Cancer-Associated Intermediate Conductance Ca2+-Activated K+ Channel KCa3.1

 ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
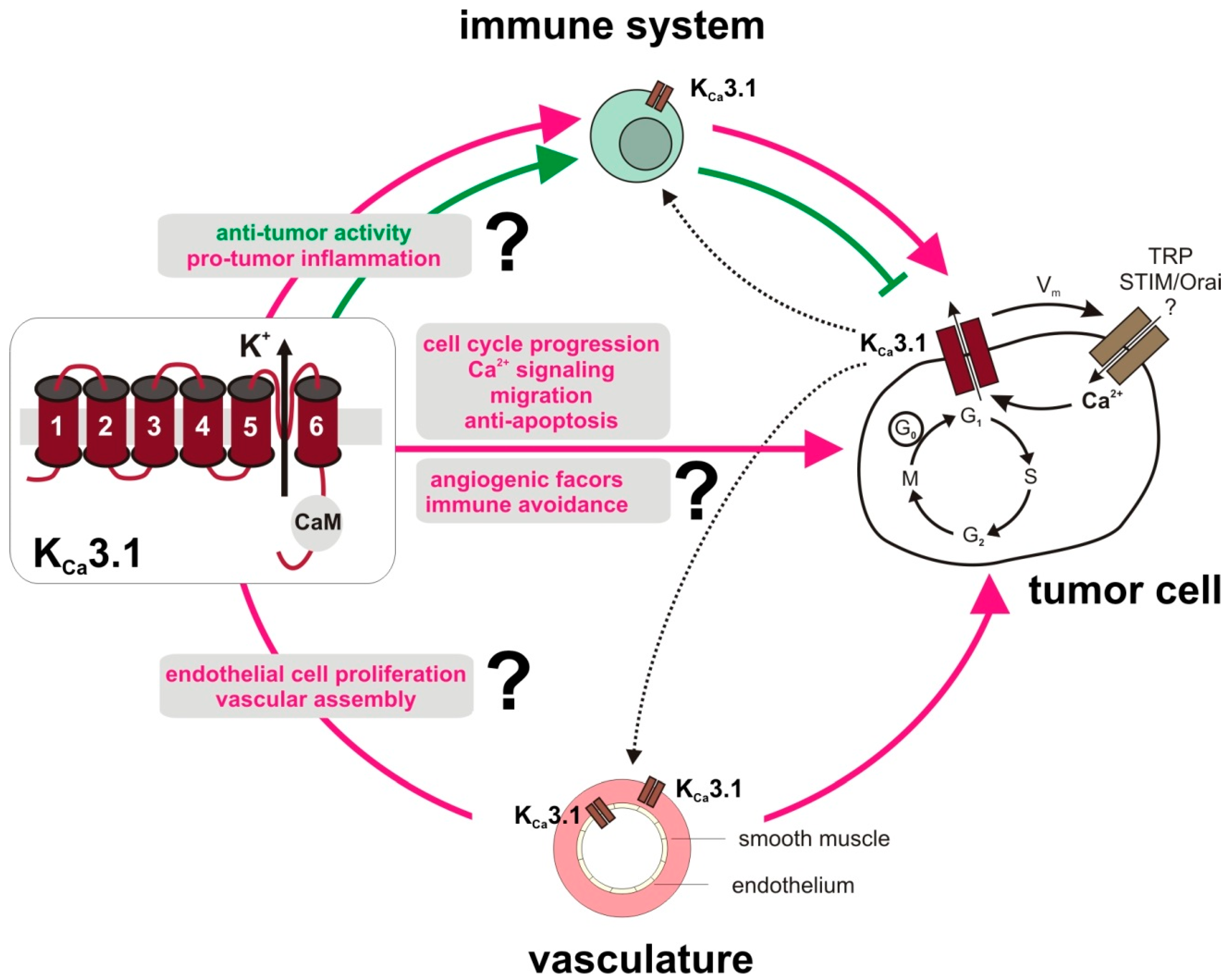
2. Tumor Cell-Specific Functions of KCa3.1
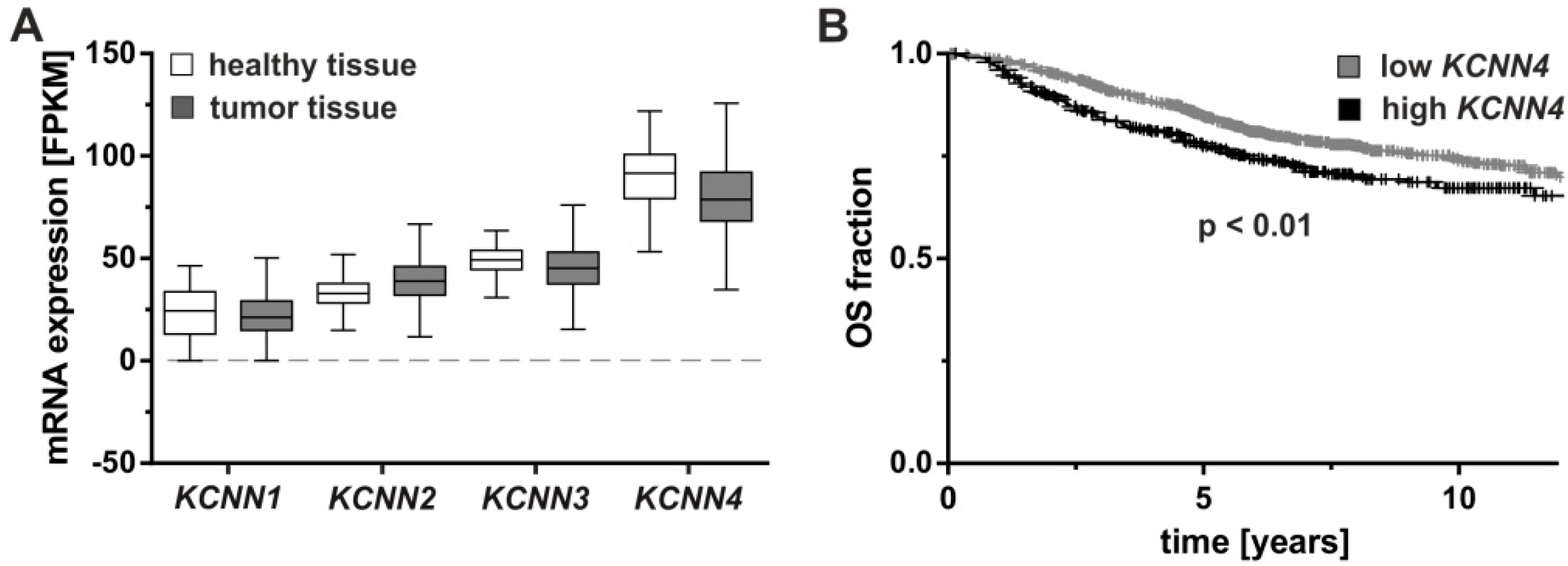
2.1. Molecular Markers and Regulation of KCNN4
2.2. Tumorigenesis
2.3. Tumor Cell Apoptosis and Survival
2.4. Cancer Invasion and Metastasis
3. KCa3.1 in the Tumor Microenvironment
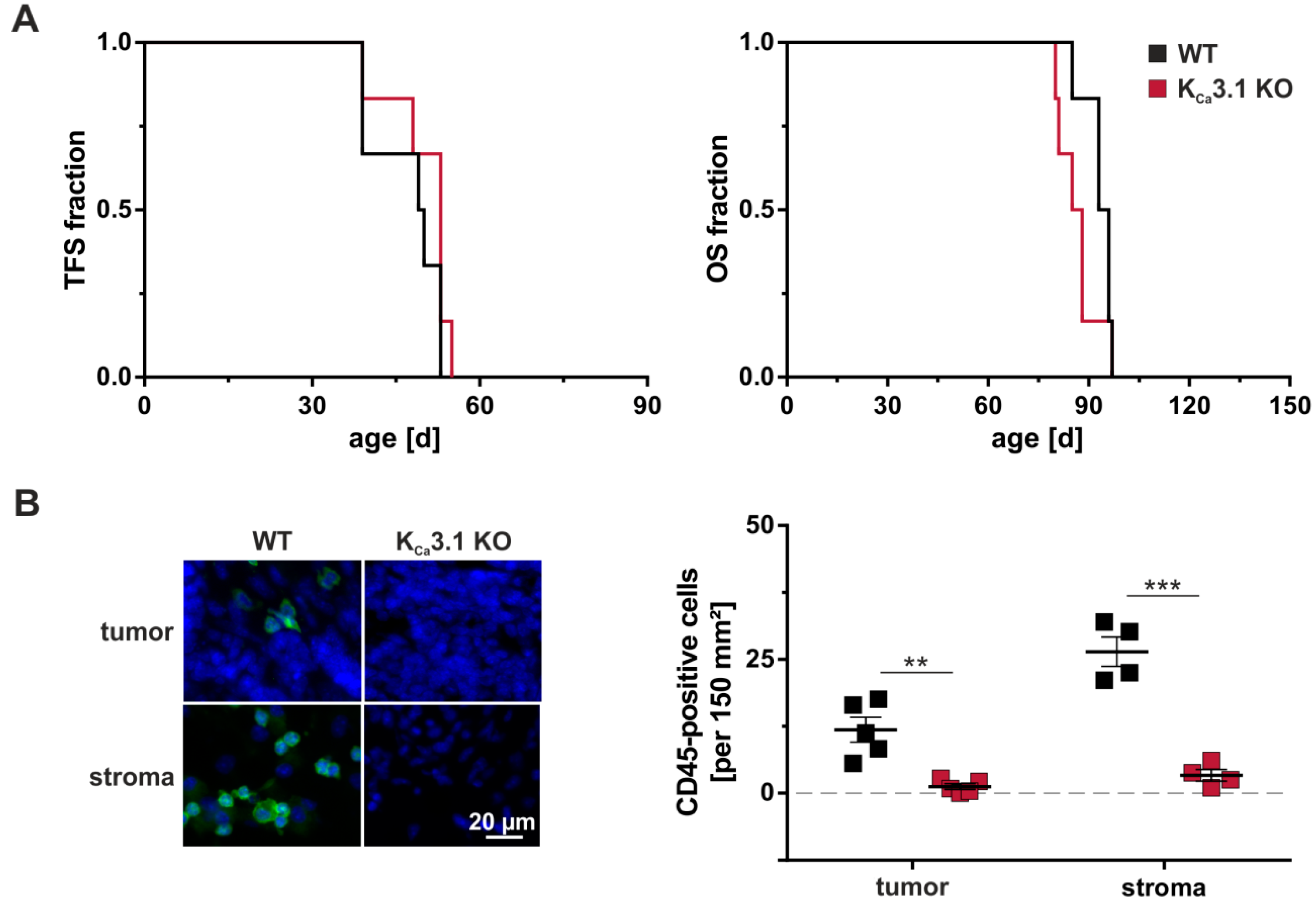
3.1. Tumor Stroma
3.2. Angiogenesis
3.3. The Immune System
3.4. Anti-Cancer Therapy with KCa3.1 Modulators
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vergara, C.; Latorre, R.; Marrion, N.V.; Adelman, J.P. Calcium-activated potassium channels. Curr. Opin. Neurobiol. 1998, 8, 321–329. [Google Scholar] [CrossRef]
- Kohler, M.; Hirschberg, B.; Bond, C.T.; Kinzie, J.M.; Marrion, N.V.; Maylie, J.; Adelman, J.P. Small-conductance, calcium-activated potassium channels from mammalian brain. Science 1996, 273, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Latorre, R.; Oberhauser, A.; Labarca, P.; Alvarez, O. Varieties of calcium-activated potassium channels. Annu. Rev. Physiol. 1989, 51, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Fanger, C.M.; Ghanshani, S.; Logsdon, N.J.; Rauer, H.; Kalman, K.; Zhou, J.; Beckingham, K.; Chandy, K.G.; Cahalan, M.D.; Aiyar, J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J. Biol. Chem. 1999, 274, 5746–5754. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Chantome, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca(2+) channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Panda, S.; Li, Z.; Fuhs, S.R.; Hunter, T.; Thiele, D.J.; Hubbard, S.R.; Skolnik, E.Y. Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.S.; Strobaek, D.; Olesen, S.P.; Christophersen, P. The Ca2+-activated K+ channel of intermediate conductance: A molecular target for novel treatments? Curr. Drug. Targets 2001, 2, 401–422. [Google Scholar] [CrossRef] [PubMed]
- Engbers, J.D.; Anderson, D.; Asmara, H.; Rehak, R.; Mehaffey, W.H.; Hameed, S.; McKay, B.E.; Kruskic, M.; Zamponi, G.W.; Turner, R.W. Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.W.; Kruskic, M.; Teves, M.; Scheidl-Yee, T.; Hameed, S.; Zamponi, G.W. Neuronal expression of the intermediate conductance calcium-activated potassium channel KCa3.1 in the mammalian central nervous system. Pflugers Arch. 2015, 467, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Weisbrod, D.; Peretz, A.; Ziskind, A.; Menaker, N.; Oz, S.; Barad, L.; Eliyahu, S.; Itskovitz-Eldor, J.; Dascal, N.; Khananshvili, D.; et al. SK4 Ca2+ activated K+ channel is a critical player in cardiac pacemaker derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1685–E1694. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.S.; Hertz, M.; Christophersen, P.; Madsen, L.S. The Ca2+-activated K+ channel of intermediate conductance: A possible target for immune suppression. Expert Opin. Ther. Targets 2002, 6, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.B.; Feng, Y.X.; Sun, Q.; Lukowski, R.; Qiu, Y.; Spiger, K.; Li, Z.; Ruth, P.; Korth, M.; Skolnik, E.Y.; et al. Nucleoside diphosphate kinase B-activated intermediate conductance potassium channels are critical for neointima formation in mouse carotid arteries. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1852–1861. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Eichler, I.; Heinau, P.; Si, H.; Brakemeier, S.; Hoyer, J.; Kohler, R. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Kohler, R.; Ruth, P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflugers Arch. 2010, 459, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Panyi, G.; Possani, L.D.; Rodriguez de la Vega, R.C.; Gaspar, R.; Varga, Z. K+ channel blockers: Novel tools to inhibit T cell activation leading to specific immunosuppression. Curr. Pharm. Des. 2006, 12, 2199–2220. [Google Scholar] [CrossRef] [PubMed]
- Grobner, S.; Lukowski, R.; Autenrieth, I.B.; Ruth, P. Lipopolysaccharide induces cell volume increase and migration of dendritic cells. Microbiol. Immunol. 2014, 58, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Shumilina, E.; Lam, R.S.; Wolbing, F.; Matzner, N.; Zemtsova, I.M.; Sobiesiak, M.; Mahmud, H.; Sausbier, U.; Biedermann, T.; Ruth, P.; et al. Blunted IgE-mediated activation of mast cells in mice lacking the Ca2+-activated K.+ channel KCa3.1. J. Immunol. 2008, 180, 8040–8047. [Google Scholar] [CrossRef]
- Yu, Z.H.; Xu, J.R.; Wang, Y.X.; Xu, G.N.; Xu, Z.P.; Yang, K.; Wu, D.Z.; Cui, Y.Y.; Chen, H.Z. Targeted inhibition of KCa3.1 channel attenuates airway inflammation and remodeling in allergic asthma. Am. J. Respir. Cell Mol. Biol. 2013, 48, 685–693. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, L.; McClafferty, H.; Lukowski, R.; MacGregor, D.; King, J.T.; Rizzi, S.; Sausbier, M.; McCobb, D.P.; Knaus, H.G.; et al. Control of hypothalamic-pituitary-adrenal stress axis activity by the intermediate conductance calcium-activated potassium channel, SK4. J. Physiol. 2011, 589, 5965–5986. [Google Scholar] [CrossRef]
