Endothelial Protein C Receptor (EPCR), Protease Activated Receptor-1 (PAR-1) and Their Interplay in Cancer Growth and Metastatic Dissemination

 , ,
, ,
Abstract
1. Introduction
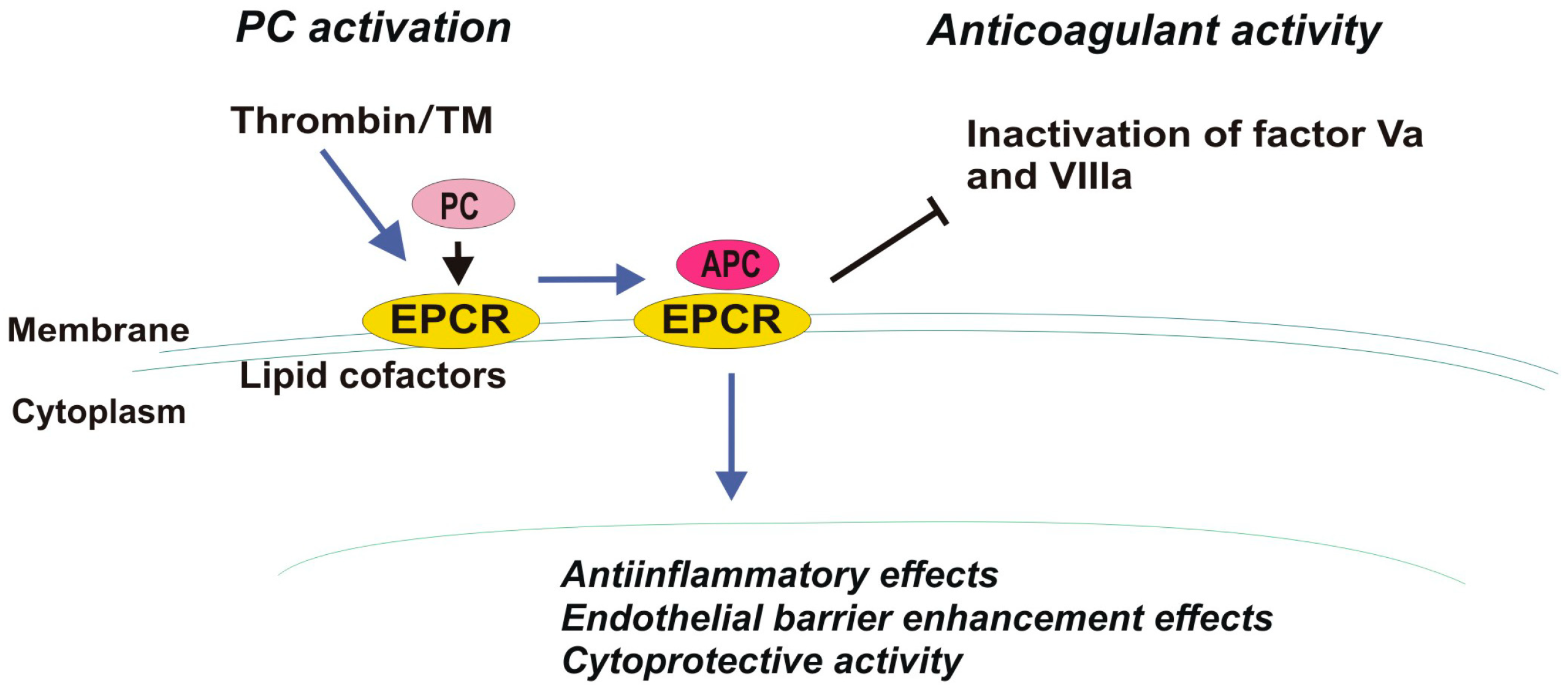
2. EPCR and Cancer
3. PAR-1 and Cancer
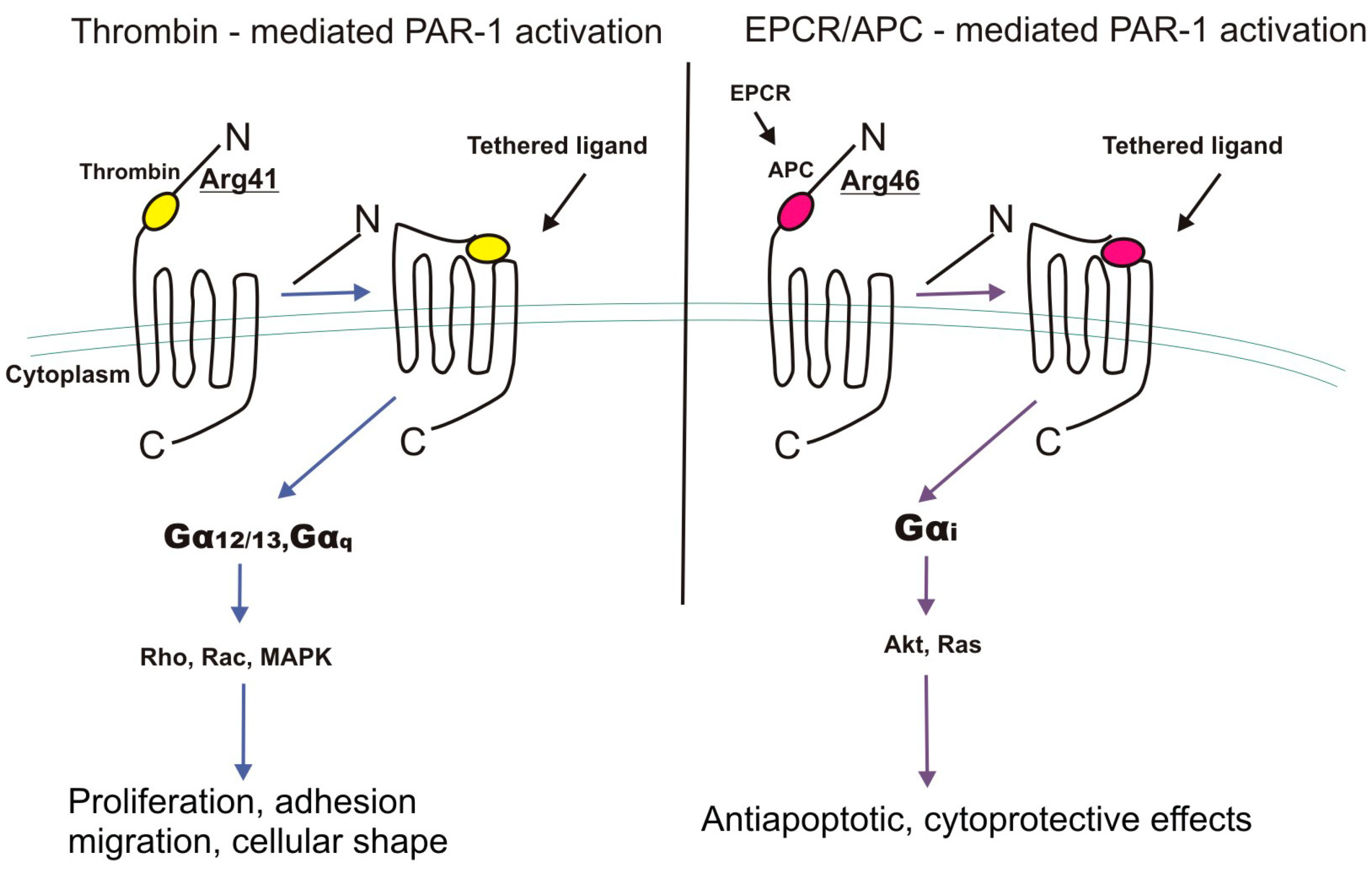
4. EPCR and PAR-1 Interactions
4.1. G-Proteins
4.2. B-Arrestin
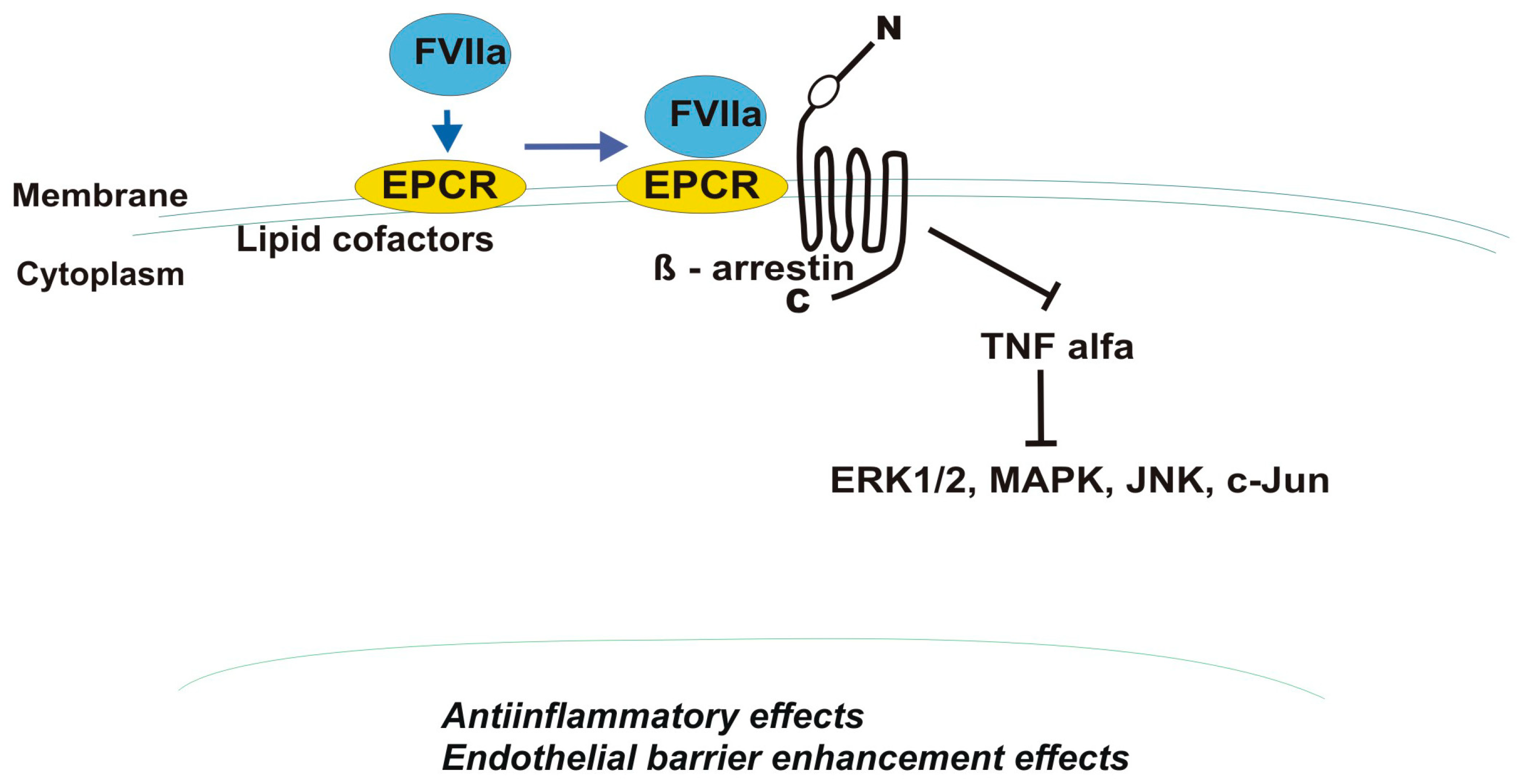
4.3. Factor VII/VIIa and Factor Xa (FXa)
4.4. Tissue Factor
4.5. Thrombomodulin, TM
4.6. Hematopoietic Stem Cells
4.7. Microbiome and EPCR/PARs Interactions
4.8. EPC/PAR-1 and Neurons
5. Treatment
6. Conclusions
Funding
Conflicts of Interest
References
- Pang, L.; Li, J.F.; Su, L.; Zang, M.; Fan, Z.; Yu, B.; Wu, X.; Li, C.; Yan, M.; Zhu, Z.G.; et al. ALEX1, a novel tumor suppressor gene, inhibits gastric cancer metastasis via the PAR-1/Rho GTPase signaling pathway. J. Gastroenterol. 2018, 53, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Pendurthi, U.R.; Rao, L.V.M. Endothelial cell protein C receptor-dependent signaling. Curr. Opin. Hematol. 2018, 25, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Mohan Rao, L.V.; Esmon, C.T.; Pendurthi, U.R. Endothelial cell protein C receptor: A multiliganded and multifunctional receptor. Blood 2014, 124, 1553–1562. [Google Scholar] [CrossRef]
- Gramling, M.W.; Beaulieu, L.M.; Church, F.C. Activated protein C enhances cell motility of endothelial cells and MDA-MB-231 breast cancer cells by intracellular signal transduction. Exp. Cell Res. 2010, 316, 314–328. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wojtukiewicz, M.Z.; Tang, D.G.; Nelson, K.K.; Walz, D.A.; Diglio, C.A.; Honn, K.V. Thrombin enhances tumor cell adhesive and metastatic properties via increased alpha IIb beta 3 expression on the cell surface. Thromb. Res. 1992, 68, 233–245. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Tang, D.G.; Ciarelli, J.J.; Nelson, K.K.; Walz, D.A.; Diglio, C.A.; Mammen, E.F.; Honn, K.V. Thrombin increases the metastatic potential of tumor cells. Int. J. Cancer 1993, 54, 793–806. [Google Scholar] [CrossRef]
- Gur-Cohen, S.; Itkin, T.; Chakrabarty, S.; Graf, C.; Kollet, O.; Ludin, A.; Golan, K.; Kalinkovich, A.; Ledergor, G.; Wong, E.; et al. PAR1 signaling regulates the retention and recruitment of EPCR-expressing bone marrow hematopoietic stem cells. Nat. Med. 2015, 21, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Gur-Cohen, S.; Kollet, O.; Graf, C.; Esmon, C.T.; Ruf, W.; Lapidot, T. Regulation of long-term repopulating hematopoietic stem cells by EPCR/PAR1 signaling. Ann. N. Y. Acad. Sci. 2016, 1370, 65–81. [Google Scholar] [CrossRef]
- Schuepbach, R.A.; Feistritzer, C.; Brass, L.F.; Riewald, M. Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood 2008, 111, 2667–2673. [Google Scholar] [CrossRef]
- Keshava, S.; Sahoo, S.; Tucker, T.A.; Idell, S.; Rao, L.V.; Pendurthi, U.R. Endothelial cell protein C receptor opposes mesothelioma growth driven by tissue factor. Cancer Res. 2013, 73, 3963–3973. [Google Scholar] [CrossRef]
- Bezuhly, M.; Cullen, R.; Esmon, C.T.; Morris, S.F.; West, K.A.; Johnston, B.; Liwski, R.S. Role of activated protein C and its receptor in inhibition of tumor metastasis. Blood 2009, 113, 3371–3374. [Google Scholar] [CrossRef]
- Smorenburg, S.M.; Vink, R.; Otten, H.M.; Swaneveld, F.; Büller, H.R. The effects of vitamin K-antagonists on survival of patients with malignancy: A systematic analysis. Thromb. Haemost. 2001, 86, 1586–1587. [Google Scholar] [PubMed]
- Suzuki, K.; Hayashi, T. Protein C and its inhibitor in malignancy. Semin. Thromb. Hemost. 2007, 33, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Kondreddy, V.; Wang, J.; Keshava, S.; Esmon, C.T.; Rao, L.V.M.; Pendurthi, U.R. Factor VIIa induces anti-inflammatory signaling via EPCR and PAR1. Blood 2018, 131, 2379–2392. [Google Scholar] [CrossRef]
