The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship


Abstract
1. Introduction
2. Composition and Role of the ECM
3. Cell Signaling via Collagens
4. Structural Regulation of the ECM
5. ECM as a Nutritional Source
6. Proteoglycans and Glycoproteins
7. Strategies to Overcome the ECM as a Barrier to Drug Delivery
7.1. Using the ECM to Target Chemotherapies to the Tumor
7.2. Inhibiting ECM Production
7.3. Preventing PDAC Cells from using the ECM as a Nutritional Source
7.4. Relieving Intratumoral Pressure
7.5. Potential Novel Therapeutic Avenues
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the eu by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Yip, D.; Karapetis, C.; Strickland, A.; Steer, C.B.; Goldstein, D. Chemotherapy and radiotherapy for inoperable advanced pancreatic cancer. Cochrane Database Syst. Rev. 2006, 19, CD002093. [Google Scholar]
- Conroy, T. Unicancer gi prodige 24/cctg pa.6 trial: A multicenter international randomized phase iii trial of adjuvant mfolfirinox versus gemcitabine (gem) in patients with resected pancreatic ductal adenocarcinomas. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 4 June 2018. [Google Scholar]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Satoh, M.; Satoh, K.; Shimosegawa, T. Rho kinase inhibitors block activation of pancreatic stellate cells. Br. J. Pharmacol. 2003, 140, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garcia, R.; Ghassemi, S.; Fabry, B.; et al. Atra mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin d receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Lohr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Muller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar] [PubMed]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Shek, F.W.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.C.; Tan, C.K.; Huang, R.L.; Tan, N.S. Matricellular proteins: A sticky affair with cancers. J. Oncol. 2012, 2012, 351089. [Google Scholar] [CrossRef] [PubMed]
- Sobel, H.; Marmorston, Y.; Moore, F.J. Collagen and hexosamine content of femurs of rats. Proc. Soc. Exp. Biol. Med. 1954, 87, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics 2012, 11, M111.014647. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hynes, R.O. Enrichment of extracellular matrix proteins from tissues and digestion into peptides for mass spectrometry analysis. J. Vis. Exp. 2015, 101, e53057. [Google Scholar] [CrossRef] [PubMed]
- Mecham, R.P. Overview of extracellular matrix. Curr. Protoc. Cell Biol. 2012, 57, 10–11. [Google Scholar]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Monboisse, J.C.; Oudart, J.B.; Ramont, L.; Brassart-Pasco, S.; Maquart, F.X. Matrikines from basement membrane collagens: A new anti-cancer strategy. Biochim. Biophys. Acta 2014, 1840, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, A.; Malfait, F. The ehlers-danlos syndrome, a disorder with many faces. Clin. Genet 2012, 82, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perrucci, G.L.; Rurali, E.; Gowran, A.; Pini, A.; Antona, C.; Chiesa, R.; Pompilio, G.; Nigro, P. Vascular smooth muscle cells in marfan syndrome aneurysm: The broken bricks in the aortic wall. Cell Mol. Life Sci. 2017, 74, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Kwak, N.; Park, C.M.; Lee, J.; Park, Y.S.; Lee, S.M.; Yim, J.J.; Yoo, C.G.; Kim, Y.W.; Han, S.K.; Lee, C.H. Lung cancer risk among patients with combined pulmonary fibrosis and emphysema. Respir Med. 2014, 108, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed]
