Platinum Resistance in Ovarian Cancer: Role of DNA Repair



Abstract
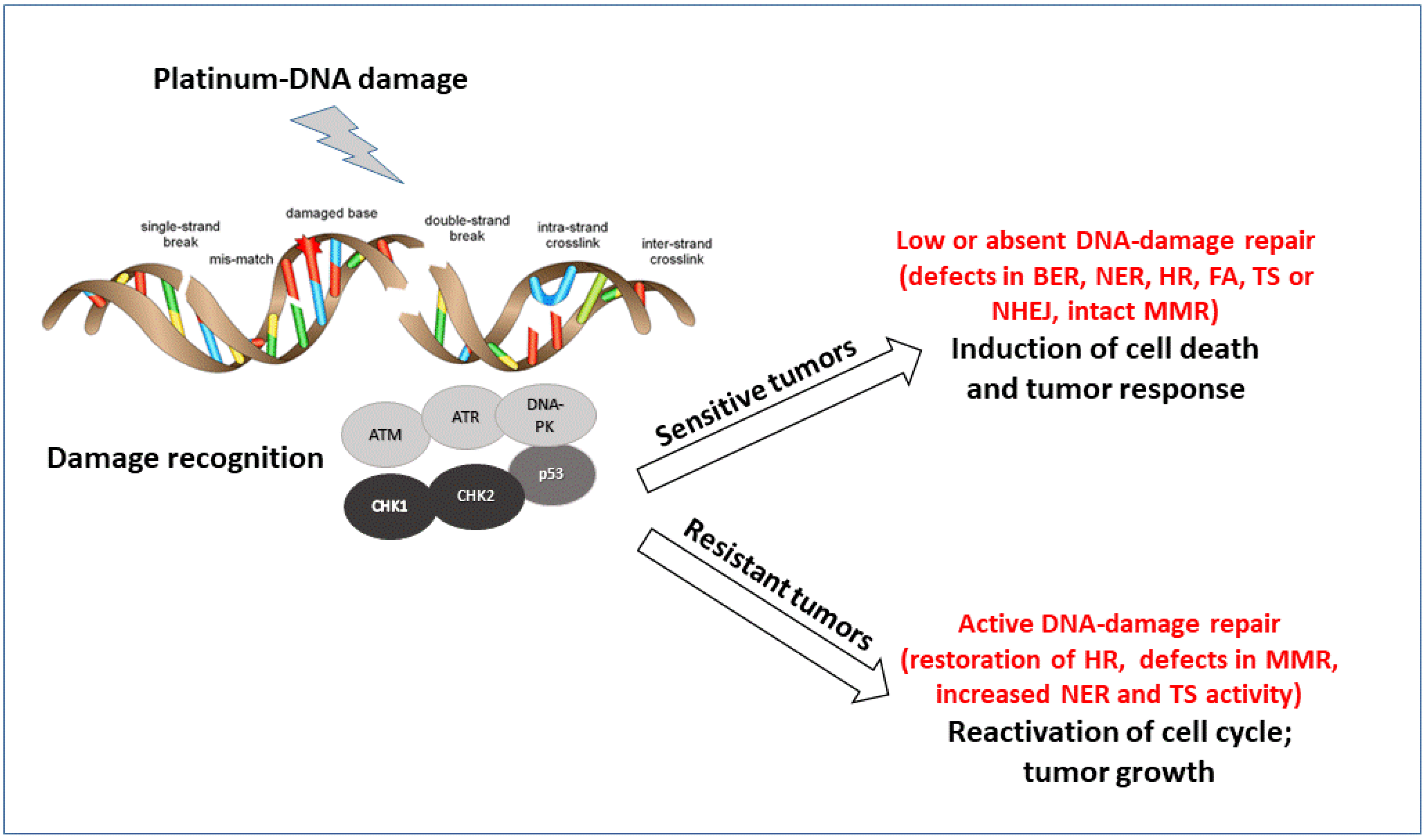
1. Introduction
2. Clinical Presentation and Management of Ovarian Cancer
3. DNA Repair Systems
4. Alterations in DNA Repair in Ovarian Cancer and Their Prognostic/Predictive Value
5. Functional Assays to Predict DNA Repair Proficiency
6. Models to Study Drug Resistance in Ovarian Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ishida:, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Peng, J.; Zeng, F.; Zhang, K.; Liu, J.; Li, X.; Ouyang, Q.; Wang, G.; Wang, L.; Liu, Z.; et al. Association between polymorphisms in CTR1, CTR2, ATP7A, and ATP7B and platinum resistance in epithelial ovarian cancer. Int. J. Clin. Pharm. 2017, 55, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Yasuda, M.; Fukushima, S. Changes in the Expression of Various Transporters as Influencing Factors of Resistance to Cisplatin. Anticancer Res. 2017, 37, 5477–5484. [Google Scholar] [PubMed]
- Öhrvik, H.; Nose, Y.; Wood, L.K.; Kim, B.-E.; Gleber, S.-C.; Ralle, M.; Thiele, D.J. Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain. Proc. Natl. Acad. Sci. USA 2013, 110, E4279–E4288. [Google Scholar] [CrossRef]
- Sørensen, B.H.; Dam, C.S.; Stürup, S.; Lambert, I.H. Dual role of LRRC8A-containing transporters on cisplatin resistance in human ovarian cancer cells. J. Inorg. Biochem. 2016, 160, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.W.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 430939. [Google Scholar] [CrossRef] [PubMed]
- Meijer, C.; Mulder, N.H.; Timmer-Bosscha, H.; Sluiter, W.J.; Meersma, G.J.; de Vries, E.G. Relationship of cellular glutathione to the cytotoxicity and resistance of seven platinum compounds. Cancer Res. 1992, 52, 6885–6889. [Google Scholar]
- Okuno, S.; Sato, H.; Kuriyama-Matsumura, K.; Tamba, M.; Wang, H.; Sohda, S.; Hamada, H.; Yoshikawa, H.; Kondo, T.; Bannai, S. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br. J. Cancer 2003, 88, 951–956. [Google Scholar] [CrossRef]
- Hagrman, D.; Goodisman, J.; Dabrowiak, J.C.; Souid, A.-K. Kinetic study on the reaction of cisplatin with metallothionein. Drug Metab. Dispos. 2003, 31, 916–923. [Google Scholar] [CrossRef]
- Beale, P.J.; Rogers, P.; Boxall, F.; Sharp, S.Y.; Kelland, L.R. BCL-2 family protein expression and platinum drug resistance in ovarian carcinoma. Br. J. Cancer 2000, 82, 436–440. [Google Scholar] [CrossRef]
- Bieg, D.; Sypniewski, D.; Nowak, E.; Bednarek, I. Morin decreases galectin-3 expression and sensitizes ovarian cancer cells to cisplatin. Arch. Gynecol. Obstet. 2018, 298, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Jin, S.; Li, X.; Wang, D. The involvement of Bcl-2 family proteins in AKT-regulated cell survival in cisplatin resistant epithelial ovarian cancer. Oncotarget 2017, 8, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Zhang, Q.; Ridgway, L.D.; Tian, L.; Claret, F.-X. Cisplatin resistance in an ovarian carcinoma is associated with a defect in programmed cell death control through XIAP regulation. Oncol. Res. 2003, 13, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; Imperatori, L.; Stefanini, M.; D’Incalci, M. Sensitivity of CHO mutant cell lines with specific defects in nucleotide excision repair to different anti-cancer agents. Int. J. Cancer 1996, 66, 779–783. [Google Scholar] [CrossRef]
- Damia, G.; D’Incalci, M. Targeting DNA repair as a promising approach in cancer therapy. Eur. J. Cancer 2007, 43, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Traganos, F.; Wlodkowic, D. Impaired DNA damage response--an Achilles’ heel sensitizing cancer to chemotherapy and radiotherapy. Eur. J. Pharmacol. 2009, 625, 143–150. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Dai, C.-H.; Li, J.; Chen, P.; Jiang, H.-G.; Wu, M.; Chen, Y.-C. RNA interferences targeting the Fanconi anemia/BRCA pathway upstream genes reverse cisplatin resistance in drug-resistant lung cancer cells. J. Biomed. Sci. 2015, 22, 77. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. Ca Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, T.; Shingleton, H.; Homesley, H.; LaGasse, L.; Blessing, J. cis-Dichlorodiammineplatinum(II) in the treatment of gynecologic malignancies: Phase II trials by the Gynecologic Oncology Group. Cancer Treat Rep. 1979, 63, 1549–1555. [Google Scholar] [PubMed]
- Rossof, A.H.; Talley, R.W.; Stephens, R.; Thigpen, T.; Samson, M.K.; Groppe, C.; Eyre, H.J.; Fisher, R. Phase II evaluation of cis-dichlorodiammineplatinum(II) in advanced malignancies of the genitourinary and gynecologic organs: A Southwest Oncology Group Study. Cancer Treat Rep. 1979, 63, 1557–1564. [Google Scholar] [PubMed]
- Tomao, F.; D’Incalci, M.; Biagioli, E.; Peccatori, F.A.; Colombo, N. Restoring platinum sensitivity in recurrent ovarian cancer by extending the platinum-free interval: Myth or reality? Cancer 2017, 123, 3450–3459. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Dizon, D.S. PARP inhibitors for targeted treatment in ovarian cancer. Lancet 2017, 390, 1929–1930. [Google Scholar] [CrossRef]
- Konecny, G.E.; Kristeleit, R.S. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: Current practice and future directions. Br. J. Cancer 2016, 115, 1157–1173. [Google Scholar] [CrossRef]
- Franzese, E.; Centonze, S.; Diana, A.; Carlino, F.; Guerrera, L.P.; Di Napoli, M.; De Vita, F.; Pignata, S.; Ciardiello, F.; Orditura, M. PARP inhibitors in ovarian cancer. Cancer Treat. Rev. 2018, 73, 1–9. [Google Scholar] [CrossRef]
- Mirza, M.R.; Pignata, S.; Ledermann, J.A. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann. Oncol. 2018, 29, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Atsushi, H.; Shuji, S.; Kosuke, A.; Takafumi, K. A comparison of in vitro platinum-DNA adduct formation between carboplatin and cisplatin. Int. J. Biochem. 1994, 26, 1009–1016. [Google Scholar] [CrossRef]
- Hah, S.S.; Stivers, K.M.; de Vere White, R.W.; Henderson, P.T. Kinetics of carboplatin-DNA binding in genomic DNA and bladder cancer cells as determined by accelerator mass spectrometry. Chem. Res. Toxicol. 2006, 19, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Sørensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008. [Google Scholar] [CrossRef]