- Soh, U.J.; Trejo, J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. USA 2011, 108, E1372–E1380. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)-biology and role in cancer invasion and metastasis. Cancer Metast. Rev. 2015, 4, 775–796. [Google Scholar] [CrossRef] [PubMed]
- Van de Wouwer, M.; Collen, D.; Conway, E.M. Thrombomodulin-protein C-EPCR system: Integrated to regulate coagulation and inflammation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1374–1383. [Google Scholar] [CrossRef]
- Montes, R.; Hurtado, V.; Alonso, A.; Foco, L.; Zonzin, P.; Mannucci, P.M.; Hermida, J. Autoantibodies against the endothelial receptor of protein C are associated with acute myocardial infarction in young women. J. Thromb. Haemost. 2005, 3, 1454–1458. [Google Scholar] [CrossRef]
- Esmon, C.T. The endothelial cell protein C receptor. Thromb. Haemost. 2000, 83, 639–643. [Google Scholar] [CrossRef]
- Joyce, D.E.; Gelbert, L.; Ciaccia, A.; DeHoff, B.; Grinnell, B.W. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J. Biol. Chem. 2001, 276, 11199–11203. [Google Scholar] [CrossRef]
- Kurosawa, S.; Stearns-Kurosawa, D.J.; Carson, C.W.; D’Angelo, A.; Della Valle, P.; Esmon, C.T. Plasma levels of endothelial cell protein C receptor are elevated in patients with sepsis and systemic lupus erythematosus: Lack of correlation with thrombomodulin suggests involvement of different pathological processes. Blood 1998, 91, 725–727. [Google Scholar] [PubMed]
- Vassiliou, A.G.; Kotanidou, A.; Mastora, Z.; Tascini, C.; Cardinali, G.; Orfanos, S.E. The H3 Haplotype of the EPCR Gene Determines High sEPCR Levels in Critically Ill Septic Patients. Infect. Dis. Ther. 2018, 7 (Suppl. 1), 3–14. [Google Scholar] [CrossRef] [PubMed]
- Lavigne-Lissalde, G.; Cochery-Nouvellon, E.; Mercier, E.; Marès, P.; Gris, J.C. High plasma levels of endothelial protein C receptor are associated with the risk of unexplained fetal death. J. Thromb. Haemost. 2005, 3, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Ireland, H.; Konstantoulas, C.K.; Cooper, J.A.; Hawe, E.; Humphries, S.E.; Mather, H.; Goodall, A.H.; Hogwood, J.; Juhan-Vague, I.; Yudkin, J.S. EPCR Ser219Gly: Elevated sEPCR, prothrombin F1+2, risk for coronary heart disease, and increased sEPCR shedding in vitro. Arteriosclerosis 2005, 183, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Uitte, D.W.; Van, M.V.; Rosendaal, F.R.; Vos, H.L.; de Visser, M.C.; Bertina, R.M. Haplotypes of the EPCR gene, plasma sEPCR levels and the risk of deep venous thrombosis. J. Thromb. Haemost. 2004, 2, 1305–1310. [Google Scholar] [CrossRef]
- Medina, P.; Navarro, S.; Bonet, E.; Martos, L.; Estellés, A.; Bertina, R.M.; Vos, H.L.; España, F. Functional analysis of two haplotypes of the human endothelial protein C receptor gene. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 684–690. [Google Scholar] [CrossRef]
- Tinholt, M.; Viken, M.K.; Dahm, A.E.; Vollan, H.K.; Sahlberg, K.K.; Garred, O.; Børresen-Dale, A.L.; Jacobsen, A.F.; Kristensen, V.; Bukholm, I.; et al. Increased coagulation activity and genetic polymorphisms in the F5, F10 and EPCR genes are associated with breast cancer: A case-control study. BMC Cancer 2014, 14, 845. [Google Scholar] [CrossRef]
- Xu, J.; Liaw, P.C.Y.; Esmon, C.T. A novel transmembrane domain of the EPCR dictates receptor localization of sphingolipid-cholesterol rich regions on plasma membrane which EPCR palmitoylation modulates intracellular trafficking patterns. Thromb. Haemost. 1999, 82 (Suppl. 1), 2195a. [Google Scholar]
- Oganesyan, V.; Oganesyan, N.; Terzyan, S.; Qu, D.; Dauter, Z.; Esmon, N.L.; Esmon, C.T. The crystal structure of the endothelial protein C receptor and a bound phospholipid. J. Biol. Chem. 2002, 277, 24851–24854.56. [Google Scholar] [CrossRef]
