Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells Via JAK-STAT3 Signaling and Inhibits Tumor Progression by Targeting Epidermal Growth Factor Receptor

Abstract
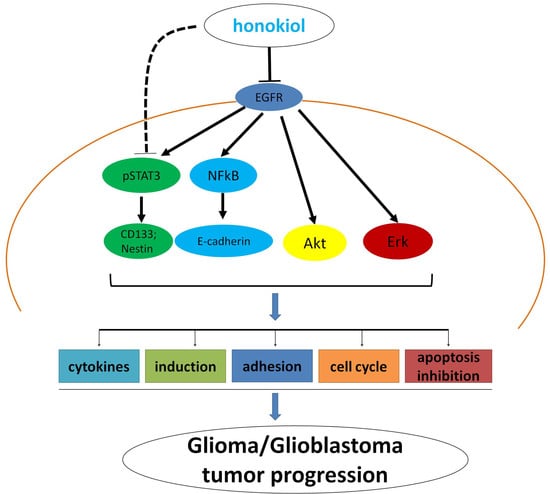
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
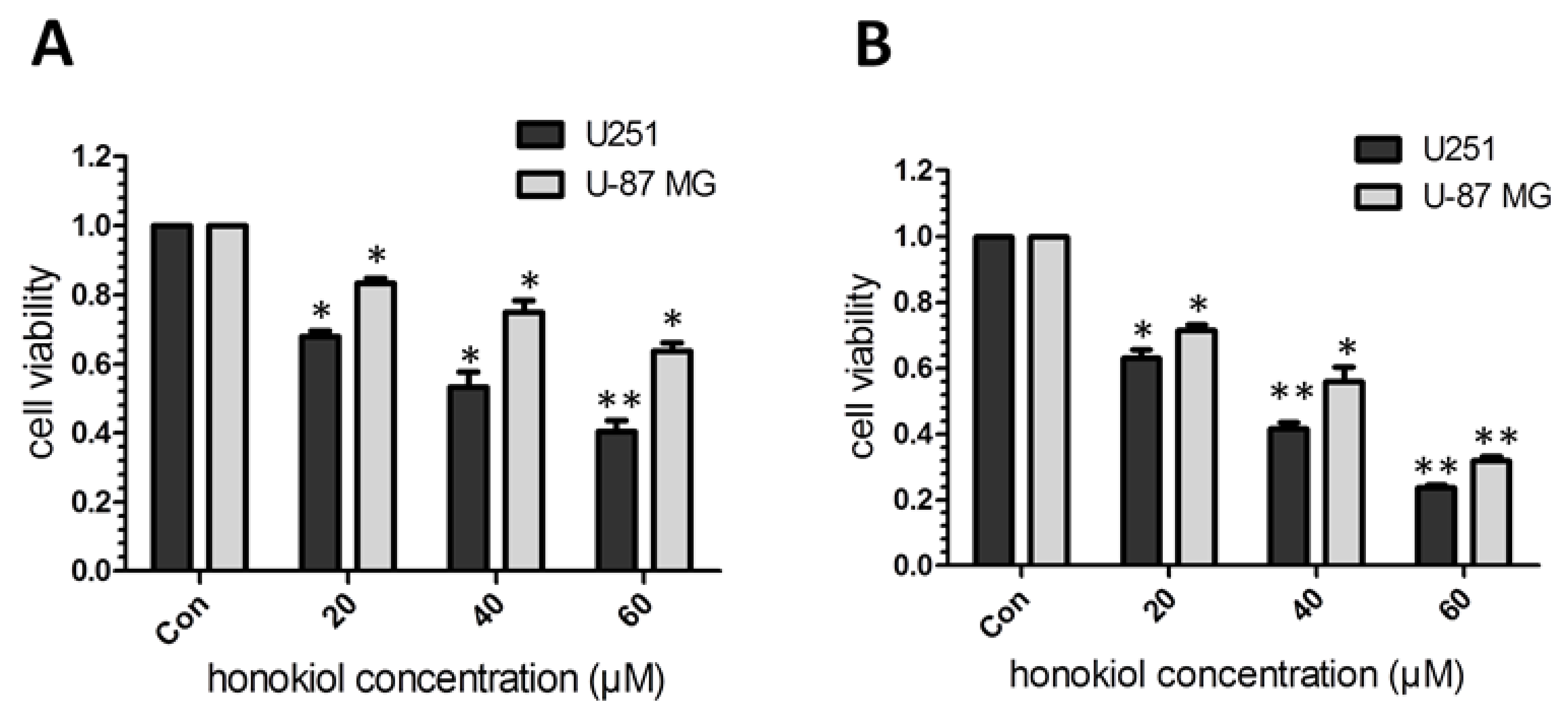
2.1. Honokiol Inhibits Glioma/Glioblatoma Cell Viability
2.2. Honokiol Inhibits Cell Migration/Proliferation and Invasion
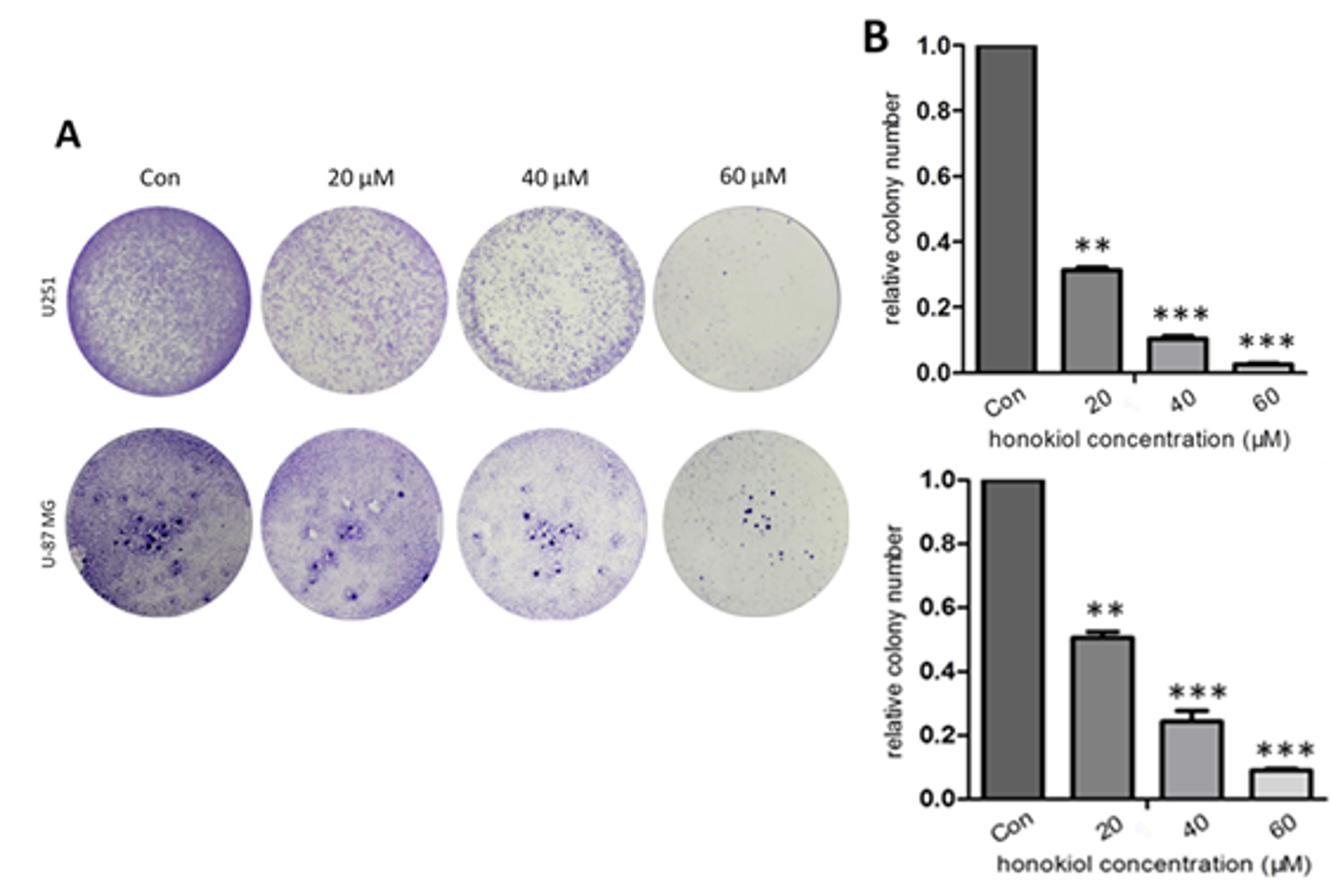
2.3. Honokiol Inhibits Colony Formation
2.4. Honokiol Promotes Apoptosis
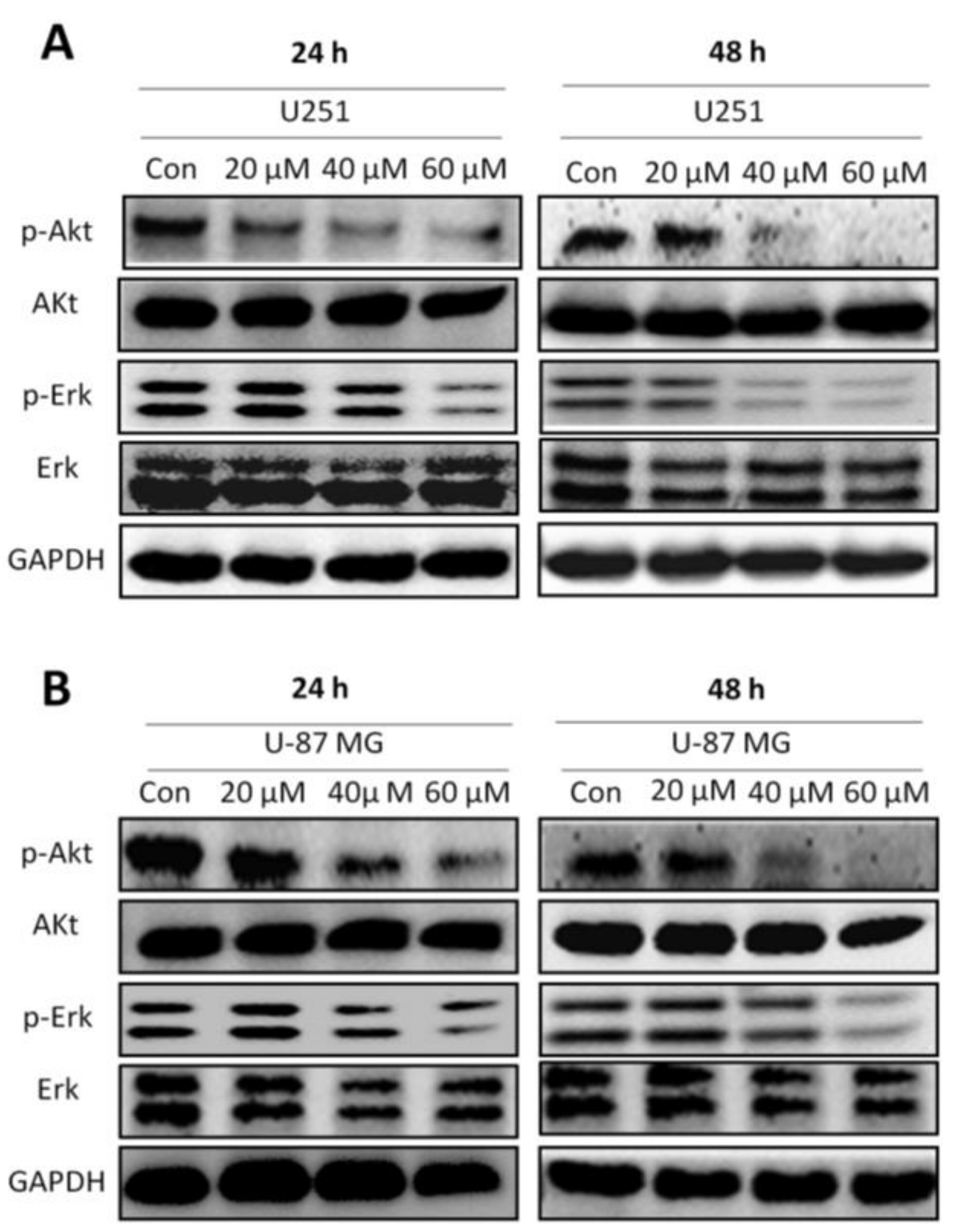
2.5. Honokiol Inhibits Erk and Akt Signaling Pathways
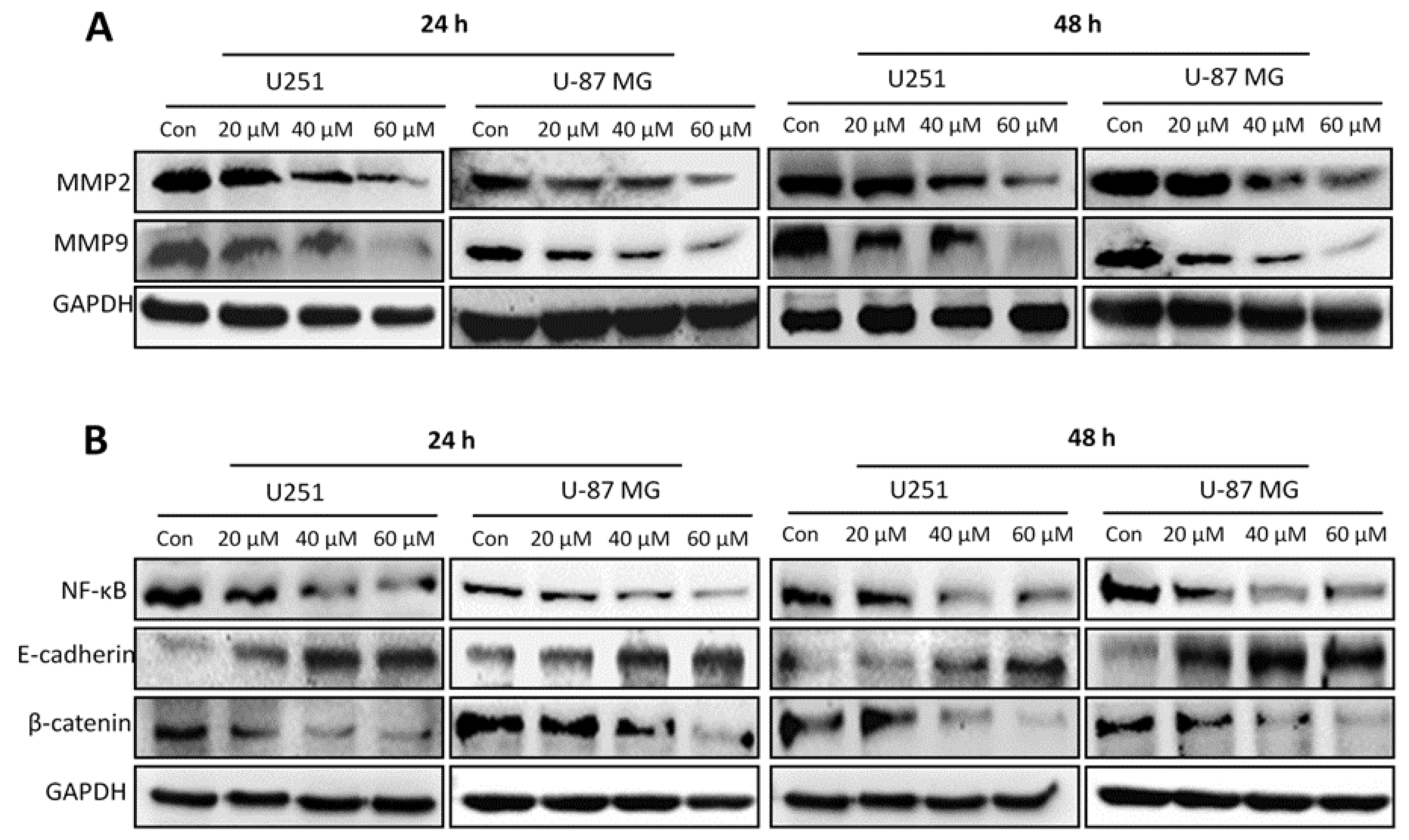
2.6. Honokiol Inhibits Expression of MMP2 and MMP9 and EMT-Related Molecules
2.7. Honokiol Inhibits the STAT3 Signaling Pathway via EGFR
2.8. Honokiol Inhibits Stem Cell Spheroid Formation
2.9. Honokiol Inhibits STAT3 Signaling in Glioma Stem Cells
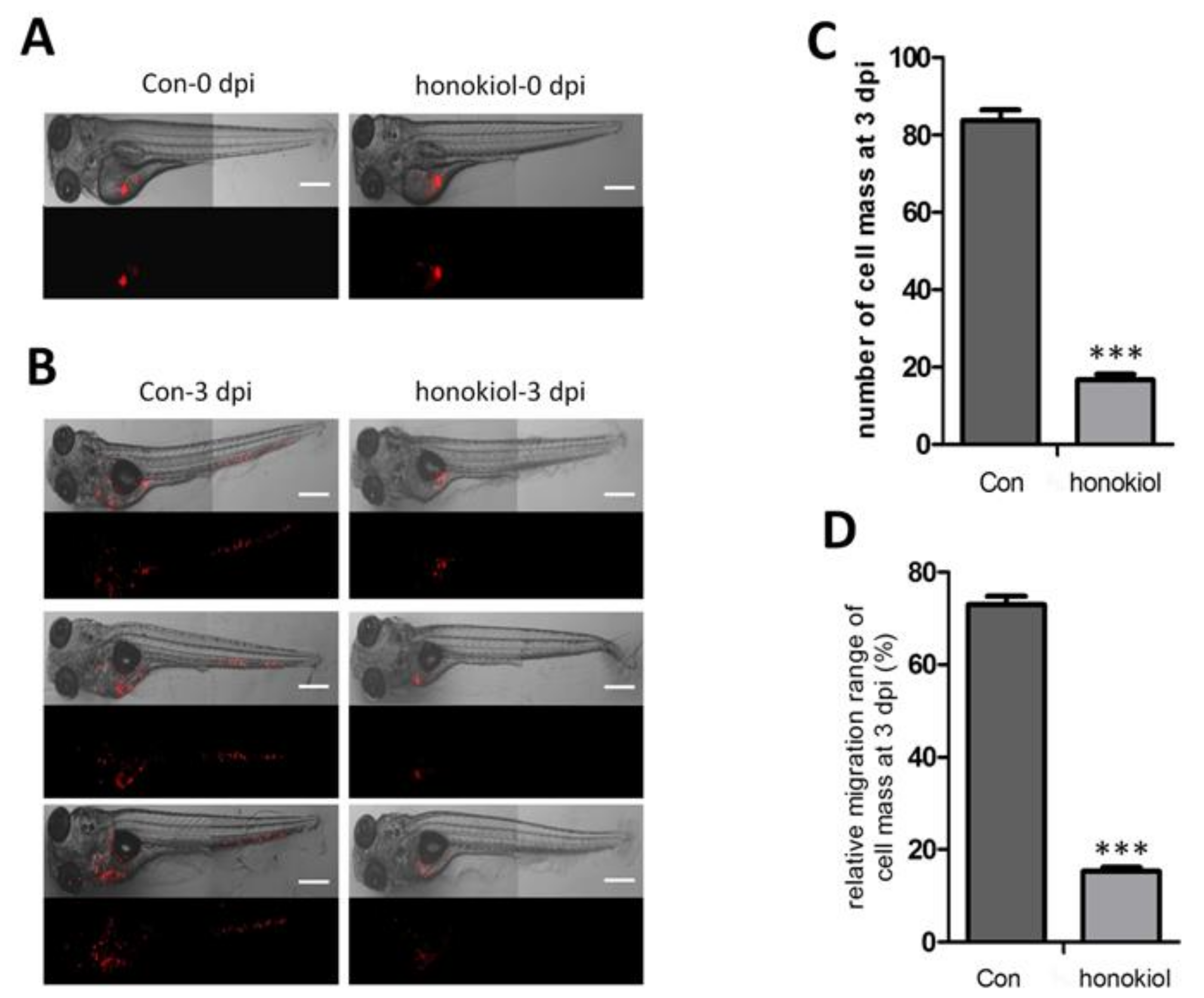
2.10. Honokiol Reduces Tumor Growth after Xenografting into Zebrafish
2.11. Honokiol Reduces Tumor Growth in Nude Mice
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. MTT Assay
4.3. Scratch Assay
4.4. Invasion Assay
4.5. Colony Formation Assay
4.6. Flow Cytometric Assay of Apoptosis
4.7. Assay for CD133-Positive Cells
4.8. Flow Cytometric Cell Sorting
4.9. Flow Cytometry of CD133+ Cells
4.10. Immunofluorescence Staining
4.11. Immunofluorescence Staining of CD133+ Cells
4.12. Western Blot Analysis
4.13. Zebrafish Xenografting
4.14. Nude Mouse Xenografting
4.15. H & E Staining and Immunofluorescence Staining of Xenografted Tumor Cells
4.16. Immunohistochemistry
4.17. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Lwin, Z.; Jiang, H.; Chung, C.; Millar, B.A.; Sahgal, A.; Laperriere, N.; Mason, W.P. Conditional probability of survival and post-progression survival in patients with glioblastoma in the temozolomide treatment era. J. Neuro-Oncol. 2014, 117, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Woolard, K.; Fine, H.A. Glioma stem cells: Better flat than round. Cell Stem Cell 2009, 4, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Ann. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Biscardi, J.S.; Ishizawar, R.C.; Silva, C.M.; Parsons, S.J. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. BCR 2000, 2, 203–210. [Google Scholar] [CrossRef]
- Klingler, S.; Guo, B.; Yao, J.; Yan, H.; Zhang, L.; Vaseva, A.V.; Chen, S.; Canoll, P.; Horner, J.W.; Wang, Y.A.; et al. Development of Resistance to EGFR-Targeted Therapy in Malignant Glioma Can Occur through EGFR-Dependent and -Independent Mechanisms. Cancer Res. 2015, 75, 2109–2119. [Google Scholar] [CrossRef]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell. Signal. 2007, 19, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Hale, A.J.; Smith, C.A.; Sutherland, L.C.; Stoneman, V.E.; Longthorne, V.L.; Culhane, A.C.; Williams, G.T. Apoptosis: Molecular regulation of cell death. Eur. J. Biochem. 1996, 236, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, E.; Joseph, A. The Use of Musculoskeletal Ultrasound for the Diagnosis of Groin and Hip Pain in Athletes. Curr. Sports Med. Rep. 2016, 15, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, X.; Shen, H.; Zhang, C. Molecular mechanisms of cell death in intervertebral disc degeneration (Review). Int. J. Mol. Med. 2016, 37, 1439–1448. [Google Scholar] [CrossRef]
- Gerdts, J.; Summers, D.W.; Milbrandt, J.; DiAntonio, A. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 2016, 89, 449–460. [Google Scholar] [CrossRef]
- Ziegler, S.A.; Loucks, C.; Madsen, S.J.; Carper, S.W. Heat shock protein 27 protects against aminolevulinic acid-mediated photodynamic therapy-induced apoptosis and necrosis in human breast cancer cells. J. Environ. Pathol. Toxicol. Oncol. Off. Organ Int. Soc. Environ. Toxicol. Cancer 2007, 26, 173–183. [Google Scholar] [CrossRef]
- Steinbach, J.P.; Weller, M. Apoptosis in gliomas: Molecular mechanisms and therapeutic implications. J. Neuro-Oncol. 2004, 70, 245–254. [Google Scholar] [CrossRef]
- Gunzburg, W.H.; Lohr, M.; Salmons, B. Novel treatments and therapies in development for pancreatic cancer. Expert Opin. Investig. Drugs 2002, 11, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Milner, J. Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev. 2003, 17, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.J.; Li, Y.B.; Wong, M.C. Expression of antisense bcl-2 cDNA abolishes tumorigenicity and enhances chemosensitivity of human malignant glioma cells. J. Neurosci. Res. 2003, 74, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.J.; Lin, C.I.; Wang, C.L.; Chao, J.I. Expression of survivin and p53 modulates honokiol-induced apoptosis in colorectal cancer cells. J. Cell. Biochem. 2014, 115, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ishii, G.; Goto, K.; Neri, S.; Hashimoto, H.; Yoh, K.; Niho, S.; Umemura, S.; Matsumoto, S.; Ohmatsu, H.; et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Servidei, T.; Meco, D.; Muto, V.; Bruselles, A.; Ciolfi, A.; Trivieri, N.; Lucchini, M.; Morosetti, R.; Mirabella, M.; Martini, M.; et al. Novel SEC61G-EGFR Fusion Gene in Pediatric Ependymomas Discovered by Clonal Expansion of Stem Cells in Absence of Exogenous Mitogens. Cancer Res. 2017, 77, 5860–5872. [Google Scholar] [CrossRef]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFkB pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells (Dayton, Ohio) 2013, 31, 248–258. [Google Scholar] [CrossRef]
- Wang, X.; Beitler, J.J.; Wang, H.; Lee, M.J.; Huang, W.; Koenig, L.; Nannapaneni, S.; Amin, A.R.; Bonner, M.; Shin, H.J.; et al. Honokiol enhances paclitaxel efficacy in multi-drug resistant human cancer model through the induction of apoptosis. PLoS ONE 2014, 9, e86369. [Google Scholar] [CrossRef]
- Kataria, H.; Lutz, D.; Chaudhary, H.; Schachner, M.; Loers, G. Small Molecule Agonists of Cell Adhesion Molecule L1 Mimic L1 Functions In Vivo. Mol. Neurobiol. 2016, 53, 4461–4483. [Google Scholar] [CrossRef]