- Van Agtmael, T.; Bruckner-Tuderman, L. Basement membranes and human disease. Cell Tissue Res. 2010, 339, 167–188. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Iguchi, H.; Manabe, T.; Ohshio, G.; Yoshimura, T.; Wang, Z.H.; Suwa, H.; Ishigami, S.; Imamura, M. Quantitative analysis of collagen and collagen subtypes i, iii, and v in human pancreatic cancer, tumor-associated chronic pancreatitis, and alcoholic chronic pancreatitis. Pancreas 1995, 11, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, J.; Roether, I.; Kern, H.F. Distribution of extracellular matrix proteins in pancreatic ductal adenocarcinoma and its influence on tumor cell proliferation in vitro. Pancreas 1987, 2, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Castanos-Velez, E.; von Rosen, A.; Biberfeld, P. Immunohistochemical expression of extracellular matrix proteins and adhesion molecules in pancreatic carcinoma. Hepatogastroenterology 2001, 48, 1321–1327. [Google Scholar] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Clementz, A.G.; Mutolo, M.J.; Leir, S.H.; Morris, K.J.; Kucybala, K.; Harris, H.; Harris, A. Collagen xv inhibits epithelial to mesenchymal transition in pancreatic adenocarcinoma cells. PLoS ONE 2013, 8, e72250. [Google Scholar] [CrossRef] [PubMed]
- Amenta, P.S.; Briggs, K.; Xu, K.; Gamboa, E.; Jukkola, A.F.; Li, D.; Myers, J.C. Type xv collagen in human colonic adenocarcinomas has a different distribution than other basement membrane zone proteins. Hum. Pathol. 2000, 31, 359–366. [Google Scholar] [CrossRef]
- Amenta, P.S.; Hadad, S.; Lee, M.T.; Barnard, N.; Li, D.; Myers, J.C. Loss of types xv and xix collagen precedes basement membrane invasion in ductal carcinoma of the female breast. J. Pathol. 2003, 199, 298–308. [Google Scholar] [CrossRef] [PubMed]
- van der Zee, J.A.; van Eijck, C.H.; Hop, W.C.; Biermann, K.; Dicheva, B.M.; Seynhaeve, A.L.; Koning, G.A.; Eggermont, A.M.; Ten Hagen, T.L. Tumour basement membrane laminin expression predicts outcome following curative resection of pancreatic head cancer. Br. J. Cancer 2012, 107, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Lundin, C.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Type iv collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 2009, 101, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Franklin, O.; Lundberg, E.; Lundin, C.; Sund, M. Type iv collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer 2013, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Tulla, M.; Pentikainen, O.T.; Viitasalo, T.; Kapyla, J.; Impola, U.; Nykvist, P.; Nissinen, L.; Johnson, M.S.; Heino, J. Selective binding of collagen subtypes by integrin alpha 1i, alpha 2i, and alpha 10i domains. J. Biol. Chem. 2001, 276, 48206–48212. [Google Scholar] [CrossRef] [PubMed]
- Grzesiak, J.J.; Bouvet, M. Determination of the ligand-binding specificities of the alpha2beta1 and alpha1beta1 integrins in a novel 3-dimensional in vitro model of pancreatic cancer. Pancreas 2007, 34, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type i collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, S.; Grunwald, B.; Kruger, A.; Reithmeier, A.; Hahl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type v promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, F.; Comte, J.; Cabanas, C.; Garrone, R. Structural requirements for alpha 1 beta 1 and alpha 2 beta 1 integrin mediated cell adhesion to collagen v. J. Cell Sci. 1996, 109 Pt 7, 1865–1874. [Google Scholar]
- Grzesiak, J.J.; Tran Cao, H.S.; Burton, D.W.; Kaushal, S.; Vargas, F.; Clopton, P.; Snyder, C.S.; Deftos, L.J.; Hoffman, R.M.; Bouvet, M. Knockdown of the beta(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int. J. Cancer 2011, 129, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhou, S.; Siech, M.; Habisch, H.; Seufferlein, T.; Bachem, M.G. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen i-mediated signalling pathway. Br. J. Cancer 2014, 110, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; Ewachiw, T.; Jung, C.; Huang, A.; Norberg, K.J.; Marchionni, L.; McMillan, R.; Penchev, V.; Rajeshkumar, N.V.; Maitra, A.; et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0180181. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: Aacr workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type i induces disruption of e-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Philippi, C.; Vogelmann, R.; Seidel, B.; Lutz, M.P.; Adler, G.; Wedlich, D. Down-regulation of e-cadherin gene expression by collagen type i and type iii in pancreatic cancer cell lines. Cancer Res. 2001, 61, 3508–3517. [Google Scholar] [PubMed]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for e-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Doi, R.; Toyoda, E.; Tsuji, S.; Wada, M.; Koizumi, M.; Tulachan, S.S.; Ito, D.; Kami, K.; Mori, T.; et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 2004, 10, 4125–4133. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Fukumoto, Y.; Chaika, N.; Svoboda, R.; Wheelock, M.J.; Johnson, K.R. Collagen i-mediated up-regulation of n-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J. Cell Biol. 2008, 180, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Valiathan, R.R.; Marco, M.; Leitinger, B.; Kleer, C.G.; Fridman, R. Discoidin domain receptor tyrosine kinases: New players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, K.Y.; Huang, H.; Du, W.; Hagopian, M.M.; Wang, Z.; Hinz, S.; Hwang, T.H.; Wang, H.; Fleming, J.B.; Castrillon, D.H.; et al. Inhibition of discoidin domain receptor 1 reduces collagen-mediated tumorigenicity in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2017, 16, 2473–2485. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Svoboda, R.A.; Lazenby, A.J.; Saowapa, J.; Chaika, N.; Ding, K.; Wheelock, M.J.; Johnson, K.R. Up-regulation of n-cadherin by collagen i-activated discoidin domain receptor 1 in pancreatic cancer requires the adaptor molecule shc1. J. Biol. Chem. 2016, 291, 23208–23223. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Bouchal, J.; Baumforth, K.; Wei, W.; Dziechciarkova, M.; Ehrmann, J.; Klein, J.; Fridman, E.; Skarda, J.; Srovnal, J.; et al. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 2007, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Zhang, Y.; He, S.J.; Li, M.M.; Cai, X.L.; Wang, H.; Xu, L.M.; Cao, J. Tm4sf1 promotes metastasis of pancreatic cancer via regulating the expression of ddr1. Sci. Rep. 2017, 7, 45895. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yang, J.C.; Ramachandran, V.; Arumugam, T.; Deng, D.F.; Li, Z.S.; Xu, L.M.; Logsdon, C.D. Tm4sf1 regulates pancreatic cancer migration and invasion in vitro and in vivo. Cell Physiol. Biochem. 2016, 39, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.K.; Fan, X.G.; Qiu, F. Tm4sf1 promotes proliferation, invasion, and metastasis in human liver cancer cells. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, Y.; Xu, J.; Lu, D.; Wang, J. Role of tm4sf1 in regulating breast cancer cell migration and apoptosis through pi3k/akt/mtor pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 9081–9088. [Google Scholar] [PubMed]
- Park, Y.R.; Lee, S.T.; Kim, S.L.; Liu, Y.C.; Lee, M.R.; Shin, J.H.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Lee, S.O.; et al. Microrna-9 suppresses cell migration and invasion through downregulation of tm4sf1 in colorectal cancer. Int. J. Oncol. 2016, 48, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chakraborty, G.; Zhang, Z.; Akalay, I.; Gadiya, M.; Gao, Y.; Sinha, S.; Hu, J.; Jiang, C.; Akram, M.; et al. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell 2016, 166, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Dangi-Garimella, S.; Sahai, V.; Ebine, K.; Kumar, K.; Munshi, H.G. Three-dimensional collagen i promotes gemcitabine resistance in vitro in pancreatic cancer cells through hmga2-dependent histone acetyltransferase expression. PLoS ONE 2013, 8, e64566. [Google Scholar] [CrossRef] [PubMed]
- Dangi-Garimella, S.; Krantz, S.B.; Barron, M.R.; Shields, M.A.; Heiferman, M.J.; Grippo, P.J.; Bentrem, D.J.; Munshi, H.G. Three-dimensional collagen i promotes gemcitabine resistance in pancreatic cancer through mt1-mmp-mediated expression of hmga2. Cancer Res. 2011, 71, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- Haage, A.; Schneider, I.C. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J. 2014, 28, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the lox/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of lox abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. Lox-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Condello, S.; Yakubov, B.; Emerson, R.; Caperell-Grant, A.; Hitomi, K.; Xie, J.; Matei, D. Tissue transglutaminase mediated tumor-stroma interaction promotes pancreatic cancer progression. Clin. Cancer Res. 2015, 21, 4482–4493. [Google Scholar] [CrossRef] [PubMed]