- Stearns-kurosawa, D.J.; Kurosawa, S.; Mollica, J.S.; Ferrellt, G.I.; Esmon, C.T. Endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc. Natl. Acad. Sci. USA 1996, 93, 10212–10216. [Google Scholar] [CrossRef]
- Nayak, R.C.; Sen, P.; Ghosh, S.; Gopalakrishnan, R.; Esmon, C.T.; Pendurthi, U.R.; Rao, L.V. Endothelial cell protein C receptor cellular localization and trafficking: Potential functional implications. Blood 2009, 114, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Montes, R.; Puy, C.; Molina, E.; Hermida, J. Is EPCR a multi-ligand receptor? Pros and cons. Thromb. Haemost. 2012, 107, 815–826. [Google Scholar] [PubMed]
- Disse, J.; Petersen, H.H.; Larsen, K.S.; Persson, E.; Esmon, N.; Esmon, C.T.; Teyton, L.; Petersen, L.C.; Ruf, W. The endothelial protein C receptor supports tissue factor ternary coagulation initiation complex signaling through protease-activated receptors. J. Biol. Chem. 2011, 286, 5756–5767. [Google Scholar] [CrossRef] [PubMed]
- Van Hylckama Vlieg, A.; Montes, R.; Rosendaal, F.R.; Hermida, J. Autoantibodies against endothelial protein C receptor and the risk of a first deep vein thrombosis. J. Thromb. Haemost. 2007, 5, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Neyrinck, A.P.; Liu, K.D.; Howard, J.P.; Matthay, M.A. Protective mechanisms of activated protein C in severe inflammatory disorders. Br. J. Pharmacol. 2009, 158, 1034–1047. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2007, 109, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. Cytoprotective-selective activated protein C therapy for ischaemic stroke. Thromb. Haemost. 2014, 112, 883–892. [Google Scholar] [CrossRef]
- Griffin, J.H.; Mosnier, L.O.; Fernández, J.A.; Zlokovic, B.V. Scientific Sessions Sol Sherry Distinguished Lecturer in Thrombosis: Thrombotic Stroke: Neuroprotective Therapy by Recombinant-Activated Protein C. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2143–2151. [Google Scholar] [CrossRef]
- Sinha, R.K.; Wang, Y.; Zhao, Z.; Xu, X.; Burnier, L.; Gupta, N.; Fernández, J.A.; Martin, G.; Kupriyanov, S.; Mosnier, L.O.; et al. PAR1 biased signaling is required for activated protein C in vivo benefits in sepsis and stroke. Blood 2018, 131, 1163–1171. [Google Scholar] [CrossRef]
- Feistritzer, C.; Schuepbach, R.A.; Mosnier, L.O.; Bush, L.A.; Di Cera, E.; Griffin, J.H.; Riewald, M. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J. Biol. Chem. 2006, 281, 20077–20084. [Google Scholar] [CrossRef]
- Lal, N.; Willcox, C.R.; Beggs, A.; Taniere, P.; Shikotra, A.; Bradding, P.; Adams, R.; Fisher, D.; Middleton, G.; Tselepis, C.; et al. Endothelial protein C receptor is overexpressed in colorectal cancer as a result of amplification and hypomethylation of chromosome 20q. J. Pathol. Clin. Res. 2017, 14, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Q.; Wang, T.; Yang, H.; Han, Z.; Zhang, P. Endothelial cell protein C receptor promotes MGC803 gastric cancer cells proliferation and migration by activating ERK1/2. Med. Oncol. 2015, 32, 162. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, F.; Yokota, N.; Carneiro-Lobo, T.; Kitano, M.; Schaffer, M.; Anderson, G.M.; Mueller, B.M.; Esmon, C.T.; Ruf, W. Endothelial protein C receptor function in murine and human breast cancer development. PLoS ONE 2013, 8, e61071. [Google Scholar] [CrossRef] [PubMed]
- Keshava, S.; Rao, L.V.; Pendurthi, U.R. Intrapleural Adenoviral-mediated Endothelial Cell Protein C Receptor Gene Transfer Suppresses the Progression of Malignant Pleural Mesothelioma in a Mouse Model. Sci. Rep. 2016, 11, 36829. [Google Scholar] [CrossRef] [PubMed]