- Lempiäinen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. Embo J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Bajrami, I.; Lord, C.J. Synthetic Lethality and Cancer—Penetrance as the Major Barrier. Trends Cancer 2018, 4, 671–683. [Google Scholar] [CrossRef]
- Haynes, B.; Saadat, N.; Myung, B.; Shekhar, M.P.V. Crosstalk between translesion synthesis, Fanconi anemia network, and homologous recombination repair pathways in interstrand DNA crosslink repair and development of chemoresistance. Mutat. Res. Rev. Mutat. Res. 2015, 763, 258–266. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6. [Google Scholar] [PubMed]
- Drummond, J.T.; Anthoney, A.; Brown, R.; Modrich, P. Cisplatin and adriamycin resistance are associated with MutLalpha and mismatch repair deficiency in an ovarian tumor cell line. J. Biol. Chem. 1996, 271, 19645–19648. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Hirst, G.L.; Gallagher, W.M.; McIlwrath, A.J.; Margison, G.P.; van der Zee, A.G.; Anthoney, D.A. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene 1997, 15, 45–52. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef]
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Hamilton, T.C.; Chaney, S.G. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998, 58, 3579–3585. [Google Scholar]
- Caiola, E.; Salles, D.; Frapolli, R.; Lupi, M.; Rotella, G.; Ronchi, A.; Garassino, M.C.; Mattschas, N.; Colavecchio, S.; Broggini, M.; et al. Base excision repair-mediated resistance to cisplatin in KRAS(G12C) mutant NSCLC cells. Oncotarget 2015, 6, 30072–30087. [Google Scholar] [CrossRef]
- Gao, D.; Hu, J.; Zhang, X.; Gao, C.; Hong, J. Effect of hOGG1 over-expression on cisplatin resistance in esophageal squamous carcinoma cells. Cancer Biother. Radiopharm. 2013, 28, 433–440. [Google Scholar] [CrossRef]
- Sawant, A.; Floyd, A.M.; Dangeti, M.; Lei, W.; Sobol, R.W.; Patrick, S.M. Differential role of base excision repair proteins in mediating cisplatin cytotoxicity. DNA Repair 2017, 51, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Tavecchio, M.; Simone, M.; Erba, E.; Chiolo, I.; Liberi, G.; Foiani, M.; D’Incalci, M.; Damia, G. Role of homologous recombination in trabectedin-induced DNA damage. Eur. J. Cancer 2008, 44, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, D.C.; Sailer, V.; Xue, H.; Cheng, H.; Collins, C.C.; Gleave, M.; Wang, Y.; Demichelis, F.; Beltran, H.; Rubin, M.A.; et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001487. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Damia, G. Unleashing Chk1 in cancer therapy. Cell Cycle 2011, 10, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Damia, G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat. Rev. 2017, 60, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Chilà, R.; Guffanti, F.; Damia, G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat. Rev. 2016, 50, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Lui, G.Y.L.; Grandori, C.; Kemp, C.J. CDK12: An emerging therapeutic target for cancer. J. Clin. Pathol. 2018, 71, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Méndez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef]
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011, 102, 663–669. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.-C.; Choi, Y.-E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.-Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, F.; Fortier, E.; Dubé, M.; Lemay, J.-F.; Buisson, R.; Masson, J.-Y.; Elsherbiny, A.; Costantino, S.; Carmona, E.; Mes-Masson, A.-M.; et al. Replication Protein A Availability during DNA Replication Stress Is a Major Determinant of Cisplatin Resistance in Ovarian Cancer Cells. Cancer Res. 2018, 78, 5561–5573. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef]
- Avraam, K.; Pavlakis, K.; Papadimitriou, C.; Vrekoussis, T.; Panoskaltsis, T.; Messini, I.; Patsouris, E. The prognostic and predictive value of ERCC-1, p53, bcl-2 and bax in epithelial ovarian cancer. Eur. J. Gynaecol. Oncol. 2011, 32, 516–520. [Google Scholar]
- Kuhlmann, J.D.; Wimberger, P.; Bankfalvi, A.; Keller, T.; Schöler, S.; Aktas, B.; Buderath, P.; Hauch, S.; Otterbach, F.; Kimmig, R.; et al. ERCC1-positive circulating tumor cells in the blood of ovarian cancer patients as a predictive biomarker for platinum resistance. Clin. Chem. 2014, 60, 1282–1289. [Google Scholar] [CrossRef]
- Rubatt, J.M.; Darcy, K.M.; Tian, C.; Muggia, F.; Dhir, R.; Armstrong, D.K.; Bookman, M.A.; Niedernhofer, L.J.; Deloia, J.; Birrer, M.; et al. Pre-treatment tumor expression of ERCC1 in women with advanced stage epithelial ovarian cancer is not predictive of clinical outcomes: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 421–426. [Google Scholar] [CrossRef]
- Zhao, M.; Li, S.; Zhou, L.; Shen, Q.; Zhu, H.; Zhu, X. Prognostic values of excision repair cross-complementing genes mRNA expression in ovarian cancer patients. Life Sci. 2018, 194, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Ganzinelli, M.; Mariani, P.; Cattaneo, D.; Fossati, R.; Fruscio, R.; Corso, S.; Ricci, F.; Broggini, M.; Damia, G. Expression of DNA repair genes in ovarian cancer samples: Biological and clinical considerations. Eur. J. Cancer 2011, 47, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Bowden, N.A. Nucleotide excision repair: Why is it not used to predict response to platinum-based chemotherapy? Cancer Lett. 2014, 346, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Olaussen, K.A.; Pignon, J.-P.; Shepherd, F.A.; Tsao, M.-S.; Graziano, S.; Kratzke, R.; Douillard, J.-Y.; Seymour, L.; Pirker, R.; et al. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N. Engl. J. Med. 2013, 368, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Caiola, E.; Broggini, M.; Marabese, M. Genetic markers for prediction of treatment outcomes in ovarian cancer. Pharm. J. 2014, 14, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Caiola, E.; Porcu, L.; Fruscio, R.; Giuliani, D.; Milani, R.; Torri, V.; Broggini, M.; Marabese, M. DNA-damage response gene polymorphisms and therapeutic outcomes in ovarian cancer. Pharm. J. 2013, 13, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, P.; Cao, Y.; Wang, G.; Wang, N.; Zhou, R. Predicting the outcome of platinum-based chemotherapies in epithelial ovarian cancer using the 8092C/A polymorphism of ERCC1: A meta-analysis. Biomarkers 2014, 19, 128–134. [Google Scholar] [PubMed]
- Yan, L.; Shu-Ying, Y.; Shan, K.; Yip, B.H.K.; Rong-Miao, Z.; Na, W.; Hai-Yan, S. Association between polymorphisms of ERCC1 and survival in epithelial ovarian cancer patients with chemotherapy. Pharmacogenomics 2012, 13, 419–427. [Google Scholar] [CrossRef]
- White, K.L.; Vierkant, R.A.; Fogarty, Z.C.; Charbonneau, B.; Block, M.S.; Pharoah, P.D.P.; Chenevix-Trench, G.; for AOCS/ACS Group; Rossing, M.A.; Cramer, D.W.; et al. Analysis of over 10,000 Cases finds no association between previously reported candidate polymorphisms and ovarian cancer outcome. Cancer Epidemiol. Biomark. Prev. 2013, 22, 987–992. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Rodriguez, G.P.; Song, J.B.; Crouse, G.F. In Vivo Bypass of 8-oxodG. PLoS Genet. 2013, 9, e1003682. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Wang, Q.-E. Targeting translesion synthesis to facilitate the eradication of ovarian cancer stem cells by platinum-based therapy. Mol. Cell Oncol. 2016, 3, e1043482. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.-E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.K.; Maddukuri, L.; Ketkar, A.; Penthala, N.R.; Reed, M.R.; Eddy, S.; Crooks, P.A.; Eoff, R.L. A Small-Molecule Inhibitor of Human DNA Polymerase η Potentiates the Effects of Cisplatin in Tumor Cells. Biochemistry 2018, 57, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Postel-Vinay, S.; Sourisseau, T.; Adam, J.; Stoclin, A.; Ponsonnailles, F.; Dorvault, N.; Commo, F.; Saulnier, P.; Salome-Desmoulez, S.; et al. ERCC1 function in nuclear excision and interstrand crosslink repair pathways is mediated exclusively by the ERCC1-202 isoform. Cell Cycle 2013, 12, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.-S.; Adam, J.; Dorvault, N.; Robin, A.; Friboulet, L.; Soria, J.-C.; Olaussen, K.A. A novel antibody-based approach to detect the functional ERCC1-202 isoform. DNA Repair 2018, 64, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Guffanti, F. Department of Oncology, IRCCS-Istituto di Ricerche Farmacologiche Mario Negri, Milan, Italy. Unpublished Work. 2018. [Google Scholar]
- Frey, M.K.; Pothuri, B. Homologous recombination deficiency (HRD) testing in ovarian cancer clinical practice: A review of the literature. Gynecol. Oncol. Res. Pr. 2017, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Elattar, A.; Cerbinskaite, A.; Wilkinson, S.J.; Drew, Y.; Kyle, S.; Los, G.; Hostomsky, Z.; Edmondson, R.J.; Curtin, N.J. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin. Cancer Res. 2010, 16, 2344–2351. [Google Scholar] [CrossRef]
- Shah, M.M.; Dobbin, Z.C.; Nowsheen, S.; Wielgos, M.; Katre, A.A.; Alvarez, R.D.; Konstantinopoulos, P.A.; Yang, E.S.; Landen, C.N. An ex vivo assay of XRT-induced Rad51 foci formation predicts response to PARP-inhibition in ovarian cancer. Gynecol. Oncol. 2014, 134, 331–337. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Tumiati, M.; Hietanen, S.; Hynninen, J.; Pietilä, E.; Färkkilä, A.; Kaipio, K.; Roering, P.; Huhtinen, K.; Alkodsi, A.; Li, Y.; et al. A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin. Cancer Res. 2018, 24, 4482–4493. [Google Scholar] [PubMed]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e20. [Google Scholar]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.W.; Laub, P.B.; Beesley, J.S.; Ozols, R.F.; Hamilton, T.C. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 1997, 57, 850–856. [Google Scholar] [PubMed]
- Kelland, L.R.; Mistry, P.; Abel, G.; Loh, S.Y.; O’Neill, C.F.; Murrer, B.A.; Harrap, K.R. Mechanism-related circumvention of acquired cis-diamminedichloroplatinum(II) resistance using two pairs of human ovarian carcinoma cell lines by ammine/amine platinum(IV) dicarboxylates. Cancer Res. 1992, 52, 3857–3864. [Google Scholar] [PubMed]
- McNeil, E.M.; Astell, K.R.; Ritchie, A.-M.; Shave, S.; Houston, D.R.; Bakrania, P.; Jones, H.M.; Khurana, P.; Wallace, C.; Chapman, T.; et al. Inhibition of the ERCC1-XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair 2015, 31, 19–28. [Google Scholar] [CrossRef]
- Perez, R.P.; Hamilton, T.C.; Ozols, R.F. Resistance to alkylating agents and cisplatin: Insights from ovarian carcinoma model systems. Pharmacol. Ther. 1990, 48, 19–27. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Xiang, D.; Wang, D.; Xin, X. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE1/Ref-1) in human ovarian cancer and indentification of the therapeutic potential of APE1/Ref-1 inhibitor. Int. J. Oncol. 2009, 35, 1069–1079. [Google Scholar]
- Yu, J.J.; Lee, K.B.; Mu, C.; Li, Q.; Abernathy, T.V.; Bostick-Bruton, F.; Reed, E. Comparison of two human ovarian carcinoma cell lines (A2780/CP70 and MCAS) that are equally resistant to platinum, but differ at codon 118 of the ERCC1 gene. Int. J. Oncol. 2000, 16, 555–560. [Google Scholar] [CrossRef]
- Ricci, F.; Bizzaro, F.; Cesca, M.; Guffanti, F.; Ganzinelli, M.; Decio, A.; Ghilardi, C.; Perego, P.; Fruscio, R.; Buda, A.; et al. Patient-derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res. 2014, 74, 6980–6990. [Google Scholar] [CrossRef]
- AlHilli, M.M.; Becker, M.A.; Weroha, S.J.; Flatten, K.S.; Hurley, R.M.; Harrell, M.I.; Oberg, A.L.; Maurer, M.J.; Hawthorne, K.M.; Hou, X.; et al. In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol. Oncol. 2016, 143, 379–388. [Google Scholar] [CrossRef] [PubMed]
- George, E.; Kim, H.; Krepler, C.; Wenz, B.; Makvandi, M.; Tanyi, J.L.; Brown, E.; Zhang, R.; Brafford, P.; Jean, S.; et al. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. Jci Insight 2017, 2, e89760. [Google Scholar] [CrossRef] [PubMed]
- Heo, E.J.; Cho, Y.J.; Cho, W.C.; Hong, J.E.; Jeon, H.-K.; Oh, D.-Y.; Choi, Y.-L.; Song, S.Y.; Choi, J.-J.; Bae, D.-S.; et al. Patient-Derived Xenograft Models of Epithelial Ovarian Cancer for Preclinical Studies. Cancer Res. Treat 2017, 49, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Palakurthi, S.; Zeng, Q.; Zhou, S.; Ivanova, E.; Huang, W.; Zervantonakis, I.K.; Selfors, L.M.; Shen, Y.; Pritchard, C.C.; et al. Establishment of Patient-Derived Tumor Xenograft Models of Epithelial Ovarian Cancer for Preclinical Evaluation of Novel Therapeutics. Clin. Cancer Res. 2017, 23, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Fratelli, M.; Guffanti, F.; Porcu, L.; Spriano, F.; Dell’Anna, T.; Fruscio, R.; Damia, G. Patient-derived ovarian cancer xenografts re-growing after a cisplatinum treatment are less responsive to a second drug re-challenge: A new experimental setting to study response to therapy. Oncotarget 2017, 8, 7441–7451. [Google Scholar] [CrossRef] [PubMed]
- Guffanti, F.; Fratelli, M.; Ganzinelli, M.; Bolis, M.; Ricci, F.; Bizzaro, F.; Chilà, R.; Sina, F.P.; Fruscio, R.; Lupia, M.; et al. Platinum sensitivity and DNA repair in a recently established panel of patient-derived ovarian carcinoma xenografts. Oncotarget 2018, 9, 24707–24717. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Guffanti, F.; Damia, G.; Broggini, M. Combination of paclitaxel, bevacizumab and MEK162 in second line treatment in platinum-relapsing patient derived ovarian cancer xenografts. Mol. Cancer 2017, 16, 97. [Google Scholar] [CrossRef] [PubMed]
- Castellón, E.A. Patient-derived organoids: New co-clinical model to predict treatment response in cancer? Oral Dis. 2018. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Nagle, P.W.; Plukker, J.T.M.; Muijs, C.T.; van Luijk, P.; Coppes, R.P. Patient-derived tumor organoids for prediction of cancer treatment response. Semin. Cancer Biol. 2018, 53, 258–264. [Google Scholar] [CrossRef]
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; van Gerwen, B.; van der Ven, M.; van de Wetering, M.; Egan, D.A.; et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. Elife 2016, 5, e18489. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Ekawa, T.; Endo, H.; Yamazaki, K.; Tanaka, N.; Kukita, Y.; Okuyama, H.; Okami, J.; Imamura, F.; Ohue, M.; et al. High-Throughput Screening in Colorectal Cancer Tissue-Originated Spheroids. Cancer Sci. 2018, 110, 345. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Platinum DNA Damage | Repair Pathway | Specific Genes Implicated in Platinum Resistance |
|---|---|---|
| Mono-adduct | Base excision repair (BER) | OGG1, PARP1 |
| Intra-strand cross-link | Nucleotide excision repair (NER) Mismatch repair (MMR) Tolerance pathway (translesion synthesis) | ERCC1, XPF, XPD, XPG MLH1, MSH2, MSH6, PMS2 Polymerase (pol) η, ζ Rev1 |
| Inter-strand cross-link | NER Famconi Anemia (FA) Homologous recombination (HR) | ERCC1, XPF, XPG, XPD FA core complex genes, FANCD2 BRCA1, BRCA2, RAD51, RAD52, XRCC2, XRCC3, RPA |
| Double-strand break | HR Non-homologous end joining (NHEJ) | BRCA1, BRCA2, RAD51, RAD52, XRCC2, XRCC3, RPA XRCC4, XRCC5, KU70, DNA-PKcs |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. https://doi.org/10.3390/cancers11010119
Damia G, Broggini M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers. 2019; 11(1):119. https://doi.org/10.3390/cancers11010119
Chicago/Turabian StyleDamia, Giovanna, and Massimo Broggini. 2019. "Platinum Resistance in Ovarian Cancer: Role of DNA Repair" Cancers 11, no. 1: 119. https://doi.org/10.3390/cancers11010119
APA StyleDamia, G., & Broggini, M. (2019). Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers, 11(1), 119. https://doi.org/10.3390/cancers11010119