FOSB–PCDHB13 Axis Disrupts the Microtubule Network in Non-Small Cell Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Mitochondrial Stress-Induced FOSB Expression Requires Ca2+ Signaling
2.2. FOSB Regulates Cellular Integrity in NSCLC
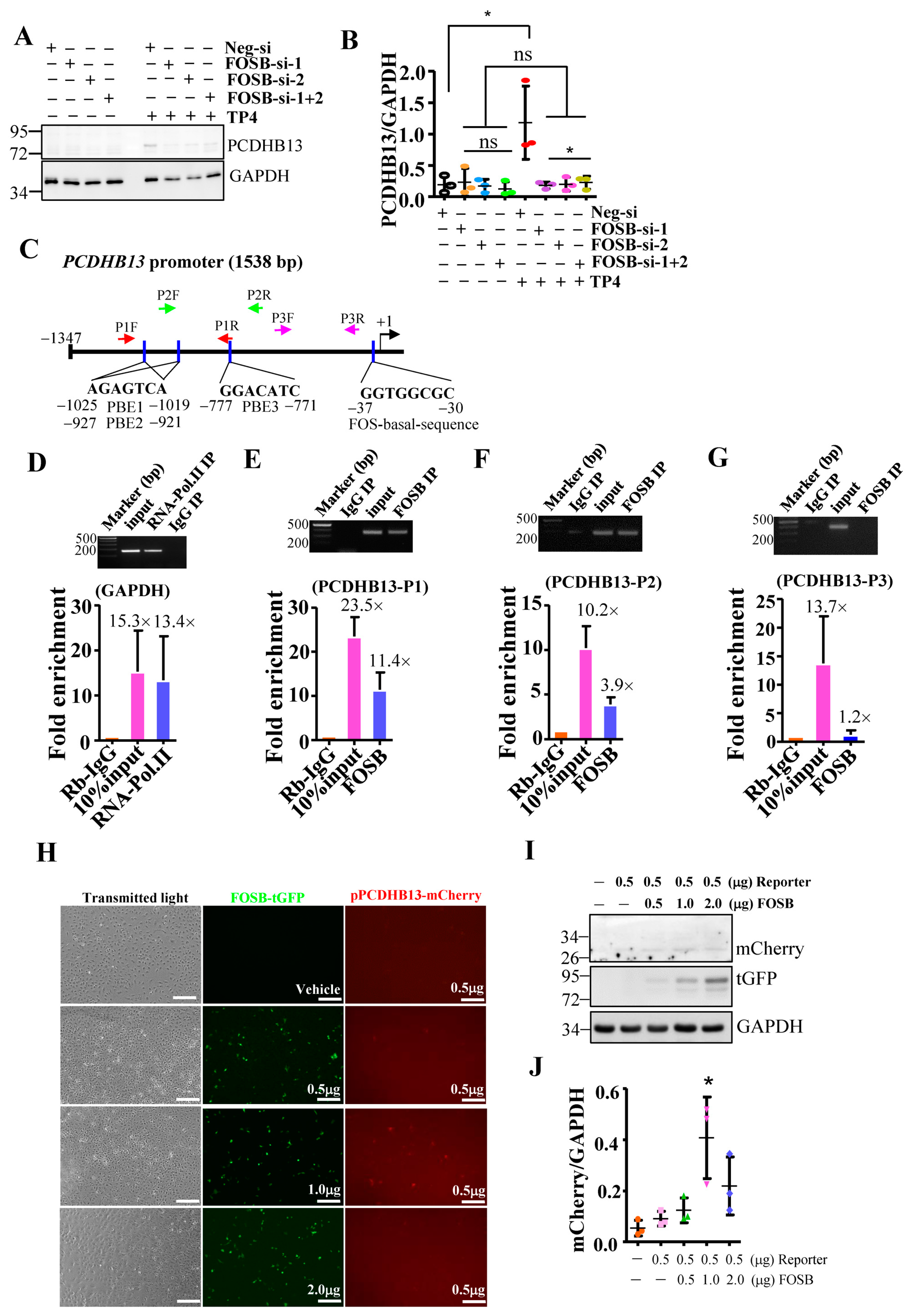
2.3. PCDHB13 as a Downstream Target of FOSB
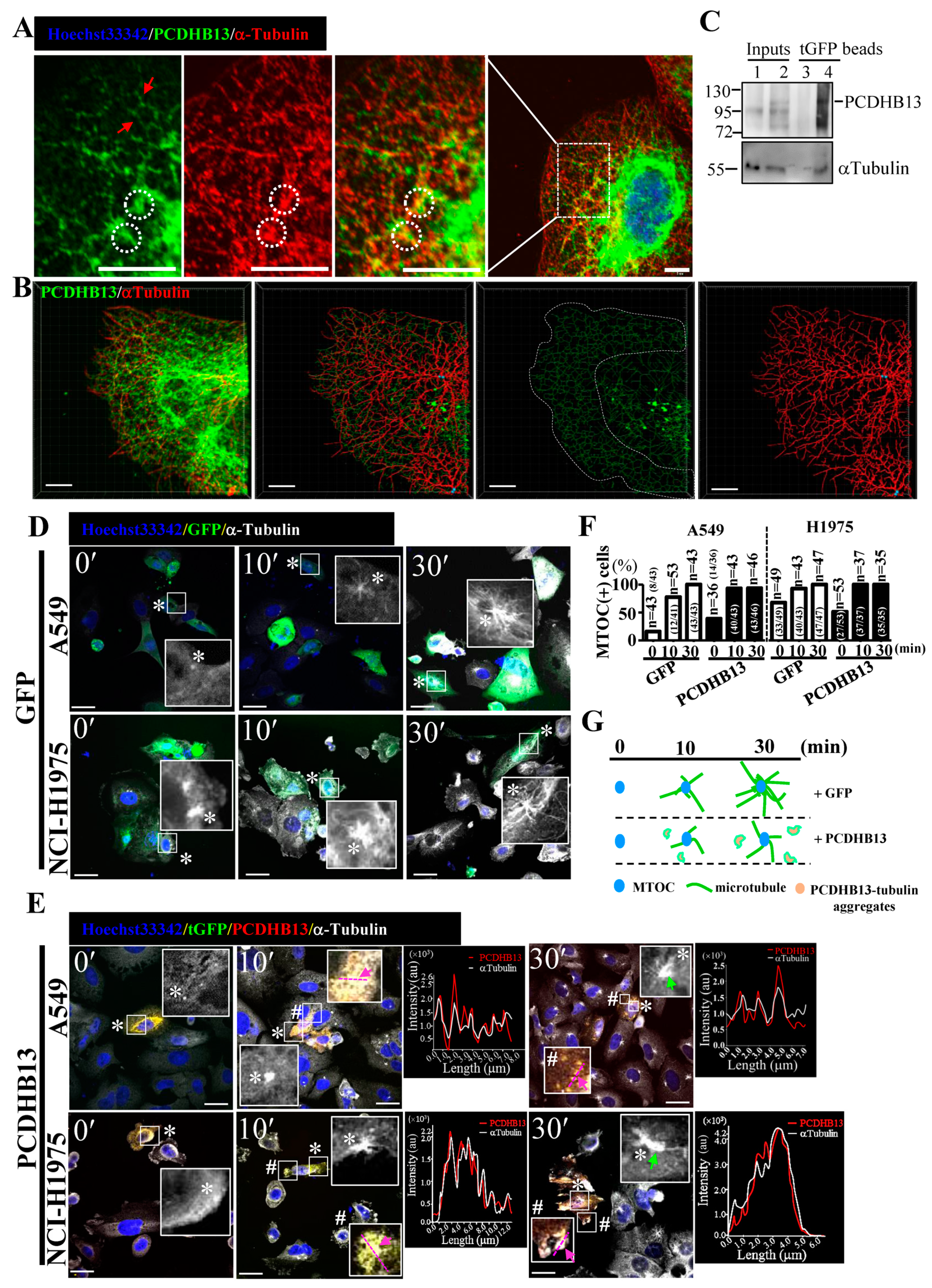
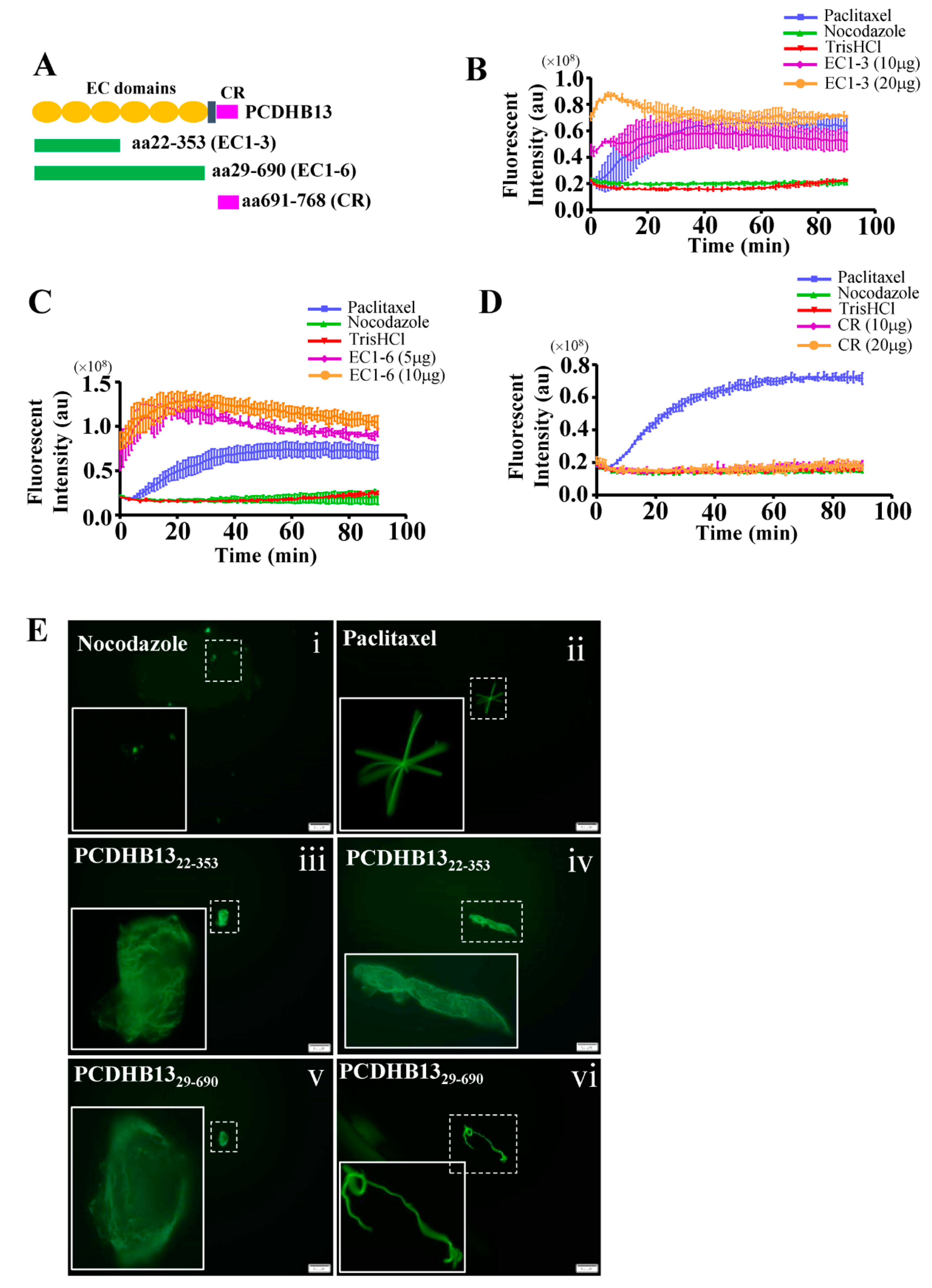
2.4. PCDHB13 Affects Microtubule Dynamics
2.5. Suppressive effects of PCDHB13 in NSCLC
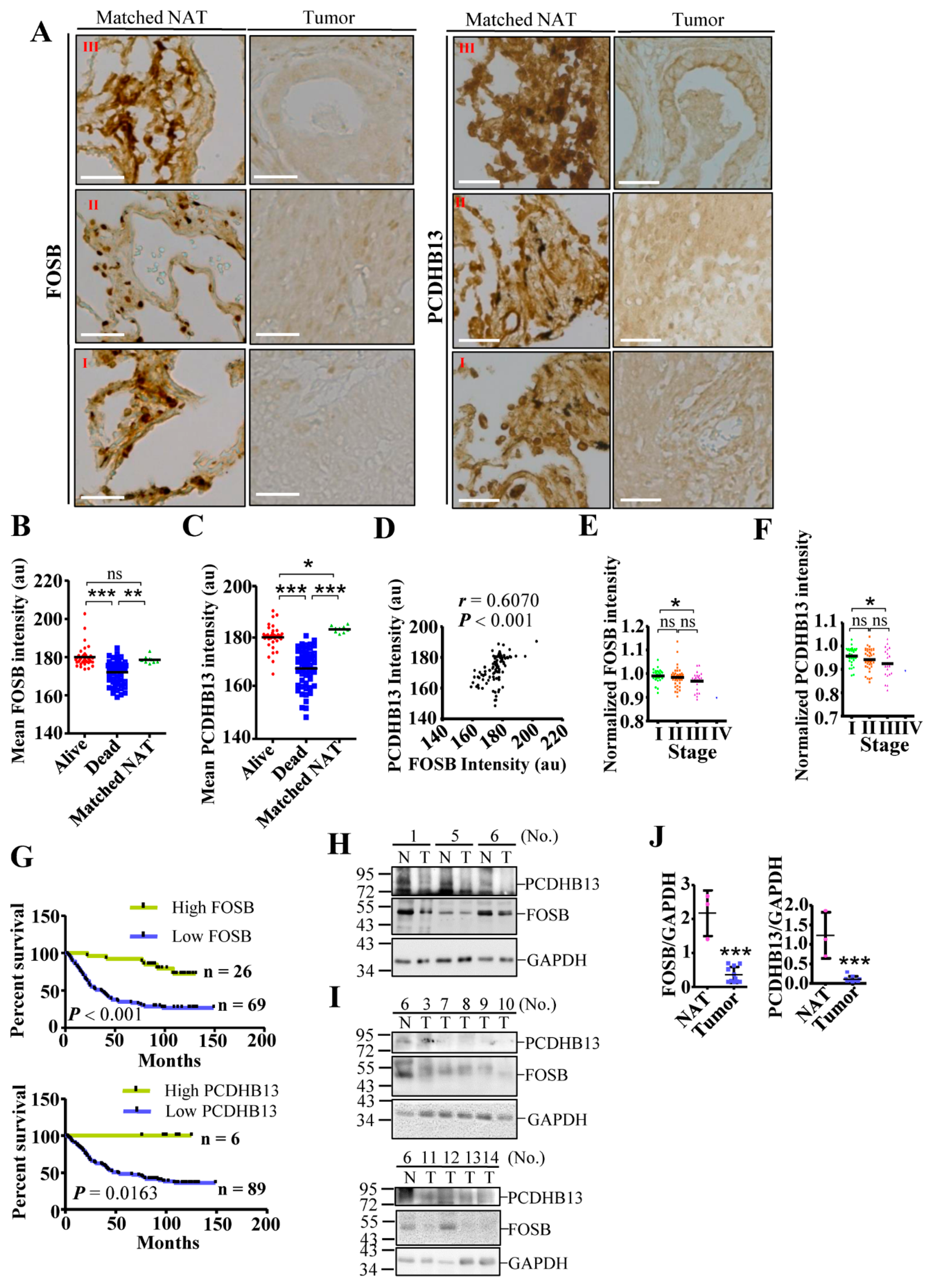
2.6. Dysregulation of PCDHB13 and FOSB in NSCLC
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. Reagents and Plasmids
4.3. Cell Culture and Knockdown Assay
4.4. Cell Viability Assay
4.5. Transcriptome Analysis
4.6. Chromatin Immunoprecipitation and Quantitative-PCR
4.7. Tissue Fractionation, Co-Immunoprecipitation, and Western Blot
4.8. Calcium Measurement
4.9. Mitochondrial Activity Assay
4.10. Reporter Analysis
4.11. Immunocytochemical and Immunohistochemical Studies
4.12. Microtubule Regrowth and In Vitro Tubulin Polymerization Assay
4.13. Cell Invasion Assay
4.14. Zebrafish Xenotransplantation Model
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Chemotherapy Resistance in Lung Cancer. Adv. Exp. Med. Biol. 2016, 893, 189–209. [Google Scholar] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Sequist, L.V.; Soria, J.C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Hilchie, A.L.; Doucette, C.D.; Pinto, D.M.; Patrzykat, A.; Douglas, S.; Hoskin, D.W. Pleurocidin-family cationic antimicrobial peptides are cytolytic for breast carcinoma cells and prevent growth of tumor xenografts. Breast Cancer Res. 2011, 13, R102. [Google Scholar] [CrossRef]
- Ting, C.H.; Huang, H.N.; Huang, T.C.; Wu, C.J.; Chen, J.Y. The mechanisms by which pardaxin, a natural cationic antimicrobial peptide, targets the endoplasmic reticulum and induces c-FOS. Biomaterials 2014, 35, 3627–3640. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.H.; Chen, Y.C.; Wu, C.J.; Chen, J.Y. Targeting FOSB with a cationic antimicrobial peptide, TP4, for treatment of triple-negative breast cancer. Oncotarget 2016, 7, 40329–40347. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.H.; Liu, Y.C.; Lyu, P.C.; Chen, J.Y. Nile Tilapia Derived Antimicrobial Peptide TP4 Exerts Antineoplastic Activity Through Microtubule Disruption. Mar. Drugs 2018, 16, 462. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Le, W.; Wang, Y.; Li, Z.; Wang, D.; Ren, L.; Lin, L.; Cui, S.; Hu, J.J.; Hu, Y.; et al. Targeting Negative Surface Charges of Cancer Cells by Multifunctional Nanoprobes. Theranostics 2016, 6, 1887–1898. [Google Scholar] [CrossRef]
- Ting, C.H.; Chen, J.Y. Nile Tilapia Derived TP4 Shows Broad Cytotoxicity Toward to Non-Small-Cell Lung Cancer Cells. Mar. Drugs 2018, 16, 506. [Google Scholar] [CrossRef]
- Kucharska-Newton, A.M.; Rosamond, W.D.; Schroeder, J.C.; McNeill, A.M.; Coresh, J.; Folsom, A.R. HDL-cholesterol and the incidence of lung cancer in the Atherosclerosis Risk in Communities (ARIC) study. Lung Cancer 2008, 61, 292–300. [Google Scholar] [CrossRef]
- Ayyub, A.; Saleem, M.; Fatima, I.; Tariq, A.; Hashmi, N.; Musharraf, S.G. Glycosylated Alpha-1-acid glycoprotein 1 as a potential lung cancer serum biomarker. Int. J. Biochem. Cell Biol. 2016, 70, 68–75. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Bamberger, A.M.; Methner, C.; Lisboa, B.W.; Stadtler, C.; Schulte, H.M.; Loning, T.; Milde-Langosch, K. Expression pattern of the AP-1 family in breast cancer: Association of fosB expression with a well-differentiated, receptor-positive tumor phenotype. Int. J. Cancer 1999, 84, 533–538. [Google Scholar] [CrossRef]
- Milde-Langosch, K.; Kappes, H.; Riethdorf, S.; Loning, T.; Bamberger, A.M. FosB is highly expressed in normal mammary epithelia, but down-regulated in poorly differentiated breast carcinomas. Breast Cancer Res. Treat. 2003, 77, 265–275. [Google Scholar] [CrossRef]
- Daraselia, N.; Wang, Y.P.; Budoff, A.; Lituev, A.; Potapova, O.; Vansant, G.; Monforte, J.; Mazo, I.; Ossovskaya, V.S. Molecular signature and pathway analysis of human primary squamous and adenocarcinoma lung cancers. Am. J. Cancer Res. 2012, 2, 93–103. [Google Scholar] [PubMed]
- Ohnishi, Y.N.; Sakumi, K.; Yamazaki, K.; Ohnishi, Y.H.; Miura, T.; Tominaga, Y.; Nakabeppu, Y. Antagonistic Regulation of Cell-Matrix Adhesion by FosB and Delta FosB/Delta 2 Delta FosB Encoded by Alternatively Spliced Forms of fosB Transcripts. Mol. Biol. Cell 2008, 19, 4717–4729. [Google Scholar] [CrossRef] [PubMed]
- Lis, R.; Karrasch, C.C.; Poulos, M.G.; Kunar, B.; Redmond, D.; Duran, J.G.B.; Badwe, C.R.; Schachterle, W.; Ginsberg, M.; Xiang, J.; et al. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature 2017, 545, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Qiao, Y.C.; Jonsson, P.; Wang, J.; Xu, L.; Rouhi, P.; Sinha, I.; Cao, Y.H.; Williams, C.; Dahlman-Wright, K. Genome-wide Profiling of AP-1-Regulated Transcription Provides Insights into the Invasiveness of Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 3983–3994. [Google Scholar] [CrossRef]
- Li, L.; Bennett, S.A.L.; Wang, L.S. Role of E-cadherin and other cell adhesion molecules in survival and differentiation of human pluripotent stem cells. Cell Adhes. Migr. 2012, 6, 59–70. [Google Scholar] [CrossRef]
- Dallosso, A.R.; Hancock, A.L.; Szemes, M.; Moorwood, K.; Chilukamarri, L.; Tsai, H.H.; Sarkar, A.; Barasch, J.; Vuononvirta, R.; Jones, C.; et al. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor. PLoS Genet. 2009, 5, e1000745. [Google Scholar] [CrossRef]
- Wang, K.H.; Lin, C.J.; Liu, C.J.; Liu, D.W.; Huang, R.L.; Ding, D.C.; Weng, C.F.; Chu, T.Y. Global methylation silencing of clustered proto-cadherin genes in cervical cancer: Serving as diagnostic markers comparable to HPV. Cancer Med. 2015, 4, 43–55. [Google Scholar] [CrossRef]
- Dallosso, A.R.; Oster, B.; Greenhough, A.; Thorsen, K.; Curry, T.J.; Owen, C.; Hancock, A.L.; Szemes, M.; Paraskeva, C.; Frank, M.; et al. Long-range epigenetic silencing of chromosome 5q31 protocadherins is involved in early and late stages of colorectal tumorigenesis through modulation of oncogenic pathways. Oncogene 2012, 31, 4409–4419. [Google Scholar] [CrossRef]
- Banelli, B.; Brigati, C.; Di Vinci, A.; Casciano, I.; Forlani, A.; Borzi, L.; Allemanni, G.; Romani, M. A pyrosequencing assay for the quantitative methylation analysis of the PCDHB gene cluster, the major factor in neuroblastoma methylator phenotype. Lab. Investig. 2012, 92, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Banelli, B.; Romani, M. Quantitative Methylation Analysis of the PCDHB Gene Cluster. Methods Mol. Biol. 2015, 1315, 189–200. [Google Scholar] [PubMed]
- Guo, H.; Nairn, A.; dela Rosa, M.; Nagy, T.; Zhao, S.; Moremen, K.; Pierce, M. Transcriptional regulation of the protocadherin beta cluster during Her-2 protein-induced mammary tumorigenesis results from altered N-glycan branching. J. Biol. Chem. 2012, 287, 24941–24954. [Google Scholar] [CrossRef] [PubMed]
- Castillo, S.D.; Matheu, A.; Mariani, N.; Carretero, J.; Lopez-Rios, F.; Lovell-Badge, R.; Sanchez-Cespedes, M. Novel transcriptional targets of the SRY-HMG box transcription factor SOX4 link its expression to the development of small cell lung cancer. Cancer Res. 2012, 72, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D. Object-oriented Transcription Factors Database (ooTFD). Nucleic Acids Res. 2000, 28, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef]
- Shimizu, Y.; Kinoshita, I.; Kikuchi, J.; Yamazaki, K.; Nishimura, M.; Birrer, M.J.; Dosaka-Akita, H. Growth inhibition of non-small cell lung cancer cells by AP-1 blockade using a cJun dominant-negative mutant. Br. J. Cancer 2008, 98, 915–922. [Google Scholar] [CrossRef]
- Tichelaar, J.W.; Yan, Y.; Tan, Q.; Wang, Y.; Estensen, R.D.; Young, M.R.; Colburn, N.H.; Yin, H.; Goodin, C.; Anderson, M.W.; et al. A dominant-negative c-jun mutant inhibits lung carcinogenesis in mice. Cancer Prev. Res. 2010, 3, 1148–1156. [Google Scholar] [CrossRef]
- Wang, L.Q.; Zhao, L.H.; Qiao, Y.Z. Identification of potential therapeutic targets for lung cancer by bioinformatics analysis. Mol. Med. Rep. 2016, 13, 1975–1982. [Google Scholar] [CrossRef]
- Risolino, M.; Mandia, N.; Iavarone, F.; Dardaei, L.; Longobardi, E.; Fernandez, S.; Talotta, F.; Bianchi, F.; Pisati, F.; Spaggiari, L.; et al. Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-beta-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, E3775–E3784. [Google Scholar] [CrossRef] [PubMed]
- Grotsch, B.; Brachs, S.; Lang, C.; Luther, J.; Derer, A.; Schlotzer-Schrehardt, U.; Bozec, A.; Fillatreau, S.; Berberich, I.; Hobeika, E.; et al. The AP-1 transcription factor Fra1 inhibits follicular B cell differentiation into plasma cells. J. Exp. Med. 2014, 211, 2199–2212. [Google Scholar] [CrossRef] [PubMed]
- Adiseshaiah, P.; Lindner, D.J.; Kalvakolanu, D.V.; Reddy, S.P. FRA-1 proto-oncogene induces lung epithelial cell invasion and anchorage-independent growth in vitro, but is insufficient to promote tumor growth in vivo. Cancer Res. 2007, 67, 6204–6211. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Takeichi, M. Cadherin superfamily genes: Functions, genomic organization, and neurologic diversity. Genes Dev. 2000, 14, 1169–1180. [Google Scholar]