- Verderio, E.A.; Johnson, T.; Griffin, M. Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids 2004, 26, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of yap/taz: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of yap/taz in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, A.J.; Sun, L.; Ananthanarayanan, V.; Munshi, H.G. Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-beta1. Cancer Res. 2006, 66, 7032–7040. [Google Scholar] [CrossRef] [PubMed]
- Benyon, R.C.; Arthur, M.J. Extracellular matrix degradation and the role of hepatic stellate cells. Semin. Liver Dis. 2001, 21, 373–384. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, Z.; Jones, K.; Azad, A.; van Stiphout, R.; Lim, S.Y.; Gomes, A.L.; Kinchesh, P.; Smart, S.C.; Gillies McKenna, W.; Buffa, F.M.; et al. Gemcitabine-induced timp1 attenuates therapy response and promotes tumor growth and liver metastasis in pancreatic cancer. Cancer Res. 2017, 77, 5952–5962. [Google Scholar] [CrossRef] [PubMed]
- Hotary, K.B.; Allen, E.D.; Brooks, P.C.; Datta, N.S.; Long, M.W.; Weiss, S.J. Membrane type i matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 2003, 114, 33–45. [Google Scholar] [CrossRef]
- Rath, N.; Olson, M.F. Rho-associated kinases in tumorigenesis: Re-considering rock inhibition for cancer therapy. EMBO Rep. 2012, 13, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. Rock signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sagi, D.; Feramisco, J.R. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 1986, 233, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Brentnall, T.A.; Chen, R. Glycoproteins and glycoproteomics in pancreatic cancer. World J. Gastroenterol. 2016, 22, 9288–9299. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Chen, R.; Tamura, Y.; Crispin, D.A.; Lai, L.A.; May, D.H.; McIntosh, M.W.; Goodlett, D.R.; Brentnall, T.A. Quantitative glycoproteomics analysis reveals changes in n-glycosylation level associated with pancreatic ductal adenocarcinoma. J. Proteome Res. 2014, 13, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Chen, R.; Reimel, B.A.; Crispin, D.A.; Mirzaei, H.; Cooke, K.; Coleman, J.F.; Lane, Z.; Bronner, M.P.; Goodlett, D.R.; et al. Quantitative proteomics investigation of pancreatic intraepithelial neoplasia. Electrophoresis 2009, 30, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Pan, S.; Ottenhof, N.A.; de Wilde, R.F.; Wolfgang, C.L.; Lane, Z.; Post, J.; Bronner, M.P.; Willmann, J.K.; Maitra, A.; et al. Stromal galectin-1 expression is associated with long-term survival in resectable pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 2012, 13, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Dawson, D.W.; Pan, S.; Ottenhof, N.A.; de Wilde, R.F.; Wolfgang, C.L.; May, D.H.; Crispin, D.A.; Lai, L.A.; Lay, A.R.; et al. Proteins associated with pancreatic cancer survival in patients with resectable pancreatic ductal adenocarcinoma. Lab. Invest. 2015, 95, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat Rev. 2014, 40, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Orozco, C.A.; Martinez-Bosch, N.; Guerrero, P.E.; Vinaixa, J.; Dalotto-Moreno, T.; Iglesias, M.; Moreno, M.; Djurec, M.; Poirier, F.; Gabius, H.J.; et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc. Natl. Acad. Sci. USA 2018, 115, E3769–E3778. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bosch, N.; Fernandez-Barrena, M.G.; Moreno, M.; Ortiz-Zapater, E.; Munne-Collado, J.; Iglesias, M.; Andre, S.; Gabius, H.J.; Hwang, R.F.; Poirier, F.; et al. Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and hedgehog signaling activation. Cancer Res. 2014, 74, 3512–3524. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Orgel, J.; Gullberg, D. Molecular composition and function of integrin-based collagen glues-introducing colinbris. Biochim. Biophys. Acta 2014, 1840, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Zeng, Z.Z.; Fay, K.S.; Veine, D.M.; Staszewski, E.D.; Morgan, M.; Wilder-Romans, K.; Williams, T.M.; Spalding, A.C.; Ben-Josef, E.; et al. Role of alpha(5)beta(1) integrin up-regulation in radiation-induced invasion by human pancreatic cancer cells. Transl. Oncol. 2011, 4, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Edderkaoui, M.; Hong, P.; Vaquero, E.C.; Lee, J.K.; Fischer, L.; Friess, H.; Buchler, M.W.; Lerch, M.M.; Pandol, S.J.; Gukovskaya, A.S. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and nadph oxidase. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1137–G1147. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Sivakumar, P.; Jones, C.J.; Chen, Q.; Peters, D.M.; Mosher, D.F.; Humphries, M.J.; Kielty, C.M. Fibronectin regulates latent transforming growth factor-beta (tgf beta) by controlling matrix assembly of latent tgf beta-binding protein-1. J. Biol. Chem. 2005, 280, 18871–18880. [Google Scholar] [CrossRef] [PubMed]
- Lohr, M.; Trautmann, B.; Gottler, M.; Peters, S.; Zauner, I.; Maillet, B.; Kloppel, G. Human ductal adenocarcinomas of the pancreas express extracellular matrix proteins. Br. J. Cancer 1994, 69, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Ryschich, E.; Khamidjanov, A.; Kerkadze, V.; Buchler, M.W.; Zoller, M.; Schmidt, J. Promotion of tumor cell migration by extracellular matrix proteins in human pancreatic cancer. Pancreas 2009, 38, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Qazi, B.S.; Tang, K.; Qazi, A. Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated inflammation and angiogenesis. Int. J. Inflam 2011, 2011, 908468. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Abbruzzese, J.L.; Huang, S.; Fidler, I.J.; Xiong, Q.; Xie, K. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin. Cancer Res. 1999, 5, 3711–3721. [Google Scholar] [PubMed]
- Patsenker, E.; Popov, Y.; Wiesner, M.; Goodman, S.L.; Schuppan, D. Pharmacological inhibition of the vitronectin receptor abrogates pdgf-bb-induced hepatic stellate cell migration and activation in vitro. J. Hepatol. 2007, 46, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Aprile, G.; Avellini, C.; Reni, M.; Mazzer, M.; Foltran, L.; Rossi, D.; Cereda, S.; Iaiza, E.; Fasola, G.; Piga, A. Biglycan expression and clinical outcome in patients with pancreatic adenocarcinoma. Tumour Biol. 2013, 34, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Katonis, P.; Tsatsakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican, a small leucine-rich proteoglycan. IUBMB Life 2008, 60, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Truty, M.A.; Kang, Y.; Chopin-Laly, X.; Zhang, R.; Roife, D.; Chatterjee, D.; Lin, E.; Thomas, R.M.; Wang, H.; et al. Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin. Cancer Res. 2014, 20, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kang, Y.; Roife, D.; Lee, Y.; Pratt, M.; Perez, M.R.; Dai, B.; Koay, E.J.; Fleming, J.B. Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene 2017, 36, 5432–5438. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.R.; Laurent, T.C.; Laurent, U.B. Hyaluronan: Its nature, distribution, functions and turnover. J. Intern. Med. 1997, 242, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Tsara, M.E.; Papageorgacopoulou, N.; Karavias, D.D.; Theocharis, D.A. Pancreatic carcinoma is characterized by elevated content of hyaluronan and chondroitin sulfate with altered disaccharide composition. Biochim. Biophys. Acta 2000, 1502, 201–206. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B.; Shepard, H.M.; O’Connor, P.M.; Kadhim, S.; Jiang, P.; Osgood, R.J.; Bookbinder, L.H.; Li, X.; Sugarman, B.J.; