The Oncogene Addiction Switch from NOTCH to PI3K Requires Simultaneous Targeting of NOTCH and PI3K Pathway Inhibition in Glioblastoma


Abstract
1. Introduction
2. Results
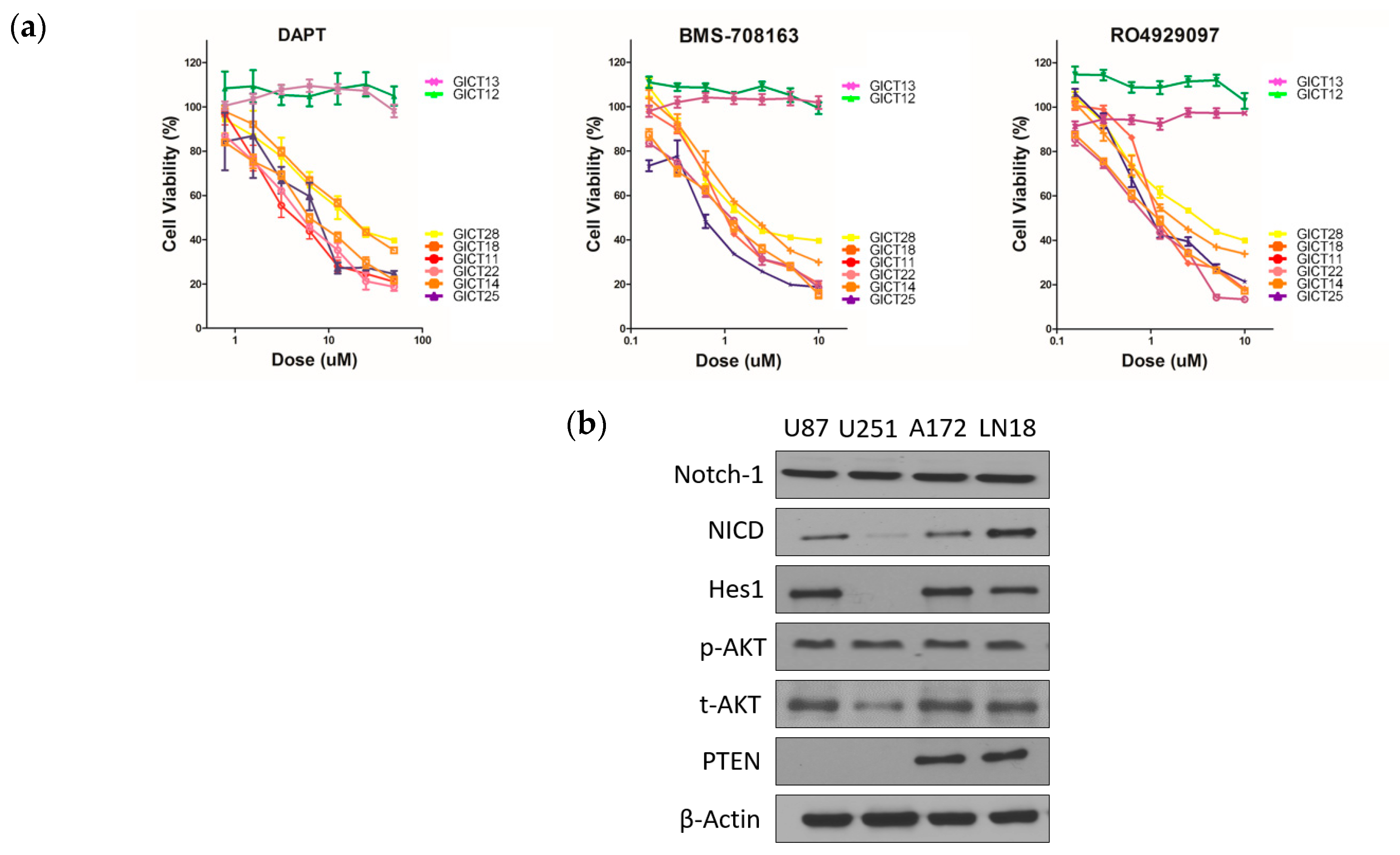
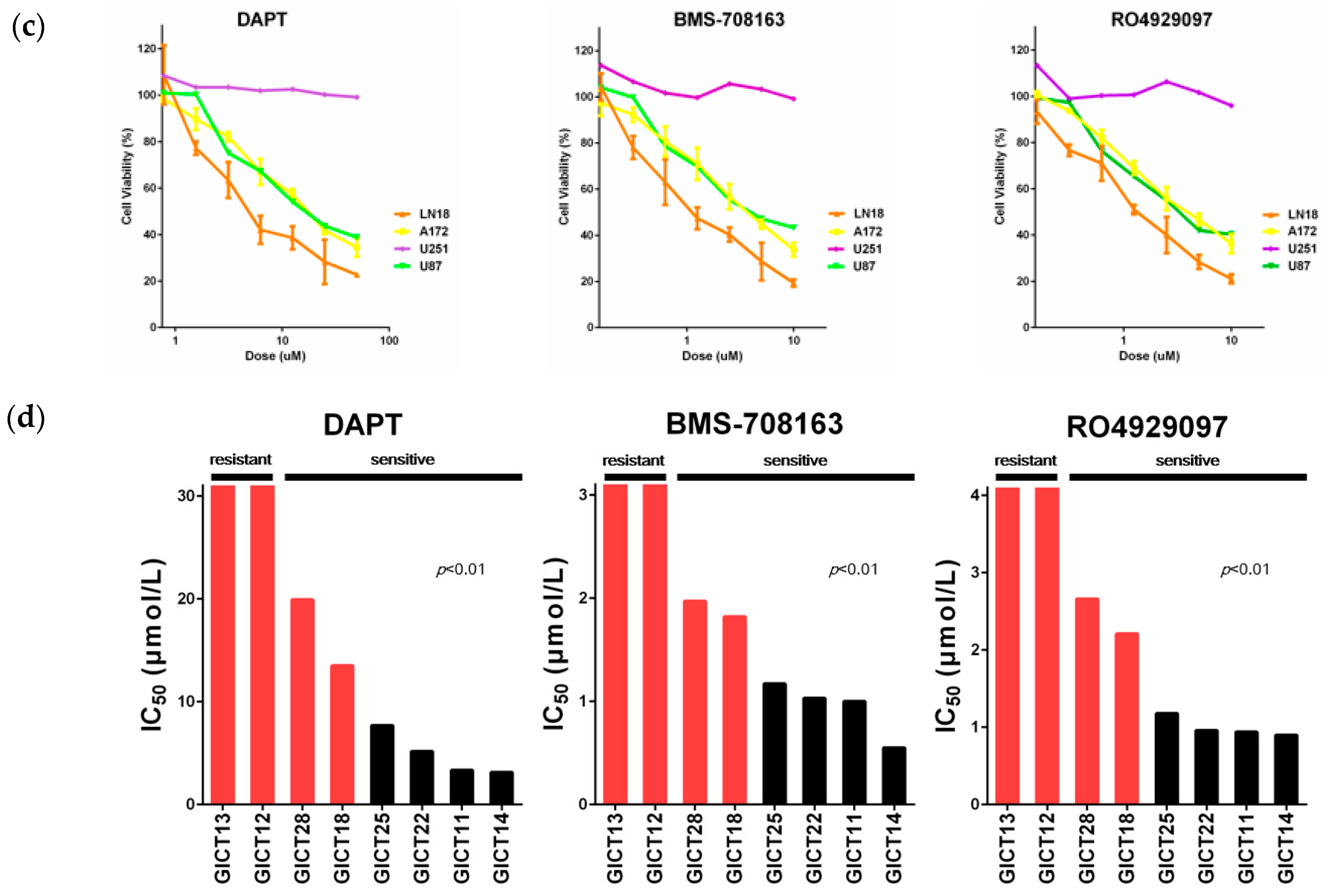
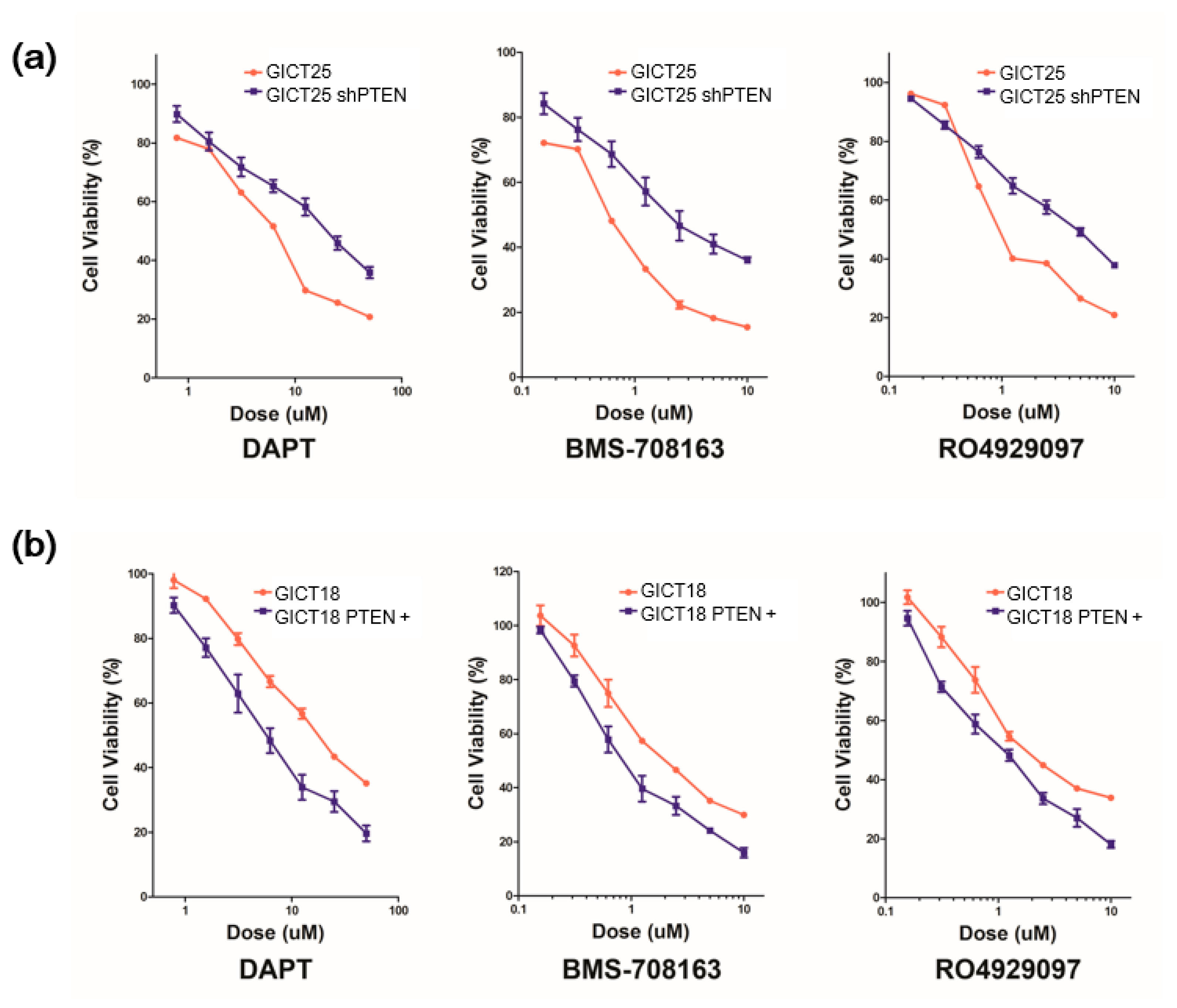
2.1. GICs Show Differential Growth in Response to GSIs
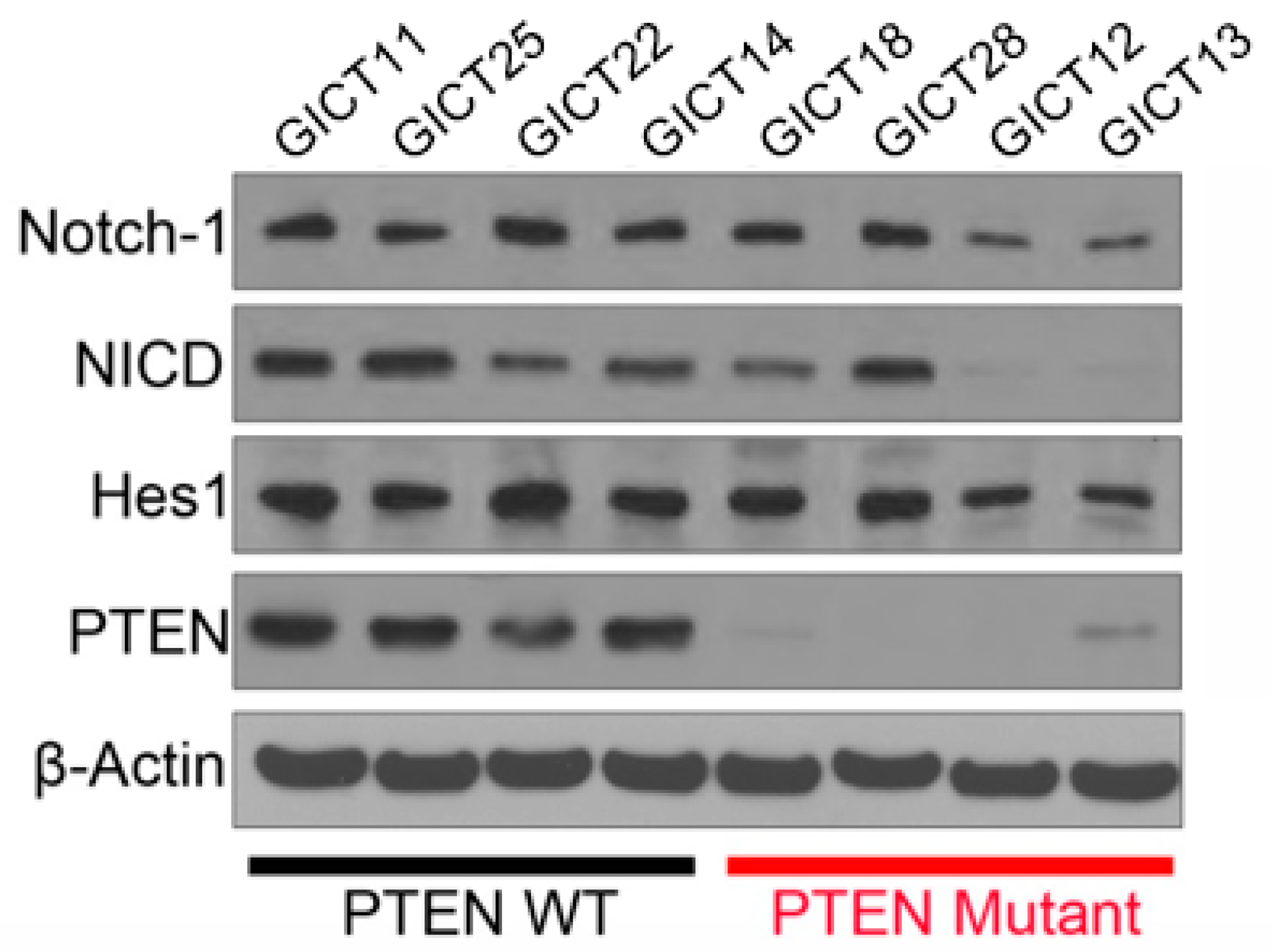
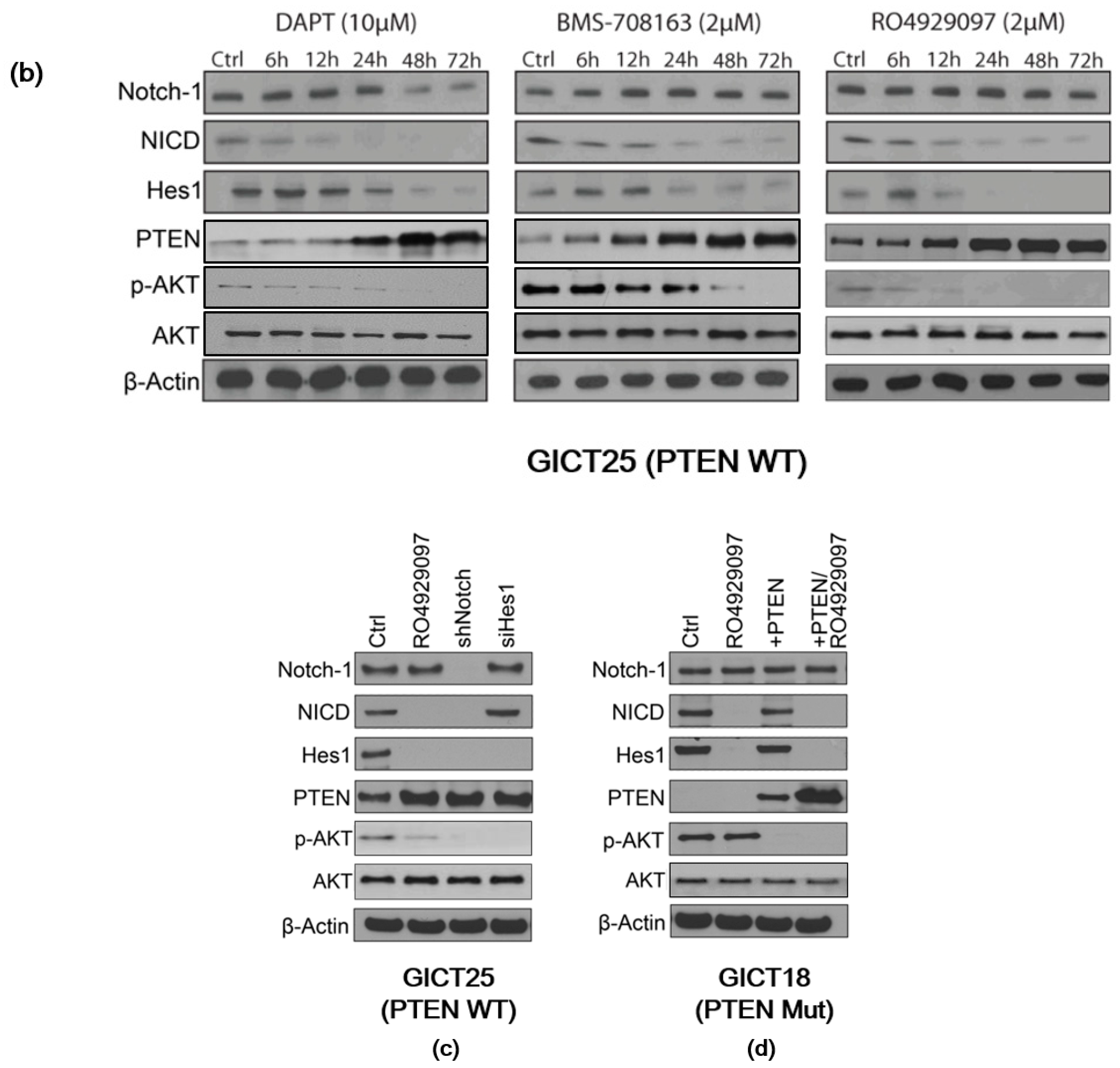
2.2. Expression of the NOTCH Signaling Pathway and PTEN Status in GICs
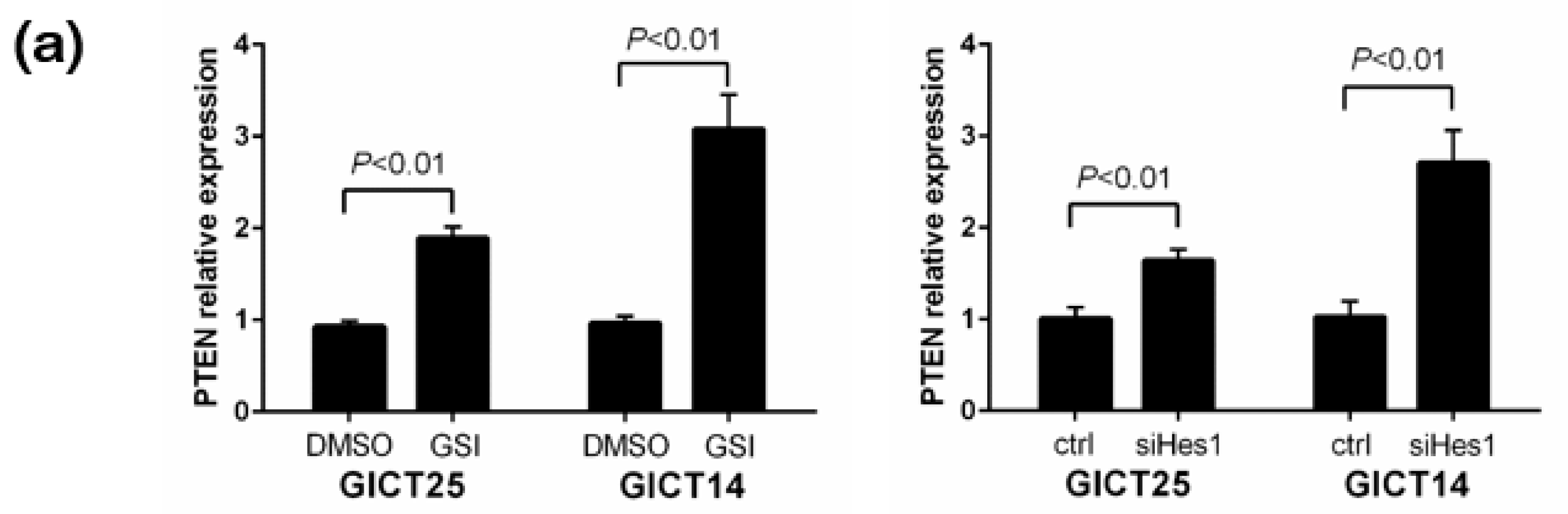
2.3. NOTCH Regulates Activity of the PI3K/AKT Pathway via Hes1 in PTEN–Wild-Type GICs
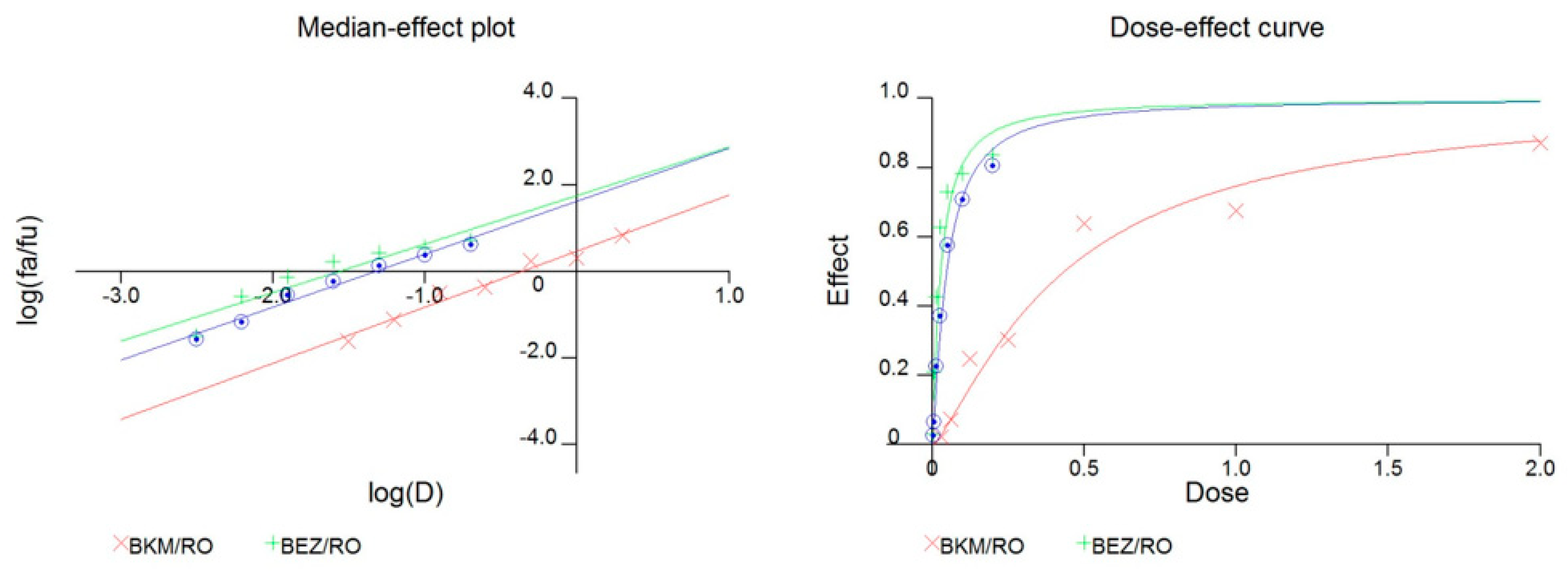
2.4. The Combination of GSI and PI3K Inhibitor had a Synergistic Effect in PTEN Mutant GICs
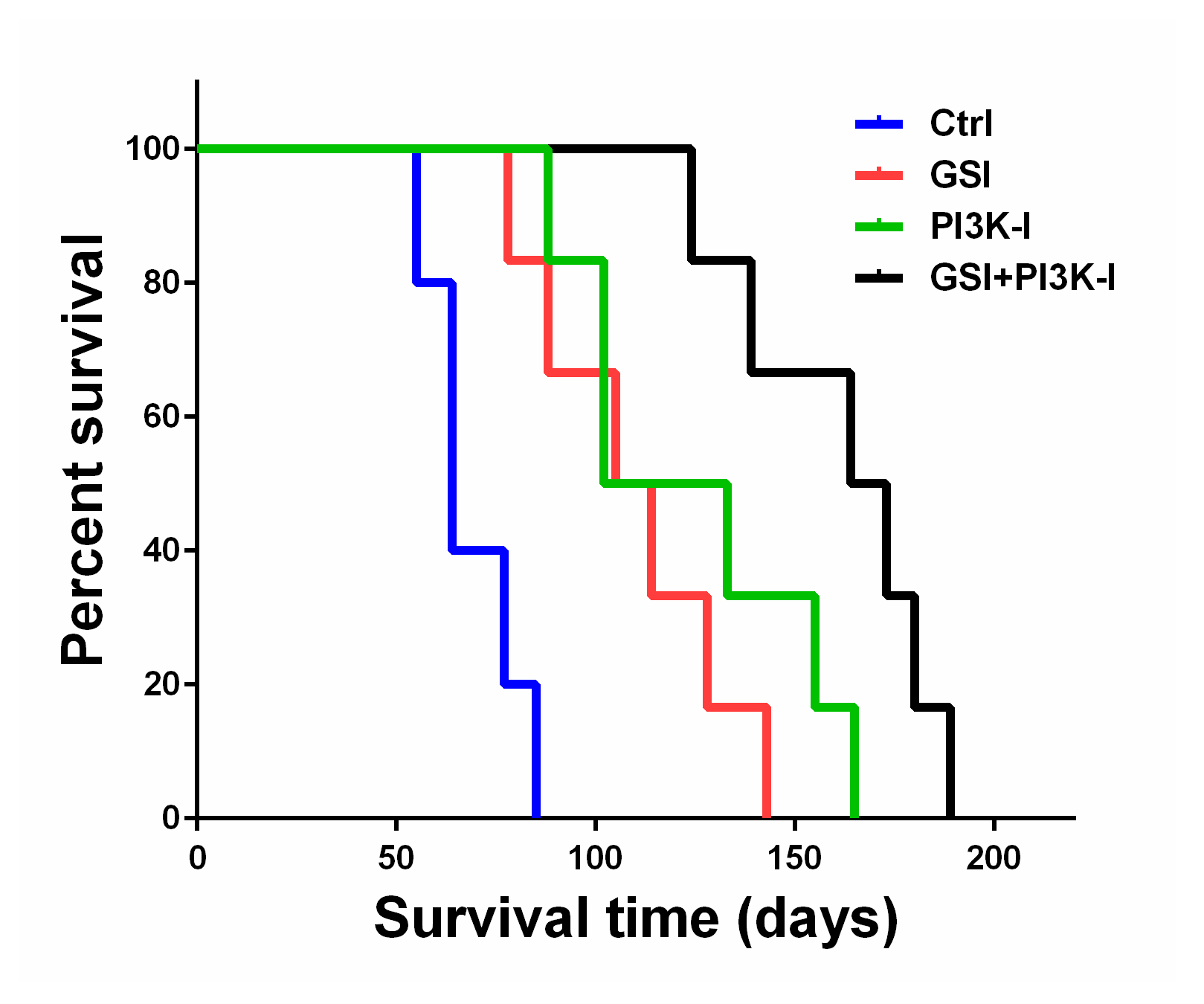
2.5. The Combination of GSI and PI3K Inhibitor Regulated Survival in an Orthotropic Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Proliferation Assay