- Chen, W.V.; Maniatis, T. Clustered protocadherins. Development 2013, 140, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Chausovsky, A.; Bershadsky, A.D.; Borisy, G.G. Cadherin-mediated regulation of microtubule dynamics. Nat. Cell Biol. 2000, 2, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Filbert, E.L.; Le Borgne, M.; Lin, J.; Heuser, J.E.; Shaw, A.S. Stathmin Regulates Microtubule Dynamics and Microtubule Organizing Center Polarization in Activated T Cells. J. Immunol. 2012, 188, 5421–5427. [Google Scholar] [CrossRef]
- Pugach, E.K.; Li, P.; White, R.; Zon, L. Retro-orbital injection in adult zebrafish. J. Vis. Exp. 2009. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ting, C.-H.; Lee, K.-Y.; Wu, S.-M.; Feng, P.-H.; Chan, Y.-F.; Chen, Y.-C.; Chen, J.-Y. FOSB–PCDHB13 Axis Disrupts the Microtubule Network in Non-Small Cell Lung Cancer. Cancers 2019, 11, 107. https://doi.org/10.3390/cancers11010107
Ting C-H, Lee K-Y, Wu S-M, Feng P-H, Chan Y-F, Chen Y-C, Chen J-Y. FOSB–PCDHB13 Axis Disrupts the Microtubule Network in Non-Small Cell Lung Cancer. Cancers. 2019; 11(1):107. https://doi.org/10.3390/cancers11010107
Chicago/Turabian StyleTing, Chen-Hung, Kang-Yun Lee, Sheng-Ming Wu, Po-Hao Feng, Yao-Fei Chan, Yi-Chun Chen, and Jyh-Yih Chen. 2019. "FOSB–PCDHB13 Axis Disrupts the Microtubule Network in Non-Small Cell Lung Cancer" Cancers 11, no. 1: 107. https://doi.org/10.3390/cancers11010107
APA StyleTing, C.-H., Lee, K.-Y., Wu, S.-M., Feng, P.-H., Chan, Y.-F., Chen, Y.-C., & Chen, J.-Y. (2019). FOSB–PCDHB13 Axis Disrupts the Microtubule Network in Non-Small Cell Lung Cancer. Cancers, 11(1), 107. https://doi.org/10.3390/cancers11010107