FOXO1 Confers Maintenance of the Dark Zone Proliferation and Survival Program and Can Be Pharmacologically Targeted in Burkitt Lymphoma

 ,
,
Abstract
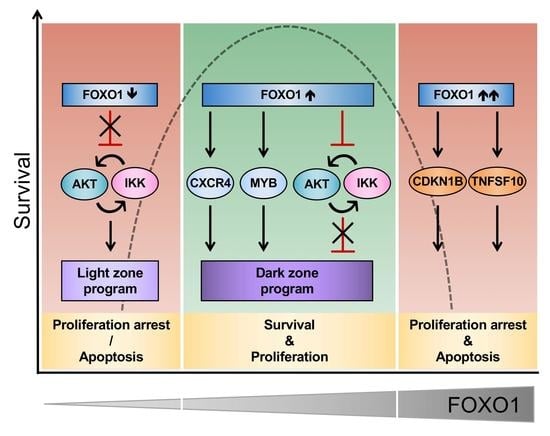
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. FOXO1 Expression is Essential for BL Survival
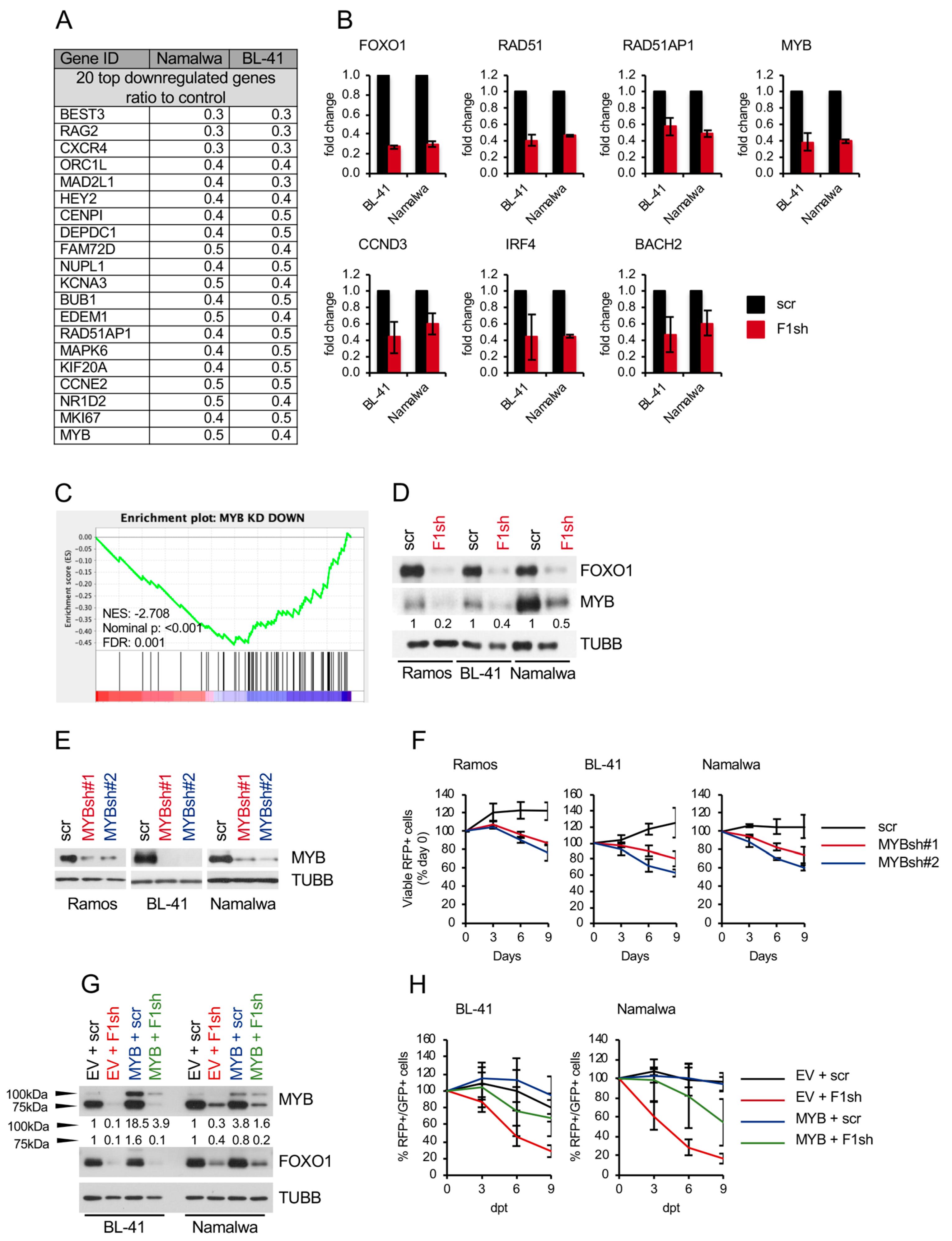
2.2. FOXO1 Knockdown Inhibits the Expression of Core-Survival Genes in BL
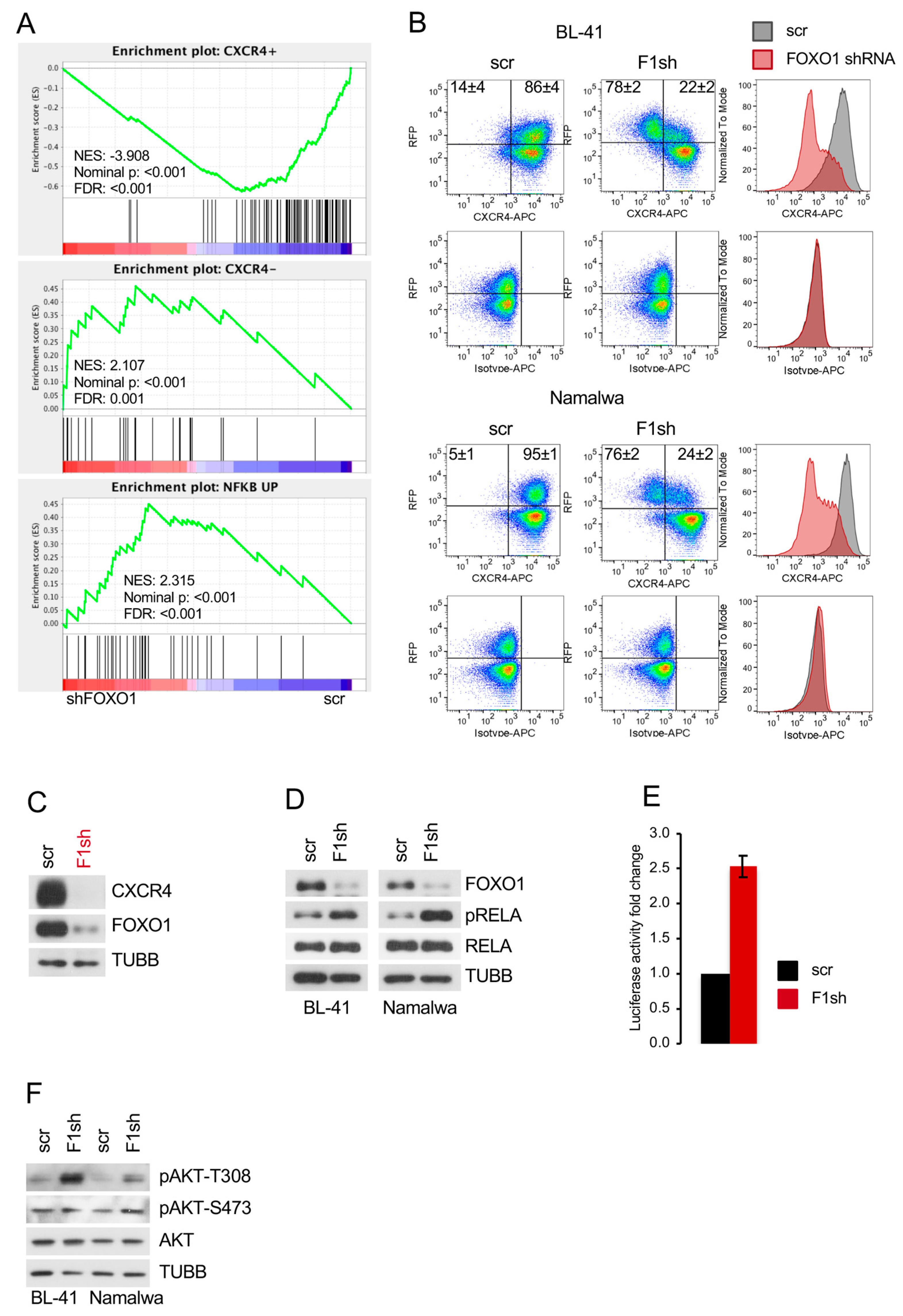
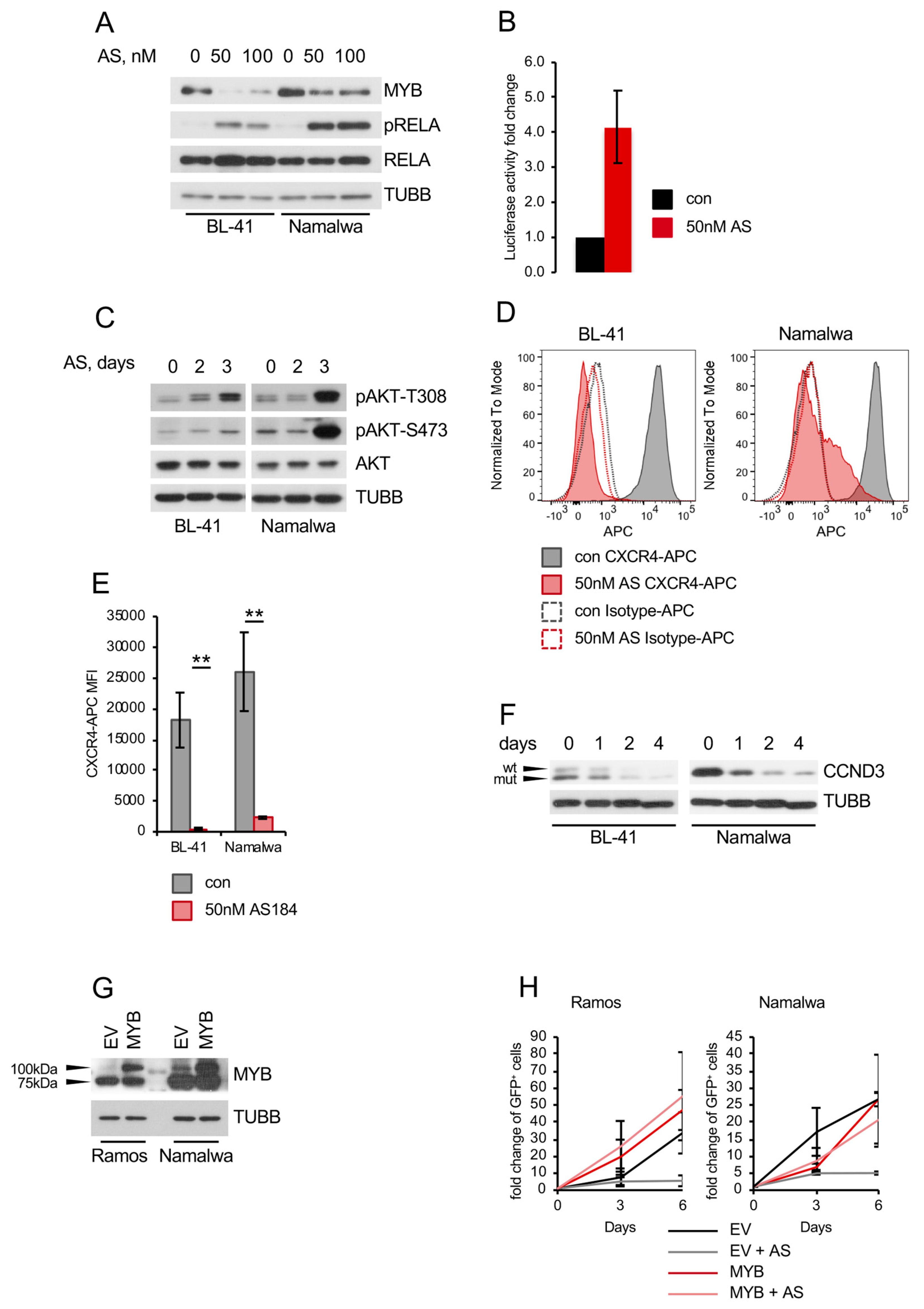
2.3. FOXO1 Is Critical for Maintenance of the DZ Phenotype and for Control of the NF-ΚB and PI3K-AKT Activity in BL
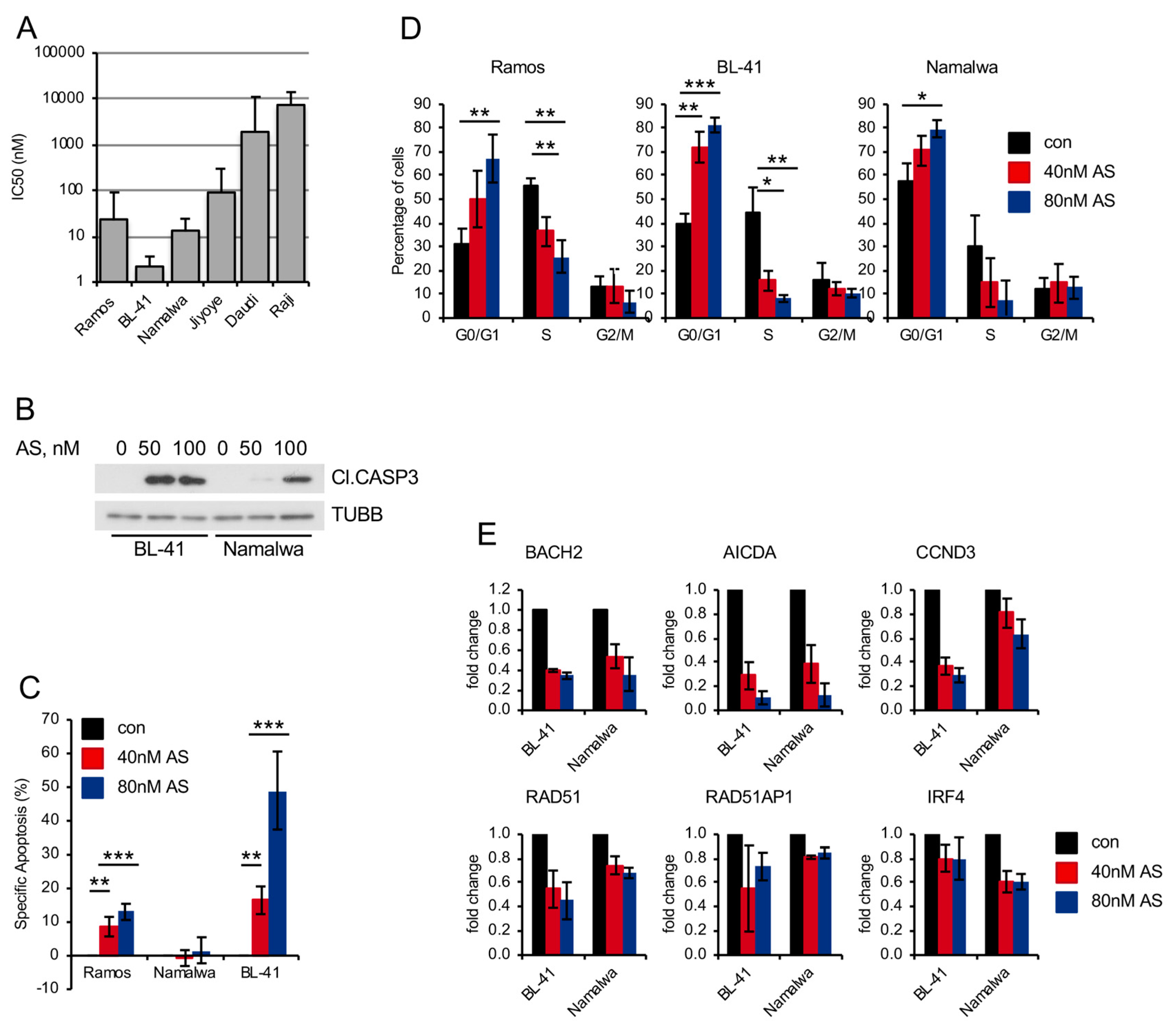
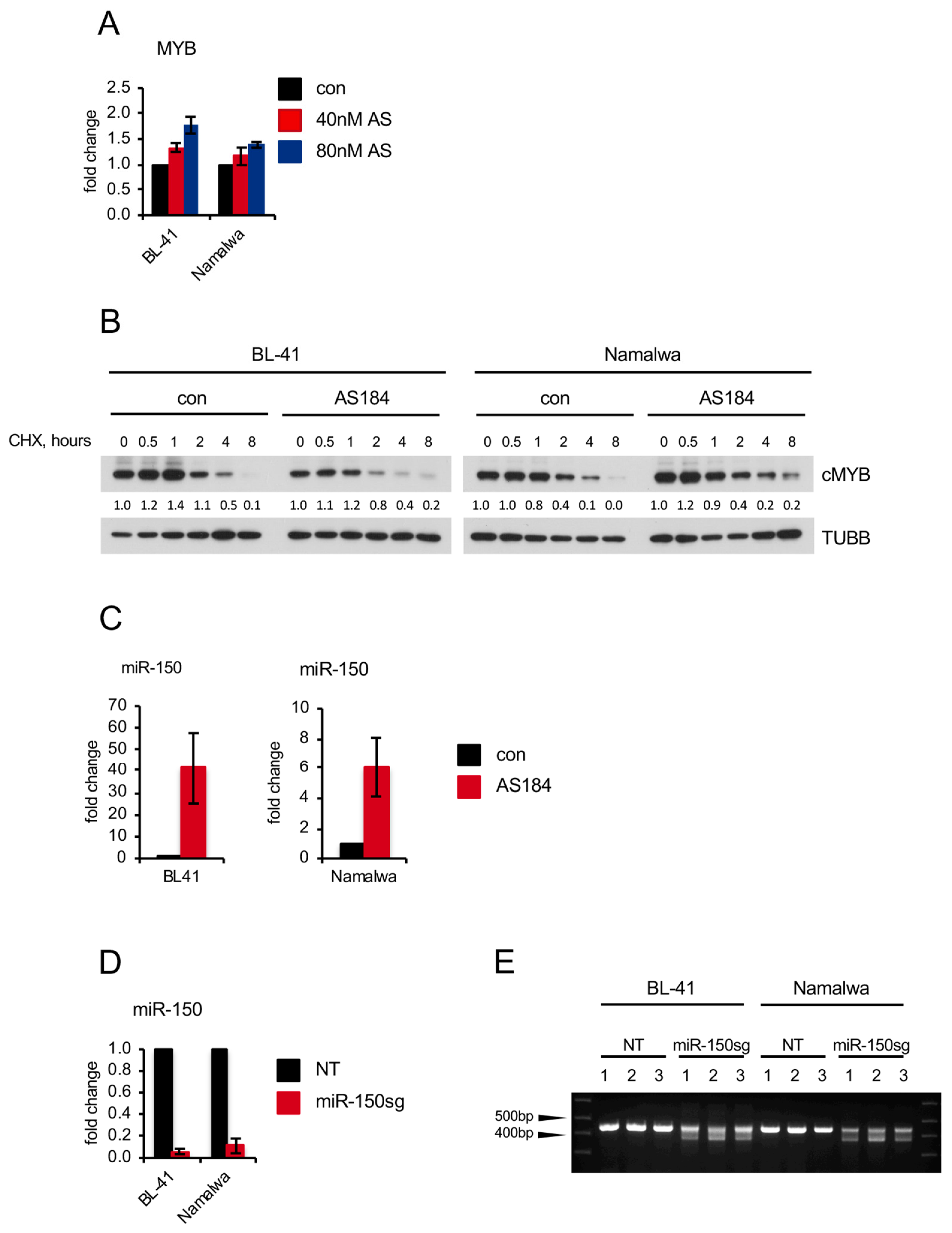
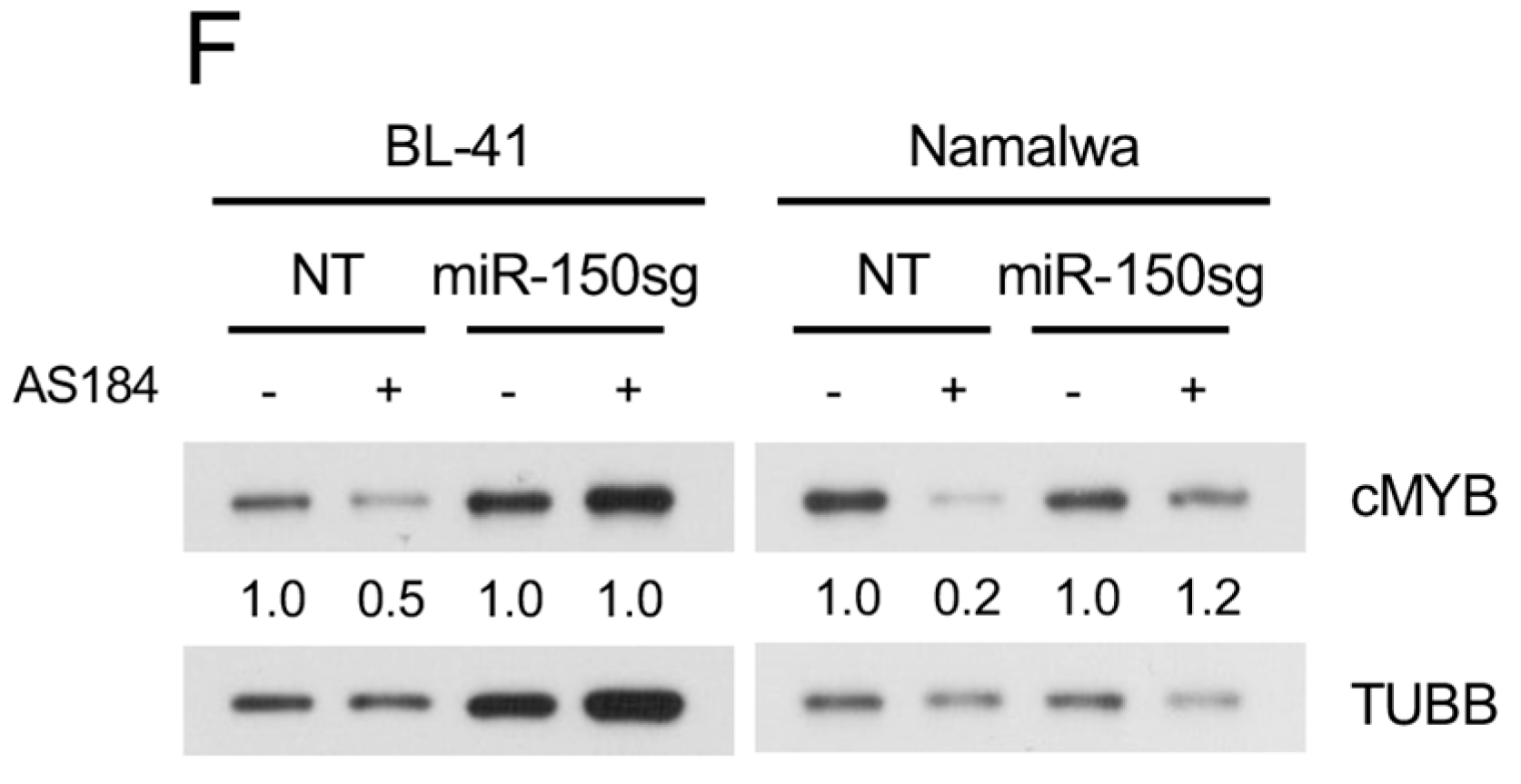
2.4. Pharmacologic FOXO1 Inactivation Inhibits Proliferation and Induces Apoptosis in BL Cell Lines
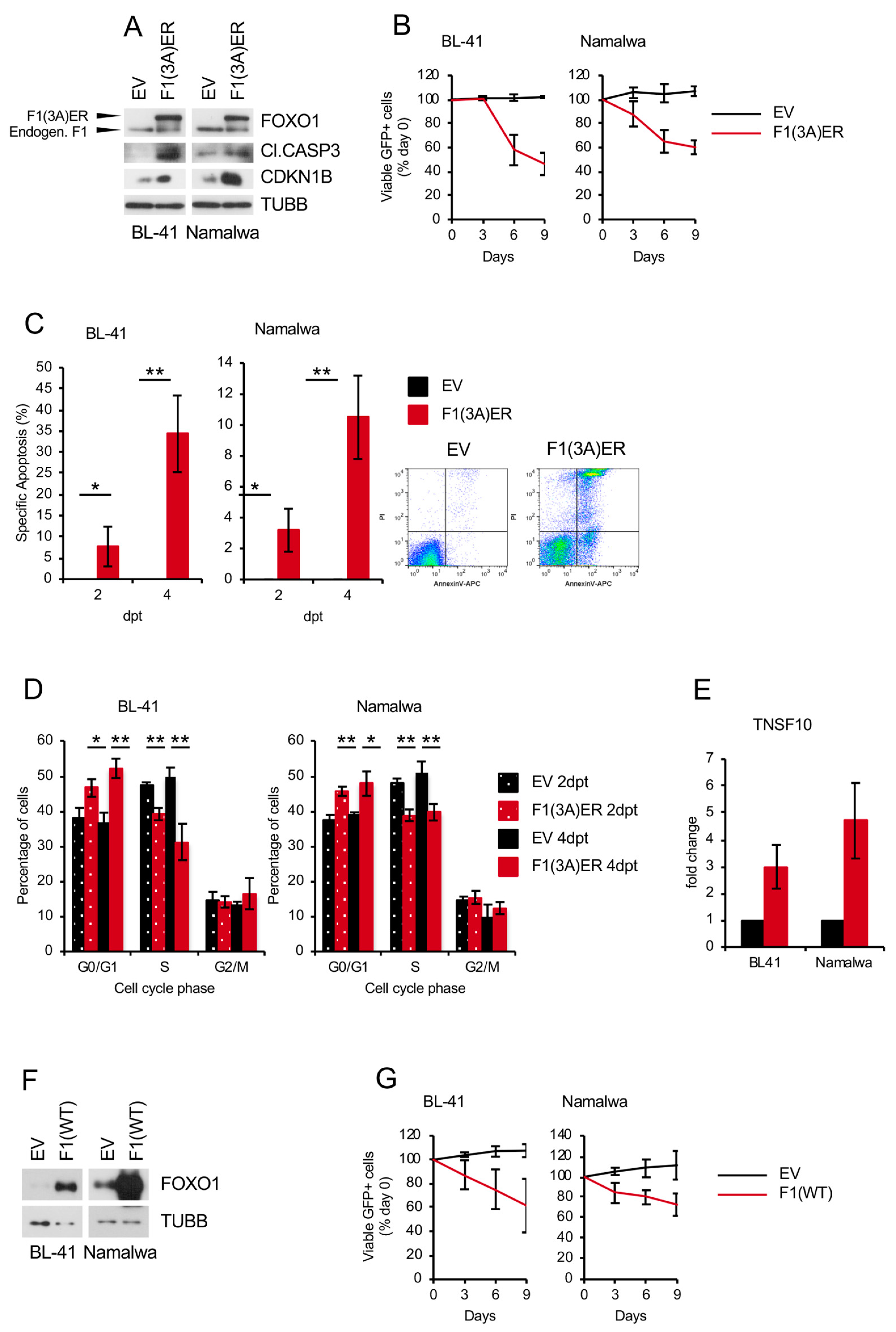
2.5. Genetic FOXO1 Hyperactivation is Toxic for BL
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Vectors and Lentiviral Transduction
4.3. Immunoblot and qRT-PCR
4.4. Flow Cytometry, Cell Sorting, and Cell Viability Analysis
4.5. Gene Expression Profiling (GEP) and Gene Set Enrichment Analysis (GSEA)
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Kretzmer, H.; Bernhart, S.H.; Wang, W.; Haake, A.; Weniger, M.A.; Bergmann, A.K.; Betts, M.J.; Carrillo-de-Santa-Pau, E.; Doose, G.; Gutwein, J.; et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat. Genet. 2015, 47, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Kung, J.; Holmes, A.B.; Wells, V.A.; Mo, T.; Basso, K.; Dalla-Favera, R. The FOXO1 Transcription Factor Instructs the Germinal Center Dark Zone Program. Immunity 2015, 43, 1064–1074. [Google Scholar] [CrossRef]
- Sander, S.; Chu, V.T.; Yasuda, T.; Franklin, A.; Graf, R.; Calado, D.P.; Li, S.; Imami, K.; Selbach, M.; Di Virgilio, M.; et al. PI3 Kinase and FOXO1 Transcription Factor Activity Differentially Control B Cells in the Germinal Center Light and Dark Zones. Immunity 2015, 43, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Victora, G.D.; Ying, C.Y.; Phan, R.T.; Saito, M.; Nussenzweig, M.C.; Dalla-Favera, R. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat. Immunol. 2012, 13, 1083–1091. [Google Scholar] [CrossRef]
- Loeffler-Wirth, H.; Kreuz, M.; Hopp, L.; Arakelyan, A.; Haake, A.; Cogliatti, S.B.; Feller, A.C.; Hansmann, M.L.; Lenze, D.; Moller, P.; et al. A modular transcriptome map of mature B cell lymphomas. Genome Med. 2019, 11, 27. [Google Scholar] [CrossRef]
- Kabrani, E.; Chu, V.T.; Tasouri, E.; Sommermann, T.; Bassler, K.; Ulas, T.; Zenz, T.; Bullinger, L.; Schultze, J.; Rajewsky, K.; et al. Nuclear FOXO1 promotes lymphomagenesis in germinal center B cells. Blood 2018. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.J.; Park, C.; Hong, M.; Ko, Y.H.; Kim, W.S.; Kim, S.J. FOXO4 expression is related to stem cell-like properties and resistance to treatment in diffuse large B cell lymphoma. Oncotarget 2017, 8, 2466–2476. [Google Scholar] [CrossRef] [PubMed]
- Kohrer, S.; Havranek, O.; Seyfried, F.; Hurtz, C.; Coffey, G.P.; Kim, E.; Ten Hacken, E.; Jager, U.; Vanura, K.; O’Brien, S.; et al. Pre-BCR signaling in precursor B cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia 2016, 30, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Szydlowski, M.; Kiliszek, P.; Sewastianik, T.; Jablonska, E.; Bialopiotrowicz, E.; Gorniak, P.; Polak, A.; Markowicz, S.; Nowak, E.; Grygorowicz, M.A.; et al. FOXO1 activation is an effector of SYK and AKT inhibition in tonic BCR signal-dependent diffuse large B cell lymphomas. Blood 2016, 127, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.J.; Xie, L.; Guan, H.; Tooze, R.M.; Maier, T.; Kostezka, U.; Maier, H.J.; Holzmann, K.; Chan, F.C.; Steidl, C.; et al. FOXO1 repression contributes to block of plasma cell differentiation in classical Hodgkin lymphoma. Blood 2014, 124, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Blain, A.E.; Newman, A.M.; Zaka, M.; Chagaluka, G.; Adlar, F.R.; Offor, U.T.; Broadbent, C.; Chaytor, L.; Whitehead, A.; et al. Sporadic and endemic Burkitt lymphoma have frequent FOXO1 mutations but distinct hotspots in the AKT recognition motif. Blood Adv. 2019, 3, 2118–2127. [Google Scholar] [CrossRef]
- Xie, L.; Ushmorov, A.; Leithauser, F.; Guan, H.; Steidl, C.; Farbinger, J.; Pelzer, C.; Vogel, M.J.; Maier, H.J.; Gascoyne, R.D.; et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood 2012, 119, 3503–3511. [Google Scholar] [CrossRef]
- Wang, F.; Demir, S.; Gehringer, F.; Osswald, C.D.; Seyfried, F.; Enzenmuller, S.; Eckhoff, S.M.; Maier, T.; Holzmann, K.; Debatin, K.M.; et al. Tight regulation of FOXO1 is essential for maintenance of B cell precursor acute lymphoblastic leukemia. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Matkar, S.; Sharma, P.; Gao, S.; Gurung, B.; Katona, B.W.; Liao, J.; Muhammad, A.B.; Kong, X.C.; Wang, L.; Jin, G.; et al. An Epigenetic Pathway Regulates Sensitivity of Breast Cancer Cells to HER2 Inhibition via FOXO/c-Myc Axis. Cancer Cell 2015, 28, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Osswald, C.D.; Xie, L.; Guan, H.; Herrmann, F.; Pick, S.M.; Vogel, M.J.; Gehringer, F.; Chan, F.C.; Steidl, C.; Wirth, T.; et al. Fine tuning of FOXO3A in cHL as a survival mechanism and a hallmark of abortive plasma cell differentiation. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.H.; Schlissel, M.S. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat. Immunol. 2008, 9, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Setty, M.; Holmes, A.B.; Maute, R.L.; Leslie, C.S.; Mussolin, L.; Rosolen, A.; Dalla-Favera, R.; Basso, K. MicroRNA 28 controls cell proliferation and is down-regulated in B cell lymphomas. Proc. Natl. Acad. Sci. USA 2014, 111, 8185–8190. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Wu, A.; Verbik, D.; Algarra, S.; Bishop, M.; Pirruccello, S.; Iversen, P.; Jackson, J.; Kessinger, M.; Sharp, J. Oligonucleotides complementary to c-myb messenger RNA inhibit growth and induce apoptosis in human Burkitt lymphoma cells. Int. J. Oncol. 1996, 8, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Rajbhandari, P.; Alvarez, M.J.; Bandaru, P.; Lim, W.K.; Sato, M.; Wang, K.; Sumazin, P.; Kustagi, M.; Bisikirska, B.C.; et al. A human B cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 2010, 6, 377. [Google Scholar] [CrossRef]
- Ricke, R.M.; Jeganathan, K.B.; van Deursen, J.M. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J. Cell Biol. 2011, 193, 1049–1064. [Google Scholar] [CrossRef]
- Zhang, T.; Kim, D.H.; Xiao, X.; Lee, S.; Gong, Z.; Muzumdar, R.; Calabuig-Navarro, V.; Yamauchi, J.; Harashima, H.; Wang, R.; et al. FoxO1 Plays an Important Role in Regulating beta-Cell Compensation for Insulin Resistance in Male Mice. Endocrinology 2016, 157, 1055–1070. [Google Scholar] [CrossRef]
- Webb, A.E.; Kundaje, A.; Brunet, A. Characterization of the direct targets of FOXO transcription factors throughout evolution. Aging Cell 2016, 15, 673–685. [Google Scholar] [CrossRef]
- Eguchi, J.; Wang, X.; Yu, S.; Kershaw, E.E.; Chiu, P.C.; Dushay, J.; Estall, J.L.; Klein, U.; Maratos-Flier, E.; Rosen, E.D. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011, 13, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.E.; Pollina, E.A.; Vierbuchen, T.; Urban, N.; Ucar, D.; Leeman, D.S.; Martynoga, B.; Sewak, M.; Rando, T.A.; Guillemot, F.; et al. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep. 2013, 4, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.P.; Ness, S.A. Alternative RNA splicing produces multiple forms of c-Myb with unique transcriptional activities. Mol. Cell Biol. 2008, 28, 2091–2101. [Google Scholar] [CrossRef] [PubMed]
- Doulatov, S.; Vo, L.T.; Chou, S.S.; Kim, P.G.; Arora, N.; Li, H.; Hadland, B.K.; Bernstein, I.D.; Collins, J.J.; Zon, L.I.; et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell 2013, 13, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Le Gallou, S.; Lamy, T.; Tarte, K.; Fest, T. CXCR4 expression functionally discriminates centroblasts versus centrocytes within human germinal center B cells. J. Immunol. 2009, 182, 7595–7602. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Dominguez-Sola, D.; Holmes, A.B.; Deroubaix, S.; Dalla-Favera, R.; Nussenzweig, M.C. Identification of human germinal center light and dark zone cells and their relationship to human B cell lymphomas. Blood 2012, 120, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef]
- Kennedy, J.E.; Marchese, A. Regulation of GPCR Trafficking by Ubiquitin. Prog. Mol. Biol. Transl. Sci. 2015, 132, 15–38. [Google Scholar] [CrossRef]
- Klapproth, K.; Sander, S.; Marinkovic, D.; Baumann, B.; Wirth, T. The IKK2/NF-κB pathway suppresses MYC-induced lymphomagenesis. Blood 2009, 114, 2448–2458. [Google Scholar] [CrossRef]
- Maier, H.J.; Marienfeld, R.; Wirth, T.; Baumann, B. Critical role of RelB serine 368 for dimerization and p100 stabilization. J. Biol. Chem. 2003, 278, 39242–39250. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Shigematsu, N.; Maruki, R.; Urano, Y.; Tanaka, H.; Shimaya, A.; Shimokawa, T.; Shibasaki, M. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: Improvement of fasting glycemia in diabetic db/db mice. Mol. Pharm. 2010, 78, 961–970. [Google Scholar] [CrossRef]
- Dzikiewicz-Krawczyk, A.; Kok, K.; Slezak-Prochazka, I.; Robertus, J.L.; Bruining, J.; Tayari, M.M.; Rutgers, B.; de Jong, D.; Koerts, J.; Seitz, A.; et al. ZDHHC11 and ZDHHC11B are critical novel components of the oncogenic MYC-miR-150-MYB network in Burkitt lymphoma. Leukemia 2017, 31, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.S.; Fu, K.; Wright, G.W.; Lam, L.T.; Kluin, P.; Boerma, E.J.; Greiner, T.C.; Weisenburger, D.D.; Rosenwald, A.; Ott, G.; et al. Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med. 2006, 354, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.C.; Gomes, A.R.; Constantinidou, D.; Costa, J.R.; Karadedou, C.T.; Fernandez de Mattos, S.; Wymann, M.P.; Brosens, J.J.; Schulze, A.; Lam, E.W. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol. Cell Biol. 2008, 28, 5886–5898. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, E.; Li, W.; Cui, J.; Li, T. MiR-3188 Inhibits Non-small Cell Lung Cancer Cell Proliferation through FOXO1-Mediated mTOR-p-PI3K/AKT-c-JUN Signaling Pathway. Front. Pharm. 2018, 9, 1362. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Xie, L.; Klapproth, K.; Weitzer, C.D.; Wirth, T.; Ushmorov, A. Decitabine represses translocated MYC oncogene in Burkitt lymphoma. J. Pathol. 2013, 229, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hron, J.D.; Peng, S.L. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 2004, 21, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Larson, M.; Vidrine, A.; Barrios, K.; Navarro, F.; Meyers, K.; Simms, P.; Prajapati, K.; Chitsike, L.; Hellman, L.M.; et al. FOXO3-NF-κB RelA Protein Complexes Reduce Proinflammatory Cell Signaling and Function. J. Immunol. 2015, 195, 5637–5647. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef]
- Vasudevan, K.M.; Gurumurthy, S.; Rangnekar, V.M. Suppression of PTEN expression by NF-κ B prevents apoptosis. Mol. Cell Biol. 2004, 24, 1007–1021. [Google Scholar] [CrossRef]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Biophys. Acta 2011, 1813, 1961–1964. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gao, M.; Yang, F.; Zhang, L.; Su, Z. Deciphering the promiscuous interactions between intrinsically disordered transactivation domains and the KIX domain. Proteins 2017, 85, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.M.; Zhou, Y.E.; Pena, J.J.; O’Rourke, J.P.; Ness, S.A. Dramatic repositioning of c-Myb to different promoters during the cell cycle observed by combining cell sorting with chromatin immunoprecipitation. PLoS ONE 2011, 6, e17362. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.M.; Liu, F.; O’Rourke, J.P.; Ness, S.A. Identification and regulation of c-Myb target genes in MCF-7 cells. BMC Cancer 2011, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lei, W.; O’Rourke, J.P.; Ness, S.A. Oncogenic mutations cause dramatic, qualitative changes in the transcriptional activity of c-Myb. Oncogene 2006, 25, 795–805. [Google Scholar] [CrossRef]
- Wang, N.; Zhou, Z.; Wu, T.; Liu, W.; Yin, P.; Pan, C.; Yu, X. TNF-alpha-induced NF-κB activation upregulates microRNA-150-3p and inhibits osteogenesis of mesenchymal stem cells by targeting beta-catenin. Open Biol. 2016, 6, 150258. [Google Scholar] [CrossRef]
- Toth, C.R.; Hostutler, R.F.; Baldwin, A.S., Jr.; Bender, T.P. Members of the nuclear factor κB family transactivate the murine c-myb gene. J. Biol. Chem. 1995, 270, 7661–7671. [Google Scholar] [CrossRef]
- He, Y.; Jiang, X.; Chen, J. The role of miR-150 in normal and malignant hematopoiesis. Oncogene 2013, 33, 3887. [Google Scholar] [CrossRef]
- Musilova, K.; Mraz, M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia 2014, 29, 1004. [Google Scholar] [CrossRef]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Langlet, F.; Haeusler, R.A.; Linden, D.; Ericson, E.; Norris, T.; Johansson, A.; Cook, J.R.; Aizawa, K.; Wang, L.; Buettner, C.; et al. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell 2017, 171, 824–835.e818. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Lane, S.W.; Bullinger, L.; Kalaitzidis, D.; Yusuf, R.; Saez, B.; Ferraro, F.; Mercier, F.; Singh, H.; Brumme, K.M.; et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell 2011, 146, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Ptasinska, A.; Chen, X.; Shrestha, M.; Assi, S.A.; Chin, P.S.; Imperato, M.R.; Aronow, B.J.; Zhang, J.; Weirauch, M.T.; et al. A FOXO1-induced oncogenic network defines the AML1-ETO preleukemic program. Blood 2017, 130, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Dharaneeswaran, H.; Abid, M.R.; Yuan, L.; Dupuis, D.; Beeler, D.; Spokes, K.C.; Janes, L.; Sciuto, T.; Kang, P.M.; Jaminet, S.S.; et al. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ. Res. 2014, 115, 238–251. [Google Scholar] [CrossRef]