- Kraus, K.; Kleene, R.; Braren, I.; Loers, G.; Lutz, D.; Schachner, M. A fragment of adhesion molecule L1 is imported into mitochondria, and regulates mitochondrial metabolism and trafficking. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, G.; Venugopal, A.; Ramamoorthy, P.; Standing, D.; Subramaniam, D.; Umar, S.; Jensen, R.A.; Anant, S.; Mammen, J.M. Honokiol inhibits melanoma stem cells by targeting notch signaling. Mol. Carcinog. 2015, 54, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Jing, J.; Lee, J.; Schedin, P.; Gilbert, S.M.; Peden, A.A.; Junutula, J.R.; Prekeris, R. Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. J. Cell Sci. 2013, 126, 4647–4658. [Google Scholar] [CrossRef] [PubMed]
- Tabouret, E.; Boudouresque, F.; Farina, P.; Barrie, M.; Bequet, C.; Sanson, M.; Chinot, O. MMP2 and MMP9 as candidate biomarkers to monitor bevacizumab therapy in high-grade glioma. Neuro-Oncology 2015, 17, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Farmakovskaya, M.; Khromova, N.; Rybko, V.; Dugina, V.; Kopnin, B.; Kopnin, P. E-Cadherin repression increases amount of cancer stem cells in human A549 lung adenocarcinoma and stimulates tumor growth. Cell Cycle (Georget. Tex.) 2016, 15, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.M.; Guo, S.M. Tim-3 facilitates osteosarcoma proliferation and metastasis through the NF-kappaB pathway and epithelial-mesenchymal transition. Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef]
- Vitucci, M.; Karpinich, N.O.; Bash, R.E.; Werneke, A.M.; Schmid, R.S.; White, K.K.; McNeill, R.S.; Huff, B.; Wang, S.; Van Dyke, T.; et al. Cooperativity between MAPK and PI3K signaling activation is required for glioblastoma pathogenesis. Neuro-Oncology 2013, 15, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, W.; Huang, C.; Lou, B.; Zhang, J.; Yu, D.; Huang, X.; Chen, B.; Zhou, M. Brusatol inhibits growth and induces apoptosis in pancreatic cancer cells via JNK/p38 MAPK/NF-kappab/Stat3/Bcl-2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 487, 820–826. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, R.F., Jr.; Pessoa, J.B.; Cruz, L.J.; Chan, A.B.; De Castro Miguel, E.; Cavalcante, R.S.; Brito, G.A.C.; Silva, H.F.O.; Gasparotto, L.H.S.; Guedes, P.M.M.; et al. Apoptosis in human liver carcinoma caused by gold nanoparticles in combination with carvedilol is mediated via modulation of MAPK/Akt/mTOR pathway and EGFR/FAAD proteins. Int. J. Oncol. 2018, 52, 189–200. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.H.; Li, S.H.; Li, H.Y.; Lin, Y.; Yang, J.X. Osthole Protects Bone Marrow-Derived Neural Stem Cells from Oxidative Damage through PI3K/Akt-1 Pathway. Neurochem. Res. 2017, 42, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.S.; Wang, W.; Chang, Y.A.; Lin, C.J.; Wang, J.J.; Chen, R.M. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer Lett. 2016, 370, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, M.Y.; Marcal, R.M.; Champagne, D.L.; van der Kooy, F.; Verpoorte, R.; Choi, Y.H. Effect of acute stresses on zebra fish (Danio rerio) metabolome measured by NMR-based metabolomics. Planta Med. 2014, 80, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.J.; Lee, J.H.; Chang, K.C.; Kim, H.J. Honokiol exerts an anticancer effect in T98G human glioblastoma cells through the induction of apoptosis and the regulation of adhesion molecules. Int. J. Oncol. 2012, 41, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Lai, I.C.; Shih, P.H.; Yao, C.J.; Yeh, C.T.; Wang-Peng, J.; Lui, T.N.; Chuang, S.E.; Hu, T.S.; Lai, T.Y.; Lai, G.M. Elimination of cancer stem-like cells and potentiation of temozolomide sensitivity by Honokiol in glioblastoma multiforme cells. PLoS ONE 2015, 10, e0114830. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Tai, Y.T.; Mohanraj, M.; Liu, S.H.; Yang, S.T.; Chen, R.M. Honokiol enhances temozolomide-induced apoptotic insults to malignant glioma cells via an intrinsic mitochondrion-dependent pathway. Phytomed. Int. J. Phytother. Phytopharmacol. 2018, 49, 41–51. [Google Scholar] [CrossRef]
- Ciaglia, E.; Laezza, C.; Abate, M.; Pisanti, S.; Ranieri, R.; D’alessandro, A.; Picardi, P.; Gazzerro, P.; Bifulco, M. Recognition by natural killer cells of N6-isopentenyladenosine-treated human glioma cell lines. Int. J. Cancer 2018, 142, 176–190. [Google Scholar] [CrossRef]