- Perurena, N.; Zandueta, C.; Martínez-Canarias, S.; Moreno, H.; Vicent, S.; Almeida, A.S.; Guruceaga, E.; Gomis, R.R.; Santisteban, M.; Egeblad, M. EPCR promotes breast cancer progression by altering SPOCK1/testican 1-mediated 3D growth. J. Hematol. Oncol. 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Besbes, S.; Attal, R.; Mirshahi, S.; Chidiac, J.; Mahé, I.; Pocard, M.; Soria, J.; Mirshahi, M. PO-47—Microparticles derived from ovarian cancer cell line contained genomic and biologically active proteins, including tissue factor involved in coagulation. Thromb. Res. 2016, 140 (Suppl. 1), S194. [Google Scholar] [CrossRef]
- Martin, F.; Long, J.C.; O’Toole, S.A.; O’Leary, J.J.; Abu Saadeh, F.; Gleeson, N.; Norris, L.A. PO-14—Tumour expression of coagulation proteases of the aPC pathway—A role in the pathogenesis of gynaecological cancers? Thromb. Res. 2016, 140 (Suppl. 1), S181. [Google Scholar] [CrossRef]
- De Oliveira, A.S.; de Almeida, V.H.; Gomes, F.G.; Rezaie, A.R.; Monteiro, R.Q. TR47, a PAR1-based peptide, inhibits melanoma cell migration in vitro and metastasis in vivo. Biochem. Biophys. Res. Commun. 2018, 495, 1300–1304. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Church, F.C. Activated protein C promotes breast cancer cell migration through interactions with EPCR and PAR-1. Exp. Cell Res. 2007, 313, 677–687. [Google Scholar] [CrossRef]
- Antón, I.; Molina, E.; Luis-Ravelo, D.; Zandueta, C.; Valencia, K.; Ormazabal, C.; Martínez-Canarias, S.; Perurena, N.; Pajares, M.J.; Agorreta, J. Receptor of activated protein C promotes metastasis and correlates with clinical outcome in lung adenocarcinoma. Am. J. Respir. Crit. Care Med. 2012, 186, 96–105. [Google Scholar] [CrossRef]
- Yan, Q.; Zhong, X.; Zhang, Z.; Bing, W.; Feng, Y.; Hong, B. Prevalence of protein C receptor (PROCR) is associated with inferior clinical outcome in Breast invasive ductal carcinoma. Pathol. Res. Pract. 2017, 213, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Keshava, S.; Kothari, H.; Rao, L.V.; Pendurthi, U.R. Influence of endothelial cell protein C receptor on breast cancer development. J. Thromb. Haemost. 2013, 11, 2062–2065. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Schaffner, F. Role of the protein C receptor in cancer progression. Thromb. Res. 2014, 133 (Suppl. 2), S85–S89. [Google Scholar] [CrossRef]
- Uchiba, M.; Okajima, K.; Oike, Y.; Ito, Y.; Fukudome, K.; Isobe, H.; Suda, T. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ. Res. 2004, 95, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Woodley-Cook, J.; Shin, L.Y.Y.; Swystun, L.; Caruso, S.; Beaudin, S.; Liaw, P.C. Effects of the chemotherapeutic agent doxorubicin on the protein C anticoagulant pathway. Mol. Cancer Ther. 2006, 5, 3303–3311. [Google Scholar] [CrossRef]
- Farah, R.A.; Jalkh, K.S.; Farhat, H.Z.; Sayad, P.E.; Kadri, A.M. Acquired protein C deficiency in a child with acute myelogenous leukemia, splenic, renal, and intestinal infarction. Blood Coagul. Fibrinolysis 2011, 22, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiao, J.; Zhang, H.; Zhou, W.; Li, Z.; Han, S.; Wang, J.; Yang, X.; Huang, Q.; Wu, Z. TGF-β induced PAR-1 expression promotes tumor progression and osteoclast differentiation in giant cell tumor of bone. Int. J. Cancer 2017, 141, 1630–1642. [Google Scholar] [CrossRef]
- Al Saleh, H.A.; Haas-Neill, S.; Al-Hashimi, A.; Kapoor, A.; Shayegan, B.; Austin, R.C.; Al-Nedawi, K. Thrombotic characteristics of extracellular vesicles derived from prostate cancer cells. Prostate 2018, 78, 953–961. [Google Scholar] [CrossRef]
- Che, S.P.Y.; Park, J.Y.; Stokol, T. Tissue Factor-Expressing Tumor-Derived Extracellular Vesicles Activate Quiescent Endothelial Cells via Protease-Activated Receptor-1. Front. Oncol. 2017, 7, 261. [Google Scholar] [CrossRef]
- Kreienbring, K.; Franz, A.; Richter, R.; Dragun, D.; Heidecke, H.; Müller, D.; Mentze, M.; Dechend, R.; Sehouli, J.; Braicu, E.I. The Role of PAR1 Autoantibodies in Patients with Primary Epithelial Ovarian Cancer. Anticancer Res. 2018, 38, 3619–3625. [Google Scholar] [CrossRef]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef] [PubMed]
- Riewald, M.; Petrovan, R.J.; Donner, A.; Mueller, B.M.; Ruf, W. Activation of endothelial cell protease activated receptor-1 by the protein C pathway. Science 2002, 296, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Van den Eshof, B.L.; Hoogendijk, A.J.; Simpson, P.J.; van Alphen, F.P.J.; Zanivan, S.; Mertens, K.; Meijer, A.B.; van den Biggelaar, M. Paradigm of Biased PAR1 (Protease-Activated Receptor-1) Activation and Inhibition in Endothelial Cells Dissected by Phosphoproteomics. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Tekin, C.; Shi, K.; Daalhuisen, J.B.; Ten Brink, M.S.; Bijlsma, M.F.; Spek, C.A. PAR1 signaling on tumor cells limits tumor growth by maintaining a mesenchymal phenotype in pancreatic cancer. Oncotarget 2018, 9, 32010–32023. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.R. The occupancy of endothelial protein C receptor by its ligand modulates the par-1 dependent signaling specificity of coagulation proteases. IUBMB Life 2011, 63, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Ludeman, M.J.; Kataoka, H.; Srivastava, Y.; Esmon, N.L.; Esmon, C.T.; Coughlin, S.R. PAR1 cleavage and signaling in response to activated protein C and thrombin. J. Biol. Chem. 2005, 280, 13122–13128. [Google Scholar] [CrossRef] [PubMed]
- Riewald, M.; Ruf, W. Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J. Biol. Chem. 2005, 280, 19808–19814. [Google Scholar] [CrossRef]
- Niessen, F.; Furlan-Freguia, C.; Fernández, J.A.; Mosnier, L.O.; Castellino, F.J.; Weiler, H.; Rosen, H.; Griffin, J.H.; Ruf, W. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood 2009, 113, 2859–2866. [Google Scholar] [CrossRef]
- Feistritzer, C.; Riewald, M. Endothelial barrier protection by activated protein C through PAR1-dependent sphinosine 1-phospate receptor-1 crossactivation. Blood 2005, 105, 3178–3184. [Google Scholar] [CrossRef]
- Finigan, J.H.; Dudek, S.M.; Singleton, P.A.; Chiang, E.T.; Jacobson, J.R.; Camp, S.M.; Ye, S.Q.; Garcia, J.G. Activated protein C mediates novel lung endothelial barrier enhancement: Role of sphingosine 1-phosphate receptor transactivation. J. Biol. Chem. 2005, 280, 17286–17293. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Campbell, D.; Sambrook, P.N.; Fukudome, K.; Jackson, C.J. Endothelial protein C receptor and protease-activated receptor-1 mediate induction of a wound-healing phenotype in human keratinocytes by activated protein C. J. Investig. Dermatol. 2005, 125, 1279–1285. [Google Scholar] [CrossRef]
- Gleeson, E.M.; McDonnell, C.J.; Soule, E.E.; Willis Fox, O.; Rushe, H.; Rehill, A.; Smith, O.P.; O’Donnell, J.S.; Preston, R.J.S. A novel protein C-factor VII chimera provides new insights into the structural requirements for cytoprotective protease-activated receptor 1 signaling. J. Thromb. Haemost. 2017, 15, 2198–2207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Ceunynck, K.; Peters, C.G.; Jain, A.; Higgins, S.J.; Aisiku, O.; Fitch-Tewfik, J.L.; Chaudhry, S.A.; Dockendorff, C.; Parikh, S.M.; Ingber, D.E.; et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc. Natl. Acad. Sci. USA 2018, 115, E982–E991. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, H.; Zhuo, Q.; Xu, Y.; Zhang, P. Knockdown of EPCR inhibits the proliferation and migration of human gastric cancer cells via the ERK1/2 pathway in a PAR-1-dependent manner. Oncol. Rep. 2018, 39, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, Y.; Wang, T.; Yang, H.L.; Wang, X.; Ma, H.; Zhang, P. EPCR promotes MGC803 human gastric cancer cell tumor angiogenesis in vitro through activating ERK1/2 and AKT in a PAR1-dependent manner. Oncol. Lett. 2018, 16, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.V.; Ardeshirylajimi, A.; Dinarvand, P.; Yang, L.; Rezaie, A.R. Occupancy of human EPCR by protein C induces β-arrestin-2 biased PAR1 signaling by both APC and thrombin. Blood 2016, 128, 1884–1893. [Google Scholar] [CrossRef]
- Schuepbach, R.A.; Riewald, M. Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endothelial protein C receptor. J. Thromb. Haemost. 2010, 8, 379–388. [Google Scholar] [CrossRef]
- Sundaram, J.; Keshava, S.; Gopalakrishan, R.; Esmon, C.T.; Pendurthi, U.R.; Rao, L.V. Factor VIIa binding to endothelial cell protein C receptor protects vascular barrier integrity in vivo. J. Thromb. Haemost. 2014, 12, 690–700. [Google Scholar] [CrossRef]
- Sen, P.; Gopalakrishnan, R.; Kothari, H.; Keshava, S.; Clark, C.A.; Esmon, C.T.; Pendurthi, U.R.; Rao, L.V. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood 2011, 117, 3199–3208. [Google Scholar] [CrossRef]
- Fager, A.M.; Machlus, K.R.; Ezban, M.; Hoffman, M. Human platelets express endothelial protein C receptor, which can be utilized to enhance localization of factor VIIa activity. J. Thromb. Haemost. 2018, 16, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Antiplatelet agents for cancer treatment: A real perspective or just an echo from the past? Cancer Metast. Rev. 2017, 36, 305–329. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhang, D.; Wu, S.; Yu, J.; Yu, L.; Sun, Y.; Du, Z.; Li, Z.; Zhou, L.; Wu, X.; et al. FVIIa prevents the progressive hemorrhaging of a brain contusion by protecting microvessels via formation of the TF–FVIIa–FXa complex. Neuroscience 2017, 348, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Reto, A.; Schuepbach, K.; Velez, M. Activated protein C up-regulates procoagulant tissue factor activity on endothelial cells by shedding the TFPI Kunitz 1 domain. Blood 2011, 117, 6338–6346. [Google Scholar] [CrossRef]
- Chang, Y.J.; Cheng, Y.W.; Lin, R.K.; Huang, C.C.; Chen, W.T.; Ke, T.W.; Wei, P.L. Thrombomodulin Influences the Survival of Patients with Non-Metastatic Colorectal Cancer through Epithelial-To-Mesenchymal Transition (EMT). PLoS ONE 2016, 11, e0160550. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Huo, Z.; Zhang, B.; Meng, M.; Cao, Z.; Wang, Z.; Zhou, Q. Thrombomodulin reduces tumorigenic and metastatic potential of lung cancer cells by up-regulation of E-cadherin and down-regulation of N-cadherin expression. Biochem. Biophys. Res. Commun. 2016, 476, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Nakamura, T.; Kakutani, H.; Ishii, H. Thrombomodulin suppresses invasiveness of HT1080 tumor cells by reducing plasminogen activation on the cell surface through activation of thrombin-activatable fibrinolysis inhibitor. Biol. Pharm. Bull. 2009, 32, 179–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, J.; Ma, D.; Liu, X.; Chen, Y.; Fang, J.; Lui, V.W.Y.; Zhao, S.; Xia, J.; Cheng, B.; Wang, Z. Thrombomodulin (TM) in tumor cell differentiation and periphery blood immune microenvironment in oral squamous cell carcinoma. Clin. Immunol. 2018, 191, 27–33. [Google Scholar] [CrossRef]
- Shirai, Y.; Uwagawa, T.; Shiba, H.; Shimada, Y.; Horiuchi, T.; Saito, N.; Furukawa, K.; Ohashi, T.; Yanaga, K. Recombinant thrombomodulin suppresses tumor growth of pancreatic cancer by blocking thrombin-induced PAR1 and NF-κB activation. Surgery 2017, 161, 1675–1682. [Google Scholar] [CrossRef]
- Goyama, S.; Shrestha, M.; Schibler, J.; Rosenfeldt, L.; Miller, W.; O’Brien, E.; Mizukawa, B.; Kitamura, T.; Palumbo, J.S.; Mulloy, J.C. Protease-activated receptor-1 inhibits proliferation but enhances leukemia stem cell activity in acute myeloid leukemia. Oncogene 2017, 36, 2589–2598. [Google Scholar] [CrossRef]
- Carroll, I.M.; Maharshak, N. Enteric bacterial proteases in inflammatory bowel disease-pathophysiology and clinical implications. World J. Gastroenterol. 2013, 19, 7531–7543. [Google Scholar] [CrossRef] [PubMed]
- Koziel, J.; Potempa, J. Protease-armed bacteria in the skin. Cell Tissue Res. 2013, 351, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Tagashira, M.; Kanda, T.; Murakami, Y.; Amano, A.; Matsumoto-Nakano, M. Apple- and Hop-Polyphenols Inhibit Porphyromonas gingivalis-Mediated Precursor of Matrix Metalloproteinase-9 Activation and Invasion of Oral Squamous Cell Carcinoma Cells. J. Periodontol. 2016, 87, 1103–1111. [Google Scholar] [CrossRef]
- Lourbakos, A.; Potempa, J.; Travis, J.; D’Andrea, M.R.; Andrade-Gordon, P.; Santulli, R.; Mackie, E.J.; Pike, R.N. Arginine-specific protease from Porphyromonas gingivalis activates protease-activated receptors on human oral epithelial cells and induces interleukin-6 secretion. Infect. Immun. 2001, 69, 5121–5130. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roberts, J.S.; Atanasova, K.R.; Chowdhury, N.; Han, K.; Yilmaz, Ö. Human Primary Epithelial Cells Acquire an Epithelial-Mesenchymal-Transition Phenotype during Long-Term Infection by the Oral Opportunistic Pathogen, Porphyromonas gingivalis. Front. Cell Infect. Microbiol. 2017, 7, 493. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhang, Q.; Zong, S.; Zhong, W.L.; Qin, Y.; Bi, Z.; Chen, S.; Liu, H.J.; Wei, J.J.; Zhou, B.J.; et al. Protease-activated receptor-1 (PAR1) promotes epithelial-endothelial transition through Twist1 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 185. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Cai, C.; Dong, X.; Yu, Q.C.; Zhang, X.O.; Yang, L.; Zeng, Y.A. Identification of multipotent mammary stem cells by protein C receptor expression. Nature 2015, 517, 81–84. [Google Scholar] [CrossRef]
- Hwang-Verslues, W.W.; Kuo, W.H.; Chang, P.H.; Pan, C.C.; Wang, H.H.; Tsai, S.T.; Jeng, Y.M.; Shew, J.Y.; Kung, J.T.; Chen, C.H.; et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS ONE 2009, 4, e8377. [Google Scholar] [CrossRef]
- Lee, W.; Ku, S.K.; Choi, H.; Bae, J.S. Inhibitory effects of three diketopiperazines from marine-derived bacteria on endothelial protein C receptor shedding in human endothelial cells and mice. Fitoterapia 2016, 110, 181–188. [Google Scholar] [CrossRef]
- Buhner, S.; Hahne, H.; Hartwig, K.; Li, Q.; Vignali, S.; Ostertag, D.; Meng, C.; Hörmannsperger, G.; Braak, B.; Pehl, C. Protease signaling through protease activated receptor 1 mediate nerve activation by mucosal supernatants from irritable bowel syndrome but not from ulcerative colitis patients. PLoS ONE 2018, 13, e0193943. [Google Scholar] [CrossRef]
- Lee, P.R.; Johnson, T.P.; Gnanapavan, S.; Giovannoni, G.; Wang, T.; Steiner, J.P.; Medynets, M.; Vaal, M.J.; Gartner, V.; Nath, A. Protease-activated receptor-1 activation by granzyme B causes neurotoxicity that is augmented by interleukin-1β. J. Neuroinflamm. 2017, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, A.G.; Sancho, R.; García-Limones, C.; Behrens, A.; ten Dijke, P.; Calzado, M.A.; Muñoz, E. Vanilloid receptor-1 regulates neurogenic inflammation in colon and protects mice from colon cancer. Cancer Res. 2012, 72, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Kwong, K.; Nassenstein, C.; de Garavilla, L.; Meeker, S.; Undem, B.J. Thrombin and trypsin directly activate vagal C-fibres in mouse lung via protease-activated receptor-1. J. Physiol. 2010, 588, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Maggio, N.; Itsekson, Z.; Ikenberg, B.; Strehl, A.; Vlachos, A.; Blatt, I.; Tanne, D.; Chapman, J. The anticoagulant activated protein C (aPC) promotes metaplasticity in the hippocampus through an EPCR-PAR1-S1P1 receptors dependent mechanism. Hippocampus 2014, 24, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, D.; Gelbard, H.; Cheng, T.; Insalaco, R.; Fernández, J.A.; Griffin, J.H.; Zlokovic, B.V. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron 2004, 41, 563–572. [Google Scholar] [CrossRef]
- Gorbacheva, L.; Pinelis, V.; Ishiwata, S.; Strukova, S.; Reiser, G. Activated protein C prevents glutamate- and thrombin-induced activation of nuclear factor-κB in cultured hippocampal neurons. Neuroscience 2010, 165, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Vincent, J.L.; Laterre, P.F.; LaRosa, S.P.; Dhainaut, J.F.; Lopez-Rodriguez, A.; Steingrub, J.S.; Garber, G.E.; Helterbrand, J.D.; Ely, E.W.; et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N. Eng. J. Med. 2001, 344, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Martí-Carvajal, A.J.; Solà, I.; Lathyris, D.; Cardona, A.F. Human recombinant activated protein C for severe sepsis. Cochrane Database Syst. Rev. 2012, 14, CD004388. [Google Scholar] [CrossRef]
- Krenzlin, H.; Lorenz, V.; Alessandri, B. The involvement of thrombin in the pathogenesis of glioblastoma. J. Neurosci. Res. 2017, 95, 2080–2085. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Damhofer, H.; Daalhuisen, J.; Ten Brink, M.; Richel, D.J.; Spek, C.A. Dabigatran potentiates gemcitabine-induced growth inhibition of pancreatic cancer in mice. Mol. Med. 2017, 23, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Chen, S.; Qin, Y.; Zhang, H.; Wang, H.; Meng, J.; Huai, L.; Zhang, Q.; Yin, T.; Lei, Y.; et al. Doxycycline inhibits breast cancer EMT and metastasis through PAR-1/NF-κB/miR-17/E-cadherin pathway. Oncotarget 2017, 8, 104855–104866. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Receptor | EPCR | PAR-1 |
|---|---|---|
| Ligand | Factor VIIa Factor Xa TF-VIIa-Xa TF-VIIa Plasmodium falciparum erythrocyte membrane protein T-cell receptor present on a subset of Vδ2neg γδ T cells | Thrombin Factor Xa TF-VIIa-Xa APC Plasmin Granzyme A Gingipains-R Trypsin MMP-1, -9, -2, -13, -14 |
| Cancer Cell Line/Xenograft | Mechanism | Cellular Effects |
|---|---|---|
| Colorectal cancer [41] | ERK/AKT-dependent signaling | Inhibition of migration |
| A375 melanoma cells B16F10 melanoma cells [11,48] | ERK1/2—dependent signaling | Reduction of metastatic foci by inhibition of transendothelial migration |
| Malignant Pleural Mesothelioma [44] | ERK/AKT-dependent signaling; BAX, BCL2 factors | Inhibition of proliferation and migration Promotion of apoptosis |
| MGC803 gastric cancer cells [42] | ERK1/2—dependent signaling | Increased proliferation and migration |
| Breast cancer cells line [43,45,49,52] | SPOCK1/testican 1-mediated signaling MAPK-signaling | Increased expression of integrins, proliferation, 3D tumor growth and cells survival Angiogenesis |
| Lung adenocarcinoma [50] | ERK/AKT-dependent signaling | Inhibition of apoptosis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Endothelial Protein C Receptor (EPCR), Protease Activated Receptor-1 (PAR-1) and Their Interplay in Cancer Growth and Metastatic Dissemination. Cancers 2019, 11, 51. https://doi.org/10.3390/cancers11010051
Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Endothelial Protein C Receptor (EPCR), Protease Activated Receptor-1 (PAR-1) and Their Interplay in Cancer Growth and Metastatic Dissemination. Cancers. 2019; 11(1):51. https://doi.org/10.3390/cancers11010051
Chicago/Turabian StyleWojtukiewicz, Marek Z., Dominika Hempel, Ewa Sierko, Stephanie C. Tucker, and Kenneth V. Honn. 2019. "Endothelial Protein C Receptor (EPCR), Protease Activated Receptor-1 (PAR-1) and Their Interplay in Cancer Growth and Metastatic Dissemination" Cancers 11, no. 1: 51. https://doi.org/10.3390/cancers11010051
APA StyleWojtukiewicz, M. Z., Hempel, D., Sierko, E., Tucker, S. C., & Honn, K. V. (2019). Endothelial Protein C Receptor (EPCR), Protease Activated Receptor-1 (PAR-1) and Their Interplay in Cancer Growth and Metastatic Dissemination. Cancers, 11(1), 51. https://doi.org/10.3390/cancers11010051