- Hornsveld, M.; Smits, L.M.; Meerlo, M.; van Amersfoort, M.; Groot Koerkamp, M.J.; van Leenen, D.; Kloet, D.E.; Holstege, F.C.; Derksen, P.W.B.; Burgering, B.M.T.; et al. FOXO transcription factors both suppress and support breast cancer progression. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mader, A.; Bruderlein, S.; Wegener, S.; Melzner, I.; Popov, S.; Muller-Hermelink, H.K.; Barth, T.F.; Viardot, A.; Moller, P. U-HO1, a new cell line derived from a primary refractory classical Hodgkin lymphoma. Cytogenet. Genome Res. 2007, 119, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Q.; Morita, Y.; Jiang, H.; Gross, A.; Lechel, A.; Hildner, K.; Guachalla, L.M.; Gompf, A.; Hartmann, D.; et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012, 148, 1001–1014. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gehringer, F.; Weissinger, S.E.; Swier, L.J.; Möller, P.; Wirth, T.; Ushmorov, A. FOXO1 Confers Maintenance of the Dark Zone Proliferation and Survival Program and Can Be Pharmacologically Targeted in Burkitt Lymphoma. Cancers 2019, 11, 1427. https://doi.org/10.3390/cancers11101427
Gehringer F, Weissinger SE, Swier LJ, Möller P, Wirth T, Ushmorov A. FOXO1 Confers Maintenance of the Dark Zone Proliferation and Survival Program and Can Be Pharmacologically Targeted in Burkitt Lymphoma. Cancers. 2019; 11(10):1427. https://doi.org/10.3390/cancers11101427
Chicago/Turabian StyleGehringer, Franziska, Stephanie E Weissinger, Lotteke JYM Swier, Peter Möller, Thomas Wirth, and Alexey Ushmorov. 2019. "FOXO1 Confers Maintenance of the Dark Zone Proliferation and Survival Program and Can Be Pharmacologically Targeted in Burkitt Lymphoma" Cancers 11, no. 10: 1427. https://doi.org/10.3390/cancers11101427
APA StyleGehringer, F., Weissinger, S. E., Swier, L. J., Möller, P., Wirth, T., & Ushmorov, A. (2019). FOXO1 Confers Maintenance of the Dark Zone Proliferation and Survival Program and Can Be Pharmacologically Targeted in Burkitt Lymphoma. Cancers, 11(10), 1427. https://doi.org/10.3390/cancers11101427