hum. Pathol. 2012, 43, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Neilson, L.M.; Peck, A.R.; Liu, C.; Tran, T.H.; Witkiewicz, A.; Hyslop, T.; Nevalainen, M.T.; Sauter, G.; Rui, H. Signal transducer and activator of transcription-3 and breast cancer prognosis. Am. J. Cancer Res. 2011, 1, 347–355. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Biomarker/Type of Study | Overall Survival | Ref |
|---|---|---|---|
| NSCLC | High p-STAT3/Meta-analysis of 9 studies | Log HR 0.67, 95% CI: 0.57–0.77, p < 0.0001 | [14] |
| NSCLC | High p-STAT3/Cox regression multivariate analysis | HR 2.45, 95% CI: 1.084–5.556, p = 0.031 | [15] |
| Lung cancer | High p-STAT3/Meta-analysis of 13 studies | HR 1.23, 95% CI: 1.04–1.46, p = 0.02 | [16] |
| Pancreatic cancer | High p-STAT3/Log-rank test | No association p > 0.05 | [17] |
| Liver cancer (HCC) | High p-STAT3/Meta-analysis of 8 studies | HR 1.69, 95% CI: 1.07–2.31, p < 0.0001 3yr HR 1.67, 95% CI: 1.18–2.15, p < 0.0001 5yr | [18] |
| Breast cancer | High p-STAT3/Meta-analysis of 12 studies | No association p > 0.05 | [19] |
| Breast cancer (ER+) | High p-STAT3/Log-rank test | No association p > 0.05 | [20] |
| GBM | High p-S727-STAT3/Cox regression multivariate analysis | HR 1.797, 95% CI: 1.028–3.142, p = 0.040 | [21] |
| RCC | High p-S727-STAT3/Cox regression multivariate analysis | HR 3.32, 95% CI: 1.26–8.71, p = 0.014 10yr | [22] |
| Colon cancer | High p-STAT3/p-STAT5 ratio/Cox regression multivariate analysis | HR 4.468, p = 0.043 5yr | [10] |
| Colon cancer | High p-STAT3/Log-rank test | Worse overall survival, p < 0.001 | [23] |
| Colon cancer | High p-STAT3/Cox regression multivariate analysis | HR 1.61, 95% CI: 1.11–2.34, p = 0.015 | [24] |
| Breast cancer | Low p-STAT5/Cox regression multivariate analysis | HR 2.49, 95% CI: 1.23–5.05, p = 0.012 5yr | [11] |
| Prostate cancer | High nuclear STAT5A/B/Cox regression multivariate analysis | HR 1.59, 95% CI: 1.04–2.44, p = 0.034 | [9] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 Activation in Solid Cancers. Cancers 2019, 11, 1428. https://doi.org/10.3390/cancers11101428
Igelmann S, Neubauer HA, Ferbeyre G. STAT3 and STAT5 Activation in Solid Cancers. Cancers. 2019; 11(10):1428. https://doi.org/10.3390/cancers11101428
Chicago/Turabian StyleIgelmann, Sebastian, Heidi A. Neubauer, and Gerardo Ferbeyre. 2019. "STAT3 and STAT5 Activation in Solid Cancers" Cancers 11, no. 10: 1428. https://doi.org/10.3390/cancers11101428
APA StyleIgelmann, S., Neubauer, H. A., & Ferbeyre, G. (2019). STAT3 and STAT5 Activation in Solid Cancers. Cancers, 11(10), 1428. https://doi.org/10.3390/cancers11101428