4.3. Western Blot Analysis
4.4. RNA Extraction and cDNA Synthesis
4.5. Quantitative Real-Time PCR Analysis
4.6. Knockdown of Notch-1 by Lentiviral shRNA
4.7. Transient RNA Interference
4.8. Combination Studies
4.9. Animal Studies
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 23, 1061–1068. [Google Scholar]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Fortini, M.E. Notch signaling: The core pathway and its posttranslational regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T. The Notch-Hes pathway in mammalian neural development. Cell Res. 1999, 9, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high Notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef]
- Chappell, W.H.; Green, T.D.; Spengeman, J.D.; McCubrey, J.A.; Akula, S.M.; Bertrand, F.E. Increased protein expression of the PTEN tumor suppressor in the presence of constitutively active Notch-1. Cell Cycle 2005, 4, 1389–1395. [Google Scholar] [CrossRef]
- Wu, X.; Xu, K.; Zhang, L.; Deng, Y.; Lee, P.; Shapiro, E.; Monaco, M.; Makarenkova, H.P.; Li, J.; Lepor, H.; et al. Differentiation of the ductal epithelium and smooth muscle in the prostate gland are regulated by the Notch/PTEN-dependent mechanism. Dev. Biol. 2011, 356, 337–349. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- Bertrand, F.E.; McCubrey, J.A.; Angus, C.W.; Nutter, J.M.; Sigounas, G. NOTCH and PTEN in prostate cancer. Adv. Biol. Regul. 2014, 56, 51–65. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, J.; Sakamoto, S.; Kageyama, R. Hes1 and Hes5 regulate the development of the cranial and spinal nerve systems. Dev. Neurosci. 2006, 28, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F. Regulatory T cells and innate immune regulation in tumor immunity. Springer Semin. Immunopathol. 2006, 28, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Hoey, T.; Yen, W.C.; Axelrod, F.; Basi, J.; Donigian, L.; Dylla, S.; Fitch-Bruhns, M.; Lazetic, S.; Park, I.K.; Sato, A.; et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 2009, 5, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef]
- Koul, D.; Fu, J.; Shen, R.; LaFortune, T.A.; Wang, S.; Tiao, N.; Kim, Y.W.; Liu, J.L.; Ramnarian, D.; Yuan, Y.; et al. Antitumor activity of NVP-BKM120--a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin. Cancer Res. 2012, 18, 184–195. [Google Scholar] [CrossRef]
- Masica, D.L.; Karchin, R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011, 71, 4550–4561. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Matsumura, I.; Tanaka, H.; Ezoe, S.; Sugahara, H.; Mizuki, M.; Shibayama, H.; Ishiko, E.; Ishiko, J.; Nakajima, K.; et al. Roles for c-Myc in self-renewal of hematopoietic stem cells. J. Biol. Chem. 2004, 279, 24986–24993. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; DelBianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response.; resistance.; and road maps toward a cure. EMBO Rep. 2015, 16, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, W.; Jiang, T. Oncogene addiction in gliomas: Implications for molecular targeted therapy. J. Exp. Clin. Cancer Res. 2011, 17, 30–58. [Google Scholar] [CrossRef] [PubMed]
- Aravindan, N.; Aravindan, S.; Manickam, K.; Natarajan, M. High Energy Particle Radiation-associated Oncogenic Transformation in Normal Mice: Insight into the Connection between Activation of Oncotargets and Oncogene Addiction. Sci. Rep. 2016, 6, 37623. [Google Scholar] [CrossRef] [PubMed]
- DeGraffenried, L.A.; Fulcher, L.; Friedrichs, W.E.; Grünwald, V.; Ray, R.B.; Hidalgo, M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann. Oncol. 2004, 15, 1510–1516. [Google Scholar] [CrossRef]
- She, Q.B.; Solit, D.; Basso, A.; Moasser, M.M. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin. Cancer Res. 2003, 9, 4340–4346. [Google Scholar]
- Bhat, K.P.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef]
- Koul, D.; Jasser, S.A.; Lu, Y.; Davies, M.A.; Shen, R.; Shi, Y.; Mills, G.B.; Yung, W.K. Motif analysis of the tumor suppressor gene MMAC/PTEN identifies tyrosines critical for tumor suppression and lipid phosphatase activity. Oncogene 2002, 21, 57–64. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008, 68, 6043–6048. [Google Scholar] [CrossRef] [PubMed]
- Damaraju, V.L.; Bouffard, D.Y.; Wong, C.K.; Clarke, M.L.; Mackey, J.R.; Leblond, L.; Cass, C.E.; Grey, M.; Gourdeau, H. Synergistic activity of troxacitabine (Troxatyl) and gemcitabine in pancreatic cancer. BMC Cancer. 2007, 7, 121. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 | GICT25 | shPTEN |
|---|---|---|
| (a) | ||
| DAPT (µM) | 7.69 | 19.8 |
| BMS-708163 (µM) | 0.55 | 2.00 |
| RO4929097 (µM) | 0.96 | 4.67 |
| (b) | ||
| DAPT (µM) | 13.50 | 5.78 |
| BMS-708163 (µM) | 1.82 | 0.84 |
| RO4929097 (µM) | 2.21 | 1.11 |
| Reagents | CI Values at ED50 |
|---|---|
| BKM120/GSI (2:1) | 0.47 |
| BEZ235/GSI (1:5) | 0.58 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, N.; Hirai, N.; Aoki, K.; Suzuki, R.; Fujita, S.; Nakayama, H.; Hayashi, M.; Ito, K.; Sakurai, T.; Iwabuchi, S. The Oncogene Addiction Switch from NOTCH to PI3K Requires Simultaneous Targeting of NOTCH and PI3K Pathway Inhibition in Glioblastoma. Cancers 2019, 11, 121. https://doi.org/10.3390/cancers11010121
Saito N, Hirai N, Aoki K, Suzuki R, Fujita S, Nakayama H, Hayashi M, Ito K, Sakurai T, Iwabuchi S. The Oncogene Addiction Switch from NOTCH to PI3K Requires Simultaneous Targeting of NOTCH and PI3K Pathway Inhibition in Glioblastoma. Cancers. 2019; 11(1):121. https://doi.org/10.3390/cancers11010121
Chicago/Turabian StyleSaito, Norihiko, Nozomi Hirai, Kazuya Aoki, Ryo Suzuki, Satoshi Fujita, Haruo Nakayama, Morito Hayashi, Keisuke Ito, Takatoshi Sakurai, and Satoshi Iwabuchi. 2019. "The Oncogene Addiction Switch from NOTCH to PI3K Requires Simultaneous Targeting of NOTCH and PI3K Pathway Inhibition in Glioblastoma" Cancers 11, no. 1: 121. https://doi.org/10.3390/cancers11010121
APA StyleSaito, N., Hirai, N., Aoki, K., Suzuki, R., Fujita, S., Nakayama, H., Hayashi, M., Ito, K., Sakurai, T., & Iwabuchi, S. (2019). The Oncogene Addiction Switch from NOTCH to PI3K Requires Simultaneous Targeting of NOTCH and PI3K Pathway Inhibition in Glioblastoma. Cancers, 11(1), 121. https://doi.org/10.3390/cancers11010121