- Lu, R.; Flauaus, C.; Kennel, L.; Petersen, J.; Drees, O.; Kallenborn-Gerhardt, W.; Ruth, P.; Lukowski, R.; Schmidtko, A. KCa3.1 channels modulate the processing of noxious chemical stimuli in mice. Neuropharmacology 2017, 125, 386–395. [Google Scholar] [CrossRef]
- Chen, Y.J.; Nguyen, H.M.; Maezawa, I.; Grossinger, E.M.; Garing, A.L.; Kohler, R.; Jin, L.W.; Wulff, H. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2016, 36, 2146–2161. [Google Scholar] [CrossRef]
- Grgic, I.; Kiss, E.; Kaistha, B.P.; Busch, C.; Kloss, M.; Sautter, J.; Muller, A.; Kaistha, A.; Schmidt, C.; Raman, G.; et al. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc. Natl. Acad. Sci. USA 2009, 106, 14518–14523. [Google Scholar] [CrossRef] [PubMed]
- Maher, A.D.; Kuchel, P.W. The Gardos channel: A review of the Ca2+-activated K+ channel in human erythrocytes. Int. J. Biochem. Cell Biol. 2003, 35, 1182–1197. [Google Scholar] [CrossRef]
- Gallagher, P.G. Disorders of erythrocyte hydration. Blood 2017, 130, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Sugunan, S.; Nampoothiri, S.S.; Garg, T.; Krishnamurthy, R.G. Role of KCa3.1 Channels in CNS Diseases: A Concise Review. CNS Neurol. Disord. Drug Targets 2016, 15, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Heyken, W.T.; Wolfle, S.E.; Tysiac, M.; Schubert, R.; Grgic, I.; Vilianovich, L.; Giebing, G.; Maier, T.; Gross, V.; et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ. Res. 2006, 99, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat.Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Lo, W.Y.; Mohr, C.; Steudel, F.; Schmidt, M.; Easton, D.; Hoppe, R.; Schroth, W.; Ruth, P.; Lukowski, R.; Brauch, H.; et al. The role of genetic variation in calcium-activated potassium channels in breast cancer patients treated with tamoxifen. Cancer Res. 2016, 76, 2030. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Nextwork; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Rabjerg, M.; Olivan-Viguera, A.; Hansen, L.K.; Jensen, L.; Sevelsted-Moller, L.; Walter, S.; Jensen, B.L.; Marcussen, N.; Kohler, R. High expression of KCa3.1 in patients with clear cell renal carcinoma predicts high metastatic risk and poor survival. PLoS ONE 2015, 10, e0122992. [Google Scholar] [CrossRef] [PubMed]
- Bulk, E.; Ay, A.S.; Hammadi, M.; Ouadid-Ahidouch, H.; Schelhaas, S.; Hascher, A.; Rohde, C.; Thoennissen, N.H.; Wiewrodt, R.; Schmidt, E.; et al. Epigenetic dysregulation of KCa 3.1 channels induces poor prognosis in lung cancer. Int. J. Cancer 2015, 137, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Jager, H.; Dreker, T.; Buck, A.; Giehl, K.; Gress, T.; Grissmer, S. Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol. Pharmacol. 2004, 65, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Roudbaraki, M.; Delcourt, P.; Ahidouch, A.; Joury, N.; Prevarskaya, N. Functional and molecular identification of intermediate-conductance Ca(2+)-activated K(+) channels in breast cancer cells: Association with cell cycle progression. Am. J. Physiol. Cell Physiol. 2004, 287, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Shen, B.; Yao, H.L.; Jia, Y.C.; Ren, J.; Feng, Y.J.; Wang, Y.Z. Blockage of intermediate-conductance-Ca(2+)-activated K(+) channels inhibits progression of human endometrial cancer. Oncogene 2007, 26, 5107–5114. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kuang, D.; Zhao, X.; Chen, D.; Wang, X.; Yang, Q.; Wan, J.; Zhu, Y.; Wang, Y.; Zhang, S.; et al. miR-497-5p inhibits cell proliferation and invasion by targeting KCa3.1 in angiosarcoma. Oncotarget 2016, 7, 58148–58161. [Google Scholar] [CrossRef] [PubMed]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and downregulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Cittelly, D.M.; Das, P.M.; Salvo, V.A.; Fonseca, J.P.; Burow, M.E.; Jones, F.E. Oncogenic HER2{Delta}16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis 2010, 31, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Venturutti, L.; Huang, Y.W.; Schillaci, R.; Huang, T.H.; Elizalde, P.V. Downregulation of the tumor-suppressor miR-16 via progestin-mediated oncogenic signaling contributes to breast cancer development. Breast Cancer Res. 2012, 14, R77. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, X.; Dhakal, I.B.; Beggs, M.; Kadlubar, S.; Luo, D. Induction of cell proliferation and survival genes by estradiol-repressed microRNAs in breast cancer cells. BMC Cancer 2012, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Pines, J. Cell proliferation and control. Curr. Opin. Cell Biol. 1992, 4, 144–148. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stuhmer, W.; Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130094. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jan, L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric controls of cell proliferation: Ion channels, membrane voltage and the cell cycle. Cell Cycle 2009, 8, 3527–3536. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.A.; Iliev, I.G.; Schwalke, M.A.; Gonzalez, E.; Marler, K.C.; Flanagan, C.A. Association between cell membrane potential and breast cancer. Tumor Biol. 1994, 15, 82–89. [Google Scholar] [CrossRef]
- Woodfork, K.A.; Wonderlin, W.F.; Peterson, V.A.; Strobl, J.S. Inhibition of ATP-sensitive potassium channels causes reversible cell-cycle arrest of human breast cancer cells in tissue culture. J. Cell. Physiol. 1995, 162, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Novel roles for hERG K(+) channels in cell proliferation and apoptosis. Cell Death Dis. 2011, 2, e193. [Google Scholar] [CrossRef] [PubMed]
- Crociani, O.; Guasti, L.; Balzi, M.; Becchetti, A.; Wanke, E.; Olivotto, M.; Wymore, R.S.; Arcangeli, A. Cell cycle-dependent expression of HERG1 and HERG1B isoforms in tumor cells. J. Biol. Chem. 2003, 278, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Roudbaraki, M.; Ahidouch, A.; Delcourt, P.; Prevarskaya, N. Cell-cycle-dependent expression of the large Ca2+-activated K+ channels in breast cancer cells. Biochem. Biophys. Res. Commun. 2004, 316, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Wonderlin, W.F.; Strobl, J.S. Potassium channels, proliferation and G1 progression. J. Membr. Biol. 1996, 154, 91–107. [Google Scholar] [CrossRef]
- Wonderlin, W.F.; Woodfork, K.A.; Strobl, J.S. Changes in membrane potential during the progression of MCF-7 human mammary tumor cells through the cell cycle. J. Cell. Physiol. 1995, 165, 177–185. [Google Scholar] [CrossRef]
- Pillozzi, S.; Brizzi, M.F.; Balzi, M.; Crociani, O.; Cherubini, A.; Guasti, L.; Bartolozzi, B.; Becchetti, A.; Wanke, E.; Bernabei, P.A.; et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 2002, 16, 1791–1798. [Google Scholar] [CrossRef]
- Parihar, A.S.; Coghlan, M.J.; Gopalakrishnan, M.; Shieh, C.C. Effects of intermediate-conductance Ca2+-activated K+ channel modulators on human prostate cancer cell proliferation. Eur. J. Pharmacol. 2003, 471, 157–164. [Google Scholar] [CrossRef]
- Lallet-Daher, H.; Roudbaraki, M.; Bavencoffe, A.; Mariot, P.; Gackiere, F.; Bidaux, G.; Urbain, R.; Gosset, P.; Delcourt, P.; Fleurisse, L.; et al. Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene 2009, 28, 1792–1806. [Google Scholar] [CrossRef]
- Freise, C.; Ruehl, M.; Seehofer, D.; Hoyer, J.; Somasundaram, R. The inhibitor of Ca(2+)-dependent K+ channels TRAM-34 blocks growth of hepatocellular carcinoma cells via downregulation of estrogen receptor alpha mRNA and nuclear factor-kappaB. Investig. New Drugs 2013, 31, 452–457. [Google Scholar] [CrossRef]
- Yang, X.W.; Liu, J.W.; Zhang, R.C.; Yin, Q.; Shen, W.Z.; Yi, J.L. Inhibitory effects of blockage of intermediate conductance Ca(2+)-activated K (+) channels on proliferation of hepatocellular carcinoma cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Thurber, A.E.; Nelson, M.; Frost, C.L.; Levin, M.; Brackenbury, W.J.; Kaplan, D.L. IK channel activation increases tumor growth and induces differential behavioral responses in two breast epithelial cell lines. Oncotarget 2017, 8, 42382–42397. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M. Oncochannels. Cell Calcium 2013, 53, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Kahl, C.R.; Means, A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signaling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Santella, L. The role of calcium in the cell cycle: Facts and hypotheses. Biochem. Biophys. Res.Commun. 1998, 244, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signaling and cell proliferation. Bioessays 1995, 17, 491–500. [Google Scholar] [CrossRef]
- Cook, S.J.; Lockyer, P.J. Recent advances in Ca(2+)-dependent Ras regulation and cell proliferation. Cell Calcium 2006, 39, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Ahidouch, A. K(+) channels and cell cycle progression in tumor cells. Front. Physiol. 2013, 4, 220. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Geerts, D.; Ay, A.S.; Potier-Cartereau, M.; Ahidouch, A.; Ouadid-Ahidouch, H. Functional cooperation between KCa3.1 and TRPC1 channels in human breast cancer: Role in cell proliferation and patient prognosis. Oncotarget 2016, 7, 36419–36435. [Google Scholar] [CrossRef]
- Steudel, F.A.; Mohr, C.J.; Stegen, B.; Nguyen, H.Y.; Barnert, A.; Steinle, M.; Beer-Hammer, S.; Koch, P.; Lo, W.Y.; Schroth, W.; et al. SK4 channels modulate Ca2+ signaling and cell cycle progression in murine breast cancer. Mol. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Butz, L.; Klumpp, L.; Zips, D.; Dittmann, K.; Ruth, P.; Huber, S.M. Ca2+-Activated IK K+ Channel Blockade Radiosensitizes Glioblastoma Cells. Mol. Cancer Res. 2015, 13, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Roles of K+ channels in regulating tumor cell proliferation and apoptosis. Pflugers Arch. 2004, 448, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Strobl, J.S.; Wonderlin, W.F.; Flynn, D.C. Mitogenic signal transduction in human breast cancer cells. Gen. Pharmacol. 1995, 26, 1643–1649. [Google Scholar] [CrossRef]
- Sheng, M.; Greenberg, M.E. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron 1990, 4, 477–485. [Google Scholar] [CrossRef]
- Millership, J.E.; Devor, D.C.; Hamilton, K.L.; Balut, C.M.; Bruce, J.I.; Fearon, I.M. Calcium-activated K+ channels increase cell proliferation independent of K+ conductance. Am. J. Physiol. Cell Physiol. 2011, 300, C792–C802. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Rudkouskaya, A.; Mongin, A.A.; Kuo, Y.H. Calcium-activated potassium channels BK and IK1 are functionally expressed in human gliomas but do not regulate cell proliferation. PLoS ONE 2010, 5, e12304. [Google Scholar] [CrossRef]
- Sassi, N.; De Marchi, U.; Fioretti, B.; Biasutto, L.; Gulbins, E.; Franciolini, F.; Szabo, I.; Zoratti, M. An investigation of the occurrence and properties of the mitochondrial intermediate-conductance Ca2+-activated K+ channel mtKCa3.1. Biochim. Biophys. Acta 2010, 1797, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.W.; Cowley, E.A.; Blay, J.; Linsdell, P. The intermediate conductance Ca2+-activated K+ channel inhibitor TRAM-34 stimulates proliferation of breast cancer cells via activation of estrogen receptors. Br. J. Pharmacol. 2010, 159, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.J.; Zhu, Y.; Zhang, Q.Y.; Mongin, A.A.; Hough, L.B. TRAM-34, a putatively selective blocker of intermediate-conductance, calcium-activated potassium channels, inhibits cytochrome P450 activity. PLoS ONE 2013, 8, e63028. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X. K+ channels in apoptosis. J. Membr. Biol. 2006, 209, 3–20. [Google Scholar] [CrossRef] [PubMed]
- McFerrin, M.B.; Turner, K.L.; Cuddapah, V.A.; Sontheimer, H. Differential role of IK and BK potassium channels as mediators of intrinsic and extrinsic apoptotic cell death. Am. J. Physiol. Cell Physiol. 2012, 303, C1070–C1078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, X.; Yin, Q.; Yi, J.; Shen, W.; Zhao, L.; Zhu, Z.; Liu, J. Inhibition of SK4 Potassium Channels Suppresses Cell Proliferation, Migration and the Epithelial-Mesenchymal Transition in Triple-Negative Breast Cancer Cells. PLoS ONE 2016, 11, e0154471. [Google Scholar] [CrossRef] [PubMed]
- Quast, S.A.; Berger, A.; Buttstadt, N.; Friebel, K.; Schonherr, R.; Eberle, J. General Sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS ONE 2012, 7, e39290. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabo, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009, 45, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.L.; Hasegawa, Y.; Shimizu, T.; Okada, Y. IK1 channel activity contributes to cisplatin sensitivity of human epidermoid cancer cells. Am. J. Physiol. Cell Physiol. 2008, 294, C1398–C1406. [Google Scholar] [CrossRef]
- Lam, J.; Wulff, H. The Lymphocyte Potassium Channels Kv1.3 and KCa3.1 as Targets for Immunosuppression. Drug. Dev. Res. 2011, 72, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Bock, J.; Grassme, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Manago, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531.e10. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Klumpp, L.; Misovic, M.; Edalat, L.; Eckert, M.; Klumpp, D.; Ruth, P.; Huber, S.M. K(+) channel signaling in irradiated tumor cells. Eur. Biophys. J. 2016, 45, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.I.; Higgins, C.F. IKCa1 activity is required for cell shrinkage, phosphatidylserine translocation and death in T lymphocyte apoptosis. EMBO Rep. 2003, 4, 189–194. [Google Scholar] [CrossRef]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ge, L.; Hoa, N.T.; Wilson, Z.; Arismendi-Morillo, G.; Kong, X.T.; Tajhya, R.B.; Beeton, C.; Jadus, M.R. Big Potassium (BK) ion channels in biology, disease and possible targets for cancer immunotherapy. Int. Immunopharmacol. 2014, 22, 427–443. [Google Scholar] [CrossRef]
- Wondergem, R.; Bartley, J.W. Menthol increases human glioblastoma intracellular Ca2+, BK channel activity and cell migration. J. Biomed. Sci. 2009, 16, 90. [Google Scholar] [CrossRef]
- Steinle, M.; Palme, D.; Misovic, M.; Rudner, J.; Dittmann, K.; Lukowski, R.; Ruth, P.; Huber, S.M. Ionizing radiation induces migration of glioblastoma cells by activating BK K(+) channels. Radiother. Oncol. 2011, 101, 122–126. [Google Scholar] [CrossRef]
- Edalat, L.; Stegen, B.; Klumpp, L.; Haehl, E.; Schilbach, K.; Lukowski, R.; Kuhnle, M.; Bernhardt, G.; Buschauer, A.; Zips, D.; et al. BK K+ channel blockade inhibits radiation-induced migration/brain infiltration of glioblastoma cells. Oncotarget 2016, 7, 14259–14278. [Google Scholar] [CrossRef]
- Rosa, P.; Catacuzzeno, L.; Sforna, L.; Mangino, G.; Carlomagno, S.; Mincione, G.; Petrozza, V.; Ragona, G.; Franciolini, F.; Calogero, A. BK channels blockage inhibits hypoxia-induced migration and chemoresistance to cisplatin in human glioblastoma cells. J. Cell Physiol. 2018, 233, 6866–6877. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Catalano, M.; Sciaccaluga, M.; Chece, G.; Cipriani, R.; Rosito, M.; Grimaldi, A.; Lauro, C.; Cantore, G.; Santoro, A.; et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013, 4, e773. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; Limatola, C.; Catalano, M. Functional Roles of the Ca2+-activated K+ Channel, KCa3.1, in Brain Tumors. Curr. Neuropharmacol. 2018, 16, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Fioretti, B.; Franciolini, F. Expression and Role of the Intermediate-Conductance Calcium-Activated Potassium Channel KCa3.1 in Glioblastoma. J. Signal Transduct. 2012, 2012, 421564. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Aiello, F.; Fioretti, B.; Sforna, L.; Castigli, E.; Ruggieri, P.; Tata, A.M.; Calogero, A.; Franciolini, F. Serum-activated K and Cl currents underlay U87-MG glioblastoma cell migration. J. Cell Physiol. 2011, 226, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Sciaccaluga, M.; Fioretti, B.; Catacuzzeno, L.; Pagani, F.; Bertollini, C.; Rosito, M.; Catalano, M.; D’Alessandro, G.; Santoro, A.; Cantore, G.; et al. CXCL12-induced glioblastoma cell migration requires intermediate conductance Ca2+-activated K+ channel activity. Am. J. Physiol. Cell Physiol. 2010, 299, C175–C184. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.L.; Honasoge, A.; Robert, S.M.; McFerrin, M.M.; Sontheimer, H. A proinvasive role for the Ca(2+)-activated K(+) channel KCa3.1 in malignant glioma. Glia 2014, 62, 971–981. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Turner, K.L.; Seifert, S.; Sontheimer, H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 1427–1440. [Google Scholar] [CrossRef]
- Watkins, S.; Sontheimer, H. Hydrodynamic cellular volume changes enable glioma cell invasion. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 17250–17259. [Google Scholar] [CrossRef]
- Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 Channels in Modulating Ca(2+) Oscillations during Glioblastoma Cell Migration and Invasion. Int. J. Mol. Sci. 2018, 19, 2970. [Google Scholar] [CrossRef]
- Catacuzzeno, L.; Caramia, M.; Sforna, L.; Belia, S.; Guglielmi, L.; D’Adamo, M.C.; Pessia, M.; Franciolini, F. Reconciling the discrepancies on the involvement of large-conductance Ca(2+)-activated K channels in glioblastoma cell migration. Front. Cell Neurosci. 2015, 9, 152. [Google Scholar] [CrossRef]
- Rosa, P.; Sforna, L.; Carlomagno, S.; Mangino, G.; Miscusi, M.; Pessia, M.; Franciolini, F.; Calogero, A.; Catacuzzeno, L. Overexpression of Large-Conductance Calcium-Activated Potassium Channels in Human Glioblastoma Stem-Like Cells and Their Role in Cell Migration. J. Cell Physiol. 2017, 232, 2478–2488. [Google Scholar] [CrossRef]
- Ruggieri, P.; Mangino, G.; Fioretti, B.; Catacuzzeno, L.; Puca, R.; Ponti, D.; Miscusi, M.; Franciolini, F.; Ragona, G.; Calogero, A. The inhibition of KCa3.1 channels activity reduces cell motility in glioblastoma derived cancer stem cells. PloS ONE 2012, 7, e47825. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 Channels and Glioblastoma: In Vitro Studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Nechyporuk-Zloy, V.; Fabian, A.; Stock, C. Cells move when ions and water flow. Pflugers Arch. 2007, 453, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Robles-Martinez, L.; Garay, E.; Martel-Gallegos, M.G.; Cisneros-Mejorado, A.; Perez-Montiel, D.; Lara, A.; Arellano, R.O. Kca3.1 Activation Via P2y2 Purinergic Receptors Promotes Human Ovarian Cancer Cell (Skov-3) Migration. Sci. Rep. 2017, 7, 4340. [Google Scholar] [CrossRef]
- Schwab, A.; Reinhardt, J.; Schneider, S.W.; Gassner, B.; Schuricht, B. K(+) channel-dependent migration of fibroblasts and human melanoma cells. Cell. Physiol. Biochem. 1999, 9, 126–132. [Google Scholar] [CrossRef]
- Qiao, A.; Gu, F.; Guo, X.; Zhang, X.; Fu, L. Breast cancer-associated fibroblasts: Their roles in tumor initiation, progression and clinical applications. Front. Med. 2016, 10, 33–40. [Google Scholar] [CrossRef]
- Pena, T.L.; Chen, S.H.; Konieczny, S.F.; Rane, S.G. Ras/MEK/ERK Upregulation of the fibroblast KCa channel FIK is a common mechanism for basic fibroblast growth factor and transforming growth factor-beta suppression of myogenesis. J. Biol. Chem. 2000, 275, 13677–13682. [Google Scholar] [CrossRef]
- Wang, L.P.; Wang, Y.; Zhao, L.M.; Li, G.R.; Deng, X.L. Angiotensin II upregulates K(Ca)3.1 channels and stimulates cell proliferation in rat cardiac fibroblasts. Biochem. Pharmacol. 2013, 85, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.M.; Zhang, W.; Wang, L.P.; Li, G.R.; Deng, X.L. Advanced glycation end products promote proliferation of cardiac fibroblasts by upregulation of KCa3.1 channels. Pflugers Arch. 2012, 464, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Friebel, K.; Schonherr, R.; Kinne, R.W.; Kunisch, E. Functional role of the KCa3.1 potassium channel in synovial fibroblasts from rheumatoid arthritis patients. J. Cell. Physiol. 2015, 230, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.; Bachelot, T.; Hudis, C.A.; Curigliano, G.; Reynolds, A.R.; Petrioli, R.; Generali, D. The role of bevacizumab in solid tumors: A literature based meta-analysis of randomised trials. Eur. J. Cancer 2017, 75, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xia, J.; Yang, X.; Huang, X.; Gao, D.; Zhou, J.; Lian, J.; Zhou, J. Intermediate-conductance Ca(2+)-activated potassium and volume-sensitive chloride channels in endothelial progenitor cells from rat bone marrow mononuclear cells. Acta Physiol. 2012, 205, 302–313. [Google Scholar] [CrossRef]
- Cheong, A.; Bingham, A.J.; Li, J.; Kumar, B.; Sukumar, P.; Munsch, C.; Buckley, N.J.; Neylon, C.B.; Porter, K.E.; Beech, D.J.; et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol. Cell 2005, 20, 45–52. [Google Scholar] [CrossRef]
- Kohler, R.; Degenhardt, C.; Kuhn, M.; Runkel, N.; Paul, M.; Hoyer, J. Expression and function of endothelial Ca(2+)-activated K(+) channels in human mesenteric artery: A single-cell reverse transcriptase-polymerase chain reaction and electrophysiological study in situ. Circ. Res. 2000, 87, 496–503. [Google Scholar] [CrossRef]
- Yang, H.; Li, X.; Ma, J.; Lv, X.; Zhao, S.; Lang, W.; Zhang, Y. Blockade of the intermediate-conductance Ca(2+)-activated K+ channel inhibits the angiogenesis induced by epidermal growth factor in the treatment of corneal alkali burn. Exp. Eye Res. 2013, 110, 76–87. [Google Scholar] [CrossRef]
- Bi, D.; Toyama, K.; Lemaitre, V.; Takai, J.; Fan, F.; Jenkins, D.P.; Wulff, H.; Gutterman, D.D.; Park, F.; Miura, H. The intermediate conductance calcium-activated potassium channel KCa3.1 regulates vascular smooth muscle cell proliferation via controlling calcium-dependent signaling. J. Biol. Chem. 2013, 288, 15843–15853. [Google Scholar] [CrossRef] [PubMed]
- Ghanshani, S.; Wulff, H.; Miller, M.J.; Rohm, H.; Neben, A.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Upregulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 2000, 275, 37137–37149. [Google Scholar] [CrossRef]
- Khanna, R.; Chang, M.C.; Joiner, W.J.; Kaczmarek, L.K.; Schlichter, L.C. hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J. Biol. Chem. 1999, 274, 14838–14849. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Knaus, H.G.; Pennington, M.; Chandy, K.G. K+ channel expression during B cell differentiation: Implications for immunomodulation and autoimmunity. J Immunol 2004, 173, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, S.A.; Neumeier, L.; Peng, Y.; Devor, D.C.; Conforti, L. The Ca(2+)-activated K(+) channel KCa3.1 compartmentalizes in the immunological synapse of human T lymphocytes. Am. J. Physiol. Cell Physiol. 2007, 292, C1431–C1439. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Calabresi, P.A.; Allie, R.; Yun, S.; Pennington, M.; Beeton, C.; Chandy, K.G. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J. Clin. Investig. 2003, 111, 1703–1713. [Google Scholar] [CrossRef]
- Varga, Z.; Csepany, T.; Papp, F.; Fabian, A.; Gogolak, P.; Toth, A.; Panyi, G. Potassium channel expression in human CD4+ regulatory and naive T cells from healthy subjects and multiple sclerosis patients. Immunol. Lett. 2009, 124, 95–101. [Google Scholar] [CrossRef]
- Di, L.; Srivastava, S.; Zhdanova, O.; Ding, Y.; Li, Z.; Wulff, H.; Lafaille, M.; Skolnik, E.Y. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc. Natl. Acad. Sci. USA 2010, 107, 1541–1546. [Google Scholar] [CrossRef]
- Kuras, Z.; Yun, Y.H.; Chimote, A.A.; Neumeier, L.; Conforti, L. KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PLoS ONE 2012, 7, e43859. [Google Scholar] [CrossRef]
- Kim, U.; Siegel, R.; Ren, X.; Gunther, C.S.; Gaasterland, T.; Roeder, R.G. Identification of transcription coactivator OCA-B-dependent genes involved in antigen-dependent B cell differentiation by cDNA array analyses. Proc. Natl. Acad. Sci. USA 2003, 100, 8868–8873. [Google Scholar] [CrossRef]
- Rodriguez-Cortez, V.C.; Del Pino-Molina, L.; Rodriguez-Ubreva, J.; Ciudad, L.; Gomez-Cabrero, D.; Company, C.; Urquiza, J.M.; Tegner, J.; Rodriguez-Gallego, C.; Lopez-Granados, E.; et al. Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naive-to-memory B-cell transition. Nat. Commun. 2015, 6, 7335. [Google Scholar] [CrossRef] [PubMed]
- Grossinger, E.M.; Weiss, L.; Zierler, S.; Rebhandl, S.; Krenn, P.W.; Hinterseer, E.; Schmolzer, J.; Asslaber, D.; Hainzl, S.; Neureiter, D.; et al. Targeting proliferation of chronic lymphocytic leukemia (CLL) cells through KCa3.1 blockade. Leukemia 2014, 28, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Makinde, T.O.; Agrawal, D.K. Calcium-activated potassium channel KCa3.1 in lung dendritic cell migration. Am. J. Respir. Cell Mol. Biol. 2011, 45, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Crottes, D.; Felix, R.; Meley, D.; Chadet, S.; Herr, F.; Audiger, C.; Soriani, O.; Vandier, C.; Roger, S.; Angoulvant, D.; et al. Immature human dendritic cells enhance their migration through KCa3.1 channel activation. Cell Calcium 2016, 59, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Hanley, P.J.; Musset, B.; Renigunta, V.; Limberg, S.H.; Dalpke, A.H.; Sus, R.; Heeg, K.M.; Preisig-Muller, R.; Daut, J. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc. Natl. Acad. Sci. USA 2004, 101, 9479–9484. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lai, W.; Zhang, Y.; Liu, L.; Luo, X.; Zeng, Y.; Wu, H.; Lan, Q.; Chu, Z. Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer 2014, 14, 330. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Koeberle, P.D.; Wang, Y.; Schlichter, L.C. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J. Neurosci. 2007, 27, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Lively, S.; Schlichter, L.C. IL-4 type 1 receptor signaling upregulates KCNN4 expression, and increases the KCa3.1 current and its contribution to migration of alternative-activated microglia. Front. Cell. Neurosci. 2014, 8, 183. [Google Scholar] [CrossRef]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011, 6, e23902. [Google Scholar] [CrossRef]
- Grimaldi, A.; D’Alessandro, G.; Golia, M.T.; Grossinger, E.M.; Di Angelantonio, S.; Ragozzino, D.; Santoro, A.; Esposito, V.; Wulff, H.; Catalano, M.; et al. KCa3.1 inhibition switches the phenotype of glioma-infiltrating microglia/macrophages. Cell Death Dis. 2016, 7, e2174. [Google Scholar] [CrossRef] [PubMed]
- Koshy, S.; Wu, D.; Hu, X.; Tajhya, R.B.; Huq, R.; Khan, F.S.; Pennington, M.W.; Wulff, H.; Yotnda, P.; Beeton, C. Blocking KCa3.1 channels increases tumor cell killing by a subpopulation of human natural killer lymphocytes. PLoS ONE 2013, 8, e76740. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, C.; Riquelme, T.T.; Vera, D.; Julio-Kalajzic, F.; Ehrenfeld, P.; Melvin, J.E.; Figueroa, C.D.; Sarmiento, J.; Flores, C.A. The calcium-activated potassium channel KCa3.1 plays a central role in the chemotactic response of mammalian neutrophils. Acta Physiol. 2016, 216, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Mark Duffy, S.; Berger, P.; Cruse, G.; Yang, W.; Bolton, S.J.; Bradding, P. The K+ channel iKCA1 potentiates Ca2+ influx and degranulation in human lung mast cells. J. Allergy Clin. Immunol. 2004, 114, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Cruse, G.; Duffy, S.M.; Brightling, C.E.; Bradding, P. Functional KCa3.1 K+ channels are required for human lung mast cell migration. Thorax 2006, 61, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Miller, M.J.; Hansel, W.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 2000, 97, 8151–8156. [Google Scholar] [CrossRef]
- McNaughton-Smith, G.A.; Burns, J.F.; Stocker, J.W.; Rigdon, G.C.; Creech, C.; Arrington, S.; Shelton, T.; de Franceschi, L. Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J. Med. Chem. 2008, 51, 976–982. [Google Scholar] [CrossRef]
- Ishii, T.M.; Silvia, C.; Hirschberg, B.; Bond, C.T.; Adelman, J.P.; Maylie, J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA 1997, 94, 11651–11656. [Google Scholar] [CrossRef]
- Benzaquen, L.R.; Brugnara, C.; Byers, H.R.; Gatton-Celli, S.; Halperin, J.A. Clotrimazole inhibits cell proliferation in vitro and in vivo. Nat. Med. 1995, 1, 534–540. [Google Scholar] [CrossRef]
- Khalid, M.H.; Shibata, S.; Hiura, T. Effects of clotrimazole on the growth, morphological characteristics, and cisplatin sensitivity of human glioblastoma cells in vitro. J. Neurosurg. 1999, 90, 918–927. [Google Scholar] [CrossRef]
- Jensen, B.S.; Odum, N.; Jorgensen, N.K.; Christophersen, P.; Olesen, S.P. Inhibition of T cell proliferation by selective block of Ca(2+)-activated K(+) channels. Proc. Natl. Acad. Sci. USA 1999, 96, 10917–10921. [Google Scholar] [CrossRef] [PubMed]
- Toyama, K.; Wulff, H.; Chandy, K.G.; Azam, P.; Raman, G.; Saito, T.; Fujiwara, Y.; Mattson, D.L.; Das, S.; Melvin, J.E.; et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J. Clin. Investig. 2008, 118, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Bonito, B.; Sauter, D.R.; Schwab, A.; Djamgoz, M.B.; Novak, I. KCa3.1 (IK) modulates pancreatic cancer cell migration, invasion and proliferation: Anomalous effects on TRAM-34. Pflugers Arch. 2016, 468, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Du, Y.; Song, W.; Chen, H.; Xuan, Z.; Zhao, L.; Chen, J.; Chen, J.; Guo, D.; Jin, C.; et al. KCa3.1 as an Effective Target for Inhibition of Growth and Progression of Intrahepatic Cholangiocarcinoma. J. Cancer 2017, 8, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Orringer, E.P.; Styles, L.; Vichinsky, E.P.; Swerdlow, P.; Davis, G.A.; Desimone, P.A.; Stocker, J.W. Dose-escalation study of ICA-17043 in patients with sickle cell disease. Pharmacotherapy 2006, 26, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Stocker, J.W.; De Franceschi, L.; McNaughton-Smith, G.A.; Corrocher, R.; Beuzard, Y.; Brugnara, C. ICA-17043, a novel Gardos channel blocker, prevents sickled red blood cell dehydration in vitro and in vivo in SAD mice. Blood 2003, 101, 2412–2418. [Google Scholar] [CrossRef]
- Chen, Y.J.; Raman, G.; Bodendiek, S.; O’Donnell, M.E.; Wulff, H. The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2011, 31, 2363–2374. [Google Scholar] [CrossRef]
- Ataga, K.I.; Smith, W.R.; De Castro, L.M.; Swerdlow, P.; Saunthararajah, Y.; Castro, O.; Vichinsky, E.; Kutlar, A.; Orringer, E.P.; Rigdon, G.C.; et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood 2008, 111, 3991–3997. [Google Scholar] [CrossRef]
- Nagalla, S.; Ballas, S.K. Drugs for preventing red blood cell dehydration in people with sickle cell disease. Cochrane Database Syst. Rev. 2018, 10, CD003426. [Google Scholar] [CrossRef]
- Ataga, K.I.; Reid, M.; Ballas, S.K.; Yasin, Z.; Bigelow, C.; James, L.S.; Smith, W.R.; Galacteros, F.; Kutlar, A.; Hull, J.H.; et al. Improvements in hemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: A phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). Br. J. Hematol. 2011, 153, 92–104. [Google Scholar] [CrossRef]
- Devor, D.C.; Singh, A.K.; Frizzell, R.A.; Bridges, R.J. Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca(2+)-dependent K+ channel. Am. J. Physiol. 1996, 271, L775–L784. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Brown, B.M.; Olivan-Viguera, A.; Singh, V.; Olmstead, M.M.; Valero, M.S.; Kohler, R.; Wulff, H. New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3.1. Mol. Pharmacol. 2014, 86, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.Z.; Park, S.W.; Kang, T.W.; Kim, K.S.; Yoo, H.Y.; Lee, J.; Hah, J.H.; Sung, M.H.; Kim, S.J. Activation of K(+) channel by 1-EBIO rescues the head and neck squamous cell carcinoma cells from Ca(2+) ionophore-induced cell death. Korean J. Physiol. Pharmacol. 2016, 20, 25–33. [Google Scholar] [CrossRef]
- King, B.; Rizwan, A.P.; Asmara, H.; Heath, N.C.; Engbers, J.D.; Dykstra, S.; Bartoletti, T.M.; Hameed, S.; Zamponi, G.W.; Turner, R.W. IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep. 2015, 11, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, A.; Raman, G.; Busch, C.; Schultz, T.; Zimin, P.I.; Hoyer, J.; Kohler, R.; Wulff, H. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol. Pharmacol. 2009, 75, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Morimura, K.; Yamamura, H.; Ohya, S.; Imaizumi, Y. Voltage-dependent Ca2+-channel block by openers of intermediate and small conductance Ca2+-activated K+ channels in urinary bladder smooth muscle cells. J. Pharmacol. Sci. 2006, 100, 237–241. [Google Scholar] [CrossRef]
- Schroder, R.L.; Jensen, B.S.; Strobaek, D.; Olesen, S.P.; Christophersen, P. Activation of the human, intermediate-conductance, Ca2+-activated K+ channel by methylxanthines. Pflugers Arch. 2000, 440, 809–818. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, L.; Ma, W.; Cao, X.; Chen, H.; Feng, D.; Liang, J.; Yin, K.; Jiang, X. The Blockage of KCa3.1 Channel Inhibited Proliferation, Migration and Promoted Apoptosis of Human Hepatocellular Carcinoma Cells. J. Cancer 2015, 6, 643–651. [Google Scholar] [CrossRef]
- Brown, B.M.; Pressley, B.; Wulff, H. KCa3.1 Channel Modulators as Potential Therapeutic Compounds for Glioblastoma. Curr. Neuropharmacol. 2018, 16, 618–626. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohr, C.J.; Steudel, F.A.; Gross, D.; Ruth, P.; Lo, W.-Y.; Hoppe, R.; Schroth, W.; Brauch, H.; Huber, S.M.; Lukowski, R. Cancer-Associated Intermediate Conductance Ca2+-Activated K+ Channel KCa3.1. Cancers 2019, 11, 109. https://doi.org/10.3390/cancers11010109
Mohr CJ, Steudel FA, Gross D, Ruth P, Lo W-Y, Hoppe R, Schroth W, Brauch H, Huber SM, Lukowski R. Cancer-Associated Intermediate Conductance Ca2+-Activated K+ Channel KCa3.1. Cancers. 2019; 11(1):109. https://doi.org/10.3390/cancers11010109
Chicago/Turabian StyleMohr, Corinna J., Friederike A. Steudel, Dominic Gross, Peter Ruth, Wing-Yee Lo, Reiner Hoppe, Werner Schroth, Hiltrud Brauch, Stephan M. Huber, and Robert Lukowski. 2019. "Cancer-Associated Intermediate Conductance Ca2+-Activated K+ Channel KCa3.1" Cancers 11, no. 1: 109. https://doi.org/10.3390/cancers11010109
APA StyleMohr, C. J., Steudel, F. A., Gross, D., Ruth, P., Lo, W.-Y., Hoppe, R., Schroth, W., Brauch, H., Huber, S. M., & Lukowski, R. (2019). Cancer-Associated Intermediate Conductance Ca2+-Activated K+ Channel KCa3.1. Cancers, 11(1), 109. https://doi.org/10.3390/cancers11010109