- Zhang, B.; Xuan, C.; Ji, Y.; Zhang, W.; Wang, D. Zebrafish xenotransplantation as a tool for in vivo cancer study. Fam. Cancer 2015, 14, 487–493. [Google Scholar] [CrossRef]
- Zhao, W.J.; Schachner, M. Neuregulin 1 enhances cell adhesion molecule l1 expression in human glioma cells and promotes their migration as a function of malignancy. J. Neuropathol. Exp. Neurol. 2013, 72, 244–255. [Google Scholar] [CrossRef]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Xue, W.; Schachner, M.; Zhao, W. Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells Via JAK-STAT3 Signaling and Inhibits Tumor Progression by Targeting Epidermal Growth Factor Receptor. Cancers 2019, 11, 22. https://doi.org/10.3390/cancers11010022
Fan Y, Xue W, Schachner M, Zhao W. Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells Via JAK-STAT3 Signaling and Inhibits Tumor Progression by Targeting Epidermal Growth Factor Receptor. Cancers. 2019; 11(1):22. https://doi.org/10.3390/cancers11010022
Chicago/Turabian StyleFan, Yipu, Weikang Xue, Melitta Schachner, and Weijiang Zhao. 2019. "Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells Via JAK-STAT3 Signaling and Inhibits Tumor Progression by Targeting Epidermal Growth Factor Receptor" Cancers 11, no. 1: 22. https://doi.org/10.3390/cancers11010022
APA StyleFan, Y., Xue, W., Schachner, M., & Zhao, W. (2019). Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells Via JAK-STAT3 Signaling and Inhibits Tumor Progression by Targeting Epidermal Growth Factor Receptor. Cancers, 11(1), 22. https://doi.org/10.3390/cancers11010022