DNA Methylation of Enhancer Elements in Myeloid Neoplasms: Think Outside the Promoters?


{kind=link}
{kind=link}
Abstract
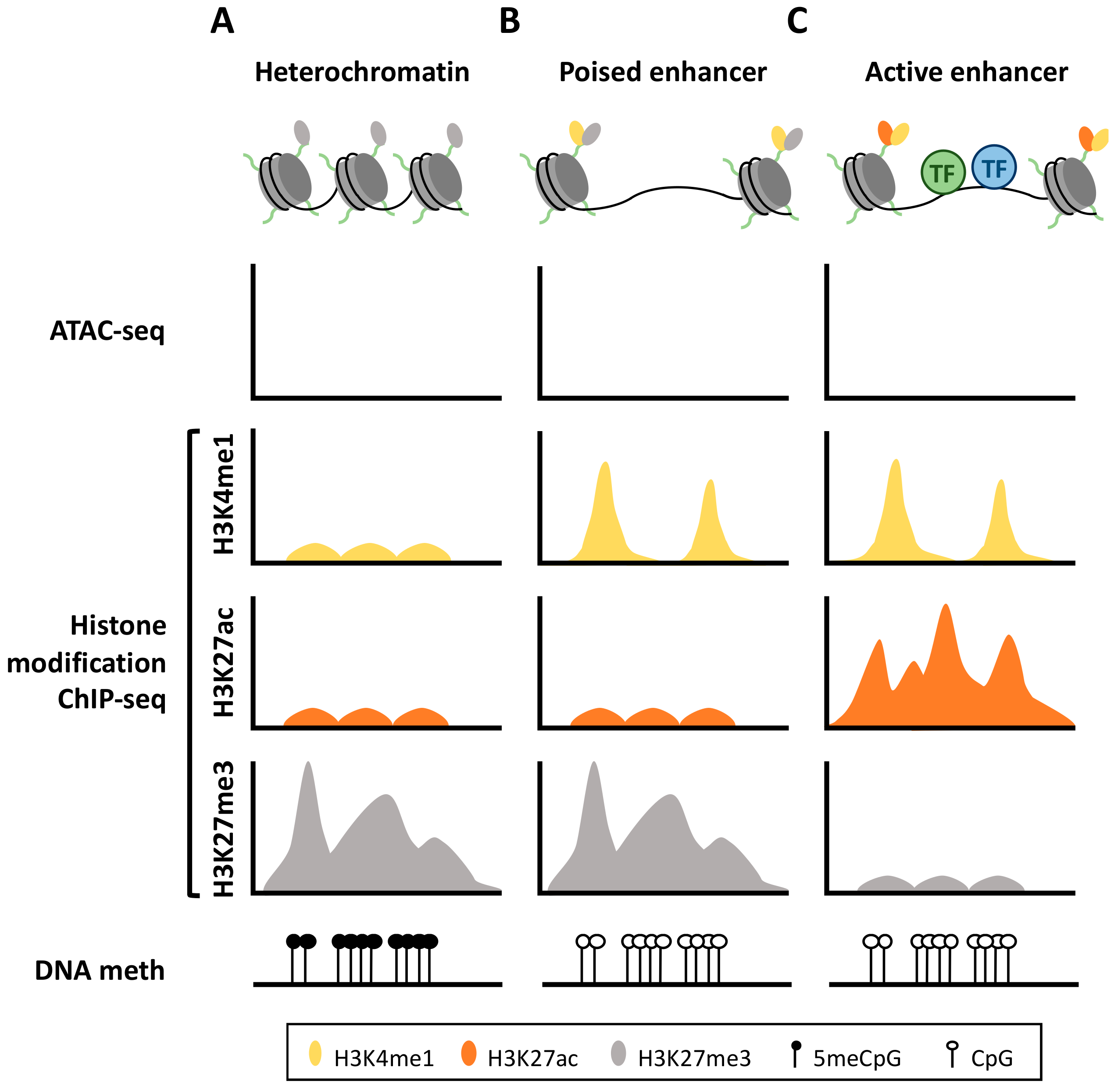
:1. Enhancer Definition
2. Enhancer DNA Methylation
3. Epigenetic Machinery Associated with Enhancer Regulation
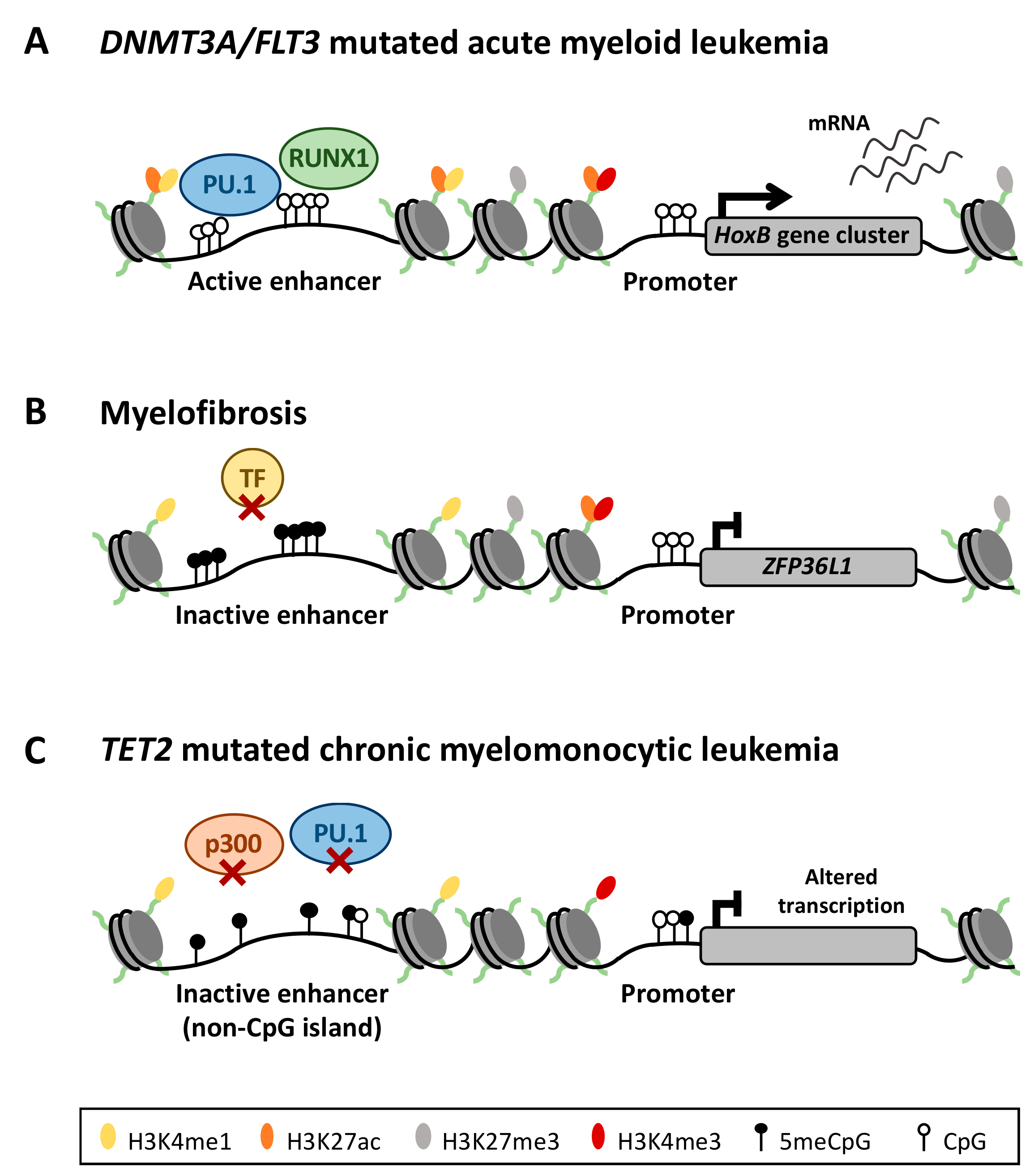
4. Enhancer DNA Methylation in Myeloid Diseases
4.1. Aberrant Enhancer DNA Methylation in Acute Myeloid Leukemia
4.2. Deregulation of the DNA Methylation Signature in Philadelphia Chromosome-Negative Myeloproliferative Neoplasms
4.3. DNA Methylation in TET2 Mutated Chronic Myelomonocytic Leukemia
5. Diagnostic and Therapeutic Implications
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Levine, M. Transcriptional enhancers in animal development and evolution. Curr. Biol. 2010, 20, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, W. Enhancers, enhancers-from their discovery to today’s universe of transcription enhancers. Biol. Chem. 2015, 396, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Coppola, C.J.; Ramaker, R.C.; Mendenhall, E.M. Identification and function of enhancers in the human genome. Hum. Mol. Genet. 2016, 25, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Local, A.; Huang, H.; Albuquerque, C.P.; Singh, N.; Lee, A.Y.; Wang, W.; Wang, C.C.; Hsia, J.E.; Shiau, A.K.; Ge, K.; et al. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat. Genet. 2018, 50, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Marsman, J.; Horsfield, J.A. Long distance relationships: Enhancer–promoter communication and dynamic gene transcription. Biochim. Biophys. Acta Bioenerg. 2012, 1819, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef]
- Gao, T.; He, B.; Liu, S.; Zhu, H.; Tan, K.; Qian, J. EnhancerAtlas: A resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics 2016, 32, 3543–3551. [Google Scholar] [CrossRef]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Jung, S. I mmunogenetics. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef]
- Agirre, X.; Meydan, C.; Jiang, Y. Long non-coding RNAs discriminate the stages and gene regulatory states of human humoral immune response. Nat. Commun. 2019, 10, 821. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, Y.; Zhu, Q.; Wang, J.; Li, G. In silico identification of enhancers on the basis of a combination of transcription factor binding motif occurrences. Sci. Rep. 2016, 6, 32476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Tesar, P.J.; Scacheri, P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Zarchi, A.; Gerovska, D.; Adachi, K. DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism. BMC Genom. 2017, 18, 964. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef]
- Lu, F.; Liu, Y.; Jiang, L.; Yamaguchi, S.; Zhang, Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014, 28, 2103–2119. [Google Scholar] [CrossRef] [Green Version]
- Nair, V.S.; Song, M.H.; Ko, M.; Oh, K.I. DNA Demethylation of the Foxp3 Enhancer Is Maintained through Modulation of Ten-Eleven-Translocation and DNA Methyltransferases. Mol. Cells 2016, 39, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ozark, P.A.; Smith, E.R. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef] [Green Version]
- Sardina, J.L.; Collombet, S.; Tian, T.V. Transcription Factors Drive Tet2-Mediated Enhancer Demethylation to Reprogram Cell Fate. Cell Stem Cell 2018, 23, 905–906. [Google Scholar] [CrossRef] [Green Version]
- Lio, C.J.; Shukla, V.; Samaniego-Castruita, D. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci. Immunol. 2019, 4, eaau7523. [Google Scholar] [CrossRef]
- Agirre, X.; Castellano, G.; Pascual, M. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 2015, 25, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.A.; Wu, X.; Li, A.X.; Hahn, T.; Pfeifer, G.P. Relationship between Gene Body DNA Methylation and Intragenic H3K9me3 and H3K36me3 Chromatin Marks. PLoS ONE 2011, 6, e18844. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Fong, C.Y.; Morison, J.; Dawson, M.A. Epigenetics in the hematologic malignancies. Haematologica 2014, 99, 1772–1783. [Google Scholar] [CrossRef] [Green Version]
- Schmidl, C.; Klug, M.; Boeld, T.J. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009, 19, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.B.; Murr, R.; Burger, L. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Thurman, R.E.; Rynes, E.; Humbert, R. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef]
- Kulis, M.; Queiros, A.C.; Beekman, R.; Martin-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta 2013, 1829, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Wiench, M.; John, S.; Baek, S. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011, 30, 3028–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Serandour, A.A.; Avner, S.; Oger, F. Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res. 2012, 40, 8255–8265. [Google Scholar] [CrossRef] [Green Version]
- Kozlenkov, A.; Roussos, P.; Timashpolsky, A. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014, 42, 109–127. [Google Scholar] [CrossRef]
- Ronnerblad, M.; Andersson, R.; Olofsson, T. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood 2014, 123, e79–e89. [Google Scholar] [CrossRef] [Green Version]
- Akhtar-Zaidi, B.; Cowper-Sal-lari, R.; Corradin, O. Epigenomic enhancer profiling defines a signature of colon cancer. Science 2012, 336, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.E.; Golan, T.; Sheinboim, D. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res. 2016, 26, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Calle, N.; Pascual, M.; Ordonez, R. Epigenomic profiling of myelofibrosis reveals widespread DNA methylation changes in enhancer elements and ZFP36L1 as a potential tumor suppressor gene epigenetically regulated. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Taberlay, P.C.; Statham, A.L.; Kelly, T.K.; Clark, S.J.; Jones, P.A. Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res. 2014, 24, 1421–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordlund, J.; Backlin, C.L.; Wahlberg, P. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.-T.; Corces, V.G. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Wang, C.; Xu, S. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, K.J.; Scelfo, A.; Jammula, S. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.M.; Smith, E.R.; Shilatifard, A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 2013, 33, 4745–4754. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, H.; Wang, Q. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 2018, 19, 18. [Google Scholar] [CrossRef] [Green Version]
- A The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [CrossRef] [PubMed]
- Brown, C.E.; Lechner, T.; Howe, L.; Workman, J.L. The many HATs of transcription coactivators. Trends Biochem. Sci. 2000, 25, 15–19. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [PubMed]
- Tie, F.; Banerjee, R.; Stratton, C.A. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, L.; Wong, E.; Li, G. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, J.; Nankumo, T.; Arita, K.; Inamoto, S.; Ariyoshi, M.; Shirakawa, M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009, 10, 1235–1241. [Google Scholar] [CrossRef]
- Ooi, S.K.; Qiu, C.; Bernstein, E. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jurkowska, R.; Soeroes, S. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef] [Green Version]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. 2015, 165, 36–47. [Google Scholar] [CrossRef]
- Li, H.; Liefke, R.; Jiang, J. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 2017, 549, 287–291. [Google Scholar] [CrossRef]
- King, A.D.; Huang, K.; Rubbi, L. Reversible Regulation of Promoter and Enhancer Histone Landscape by DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep. 2016, 17, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucie, E.L.; Weng, Z.; Geirsdottir, L. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 2016, 351, aad5510. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Benner, C.; Lichtinger, M. Dynamic epigenetic enhancer signatures reveal key transcription factors associated with monocytic differentiation states. Blood 2012, 119, e161–e171. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.L.; Hassane, D.; Wouters, B.J. Epigenetic Identity in AML Depends on Disruption of Nonpromoter Regulatory Elements and Is Affected by Antagonistic Effects of Mutations in Epigenetic Modifiers. Cancer Discov. 2017, 7, 868–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Siggens, L.; Cordeddu, L. Cancer-specific changes in DNA methylation reveal aberrant silencing and activation of enhancers in leukemia. Blood 2017, 129, e13–e25. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, A.S.; Lu, B.; Vakoc, C.R. Enhancer dysfunction in leukemia. Blood 2018, 131, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Shivarov, V.; Gueorguieva, R.; Stoimenov, A.; Tiu, R. DNMT3A mutation is a poor prognosis biomarker in AML: Results of a meta-analysis of 4500 AML patients. Leuk. Res. 2013, 37, 1445–1450. [Google Scholar] [CrossRef]
- Sun, Y.; Shen, H.; Xu, T. Persistent DNMT3A mutation burden in DNMT3A mutated adult cytogenetically normal acute myeloid leukemia patients in long-term remission. Leuk. Res. 2016, 49, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Tie, R.; Zhang, T.; Fu, H. Association between DNMT3A mutations and prognosis of adults with de novo acute myeloid leukemia: A systematic review and meta-analysis. PLoS ONE 2014, 9, e93353. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rodriguez, B.; Mayle, A. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer Cell 2016, 29, 922–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, P.; De Kumar, B.; He, X.C. Retinoid-Sensitive Epigenetic Regulation of the Hoxb Cluster Maintains Normal Hematopoiesis and Inhibits Leukemogenesis. Cell Stem Cell 2018, 22, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Skvarova Kramarzova, K.; Fiser, K.; Mejstrikova, E. Homeobox gene expression in acute myeloid leukemia is linked to typical underlying molecular aberrations. J. Hematol. Oncol. 2014, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Lee, C.; Zichner, T. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Groschel, S.; Sanders, M.A.; Hoogenboezem, R. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Suzuki, M.; Otsuki, A. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3) (q21;q26) by activating EVI1 expression. Cancer Cell 2014, 25, 415–427. [Google Scholar] [CrossRef]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 377, 895–896. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Andersen, C.L.; Westman, M. Epigenetic changes in myelofibrosis: Distinct methylation changes in the myeloid compartments and in cases with ASXL1 mutations. Sci. Rep. 2017, 7, 6774. [Google Scholar] [CrossRef]
- Perez, C.; Pascual, M.; Martin-Subero, J.I. Aberrant DNA methylation profile of chronic and transformed classic Philadelphia-negative myeloproliferative neoplasms. Haematologica 2013, 98, 1414–1420. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2018 update on diagnosis, risk stratification and management. Am. J. Hematol. 2018, 93, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Martinez-Calle, N.; Martin-Subero, J.I. TET2 mutations are associated with specific 5-methylcytosine and 5-hydroxymethylcytosine profiles in patients with chronic myelomonocytic leukemia. PLoS ONE 2012, 7, e31605. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Lugthart, S.; Li, Y. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, X.; Cassady, K.; Zou, Z.; Zhang, X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol. 2019, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo, L.; Malinverni, R.; Cabezon, M. DNA methylation profile in chronic myelomonocytic leukemia associates with distinct clinical, biological and genetic features. Epigenetics 2018, 13, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, J.; Jelinek, J.; Lu, Y. TET2 Mutations Affect Non-CpG Island DNA Methylation at Enhancers and Transcription Factor-Binding Sites in Chronic Myelomonocytic Leukemia. Cancer Res. 2015, 75, 2833–2843. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, J.; Taby, R.; Vasanthakumar, A. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 2012, 7, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, X.; Cai, C.L. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Kihslinger, J.E.; Godley, L.A. The use of hypomethylating agents in the treatment of hematologic malignancies. Leuk. Lymphoma 2007, 48, 1676–1695. [Google Scholar] [CrossRef]
- Yun, S.; Vincelette, N.D.; Abraham, I.; Robertson, K.D.; Fernandez-Zapico, M.E.; Patnaik, M.M. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: A systematic review of hypomethylating agents trials. Clin. Epigenet. 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Yang, W.; Gegonne, A. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J. 2019, 38, e100293. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Coude, M.M.; Braun, T.; Berrou, J. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Huang, B.; Wu, X. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33, 2395–2404. [Google Scholar] [CrossRef]
- Chen, H.; Kazemier, H.G.; de Groote, M.L.; Ruiters, M.H.; Xu, G.L.; Rots, M.G. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014, 42, 1563–1574. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; Stolzenburg, S.; Beltran, A.S. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012, 7, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Grimmer, M.R.; Farnham, P.J. Can genome engineering be used to target cancer-associated enhancers? Epigenomics 2014, 6, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.; Itzykson, R.; Renneville, A. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: A phase 2 trial. Blood 2011, 118, 3824–3831. [Google Scholar] [CrossRef]
- Meldi, K.; Qin, T.; Buchi, F. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J. Clin. Investig. 2015, 125, 1857–1872. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Alvarez, S.; Suela, J.; Valencia, A. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PLoS ONE 2010, 5, e12197. [Google Scholar] [CrossRef]
- Shirahata, A.; Hibi, K. Serum vimentin methylation as a potential marker for colorectal cancer. Anticancer Res. 2014, 34, 4121–4125. [Google Scholar]
- Hollink, I.H.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T. Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica 2011, 96, 384–392. [Google Scholar] [CrossRef]
- Lin, T.C.; Jiang, S.S.; Chou, W.C. Rapid assessment of the heterogeneous methylation status of CEBPA in patients with acute myeloid leukemia by using high-resolution melting profile. J. Mol. Diagn. 2011, 13, 514–519. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ordoñez, R.; Martínez-Calle, N.; Agirre, X.; Prosper, F. DNA Methylation of Enhancer Elements in Myeloid Neoplasms: Think Outside the Promoters? Cancers 2019, 11, 1424. https://doi.org/10.3390/cancers11101424
Ordoñez R, Martínez-Calle N, Agirre X, Prosper F. DNA Methylation of Enhancer Elements in Myeloid Neoplasms: Think Outside the Promoters? Cancers. 2019; 11(10):1424. https://doi.org/10.3390/cancers11101424
Chicago/Turabian StyleOrdoñez, Raquel, Nicolás Martínez-Calle, Xabier Agirre, and Felipe Prosper. 2019. "DNA Methylation of Enhancer Elements in Myeloid Neoplasms: Think Outside the Promoters?" Cancers 11, no. 10: 1424. https://doi.org/10.3390/cancers11101424
APA StyleOrdoñez, R., Martínez-Calle, N., Agirre, X., & Prosper, F. (2019). DNA Methylation of Enhancer Elements in Myeloid Neoplasms: Think Outside the Promoters? Cancers, 11(10), 1424. https://doi.org/10.3390/cancers11101424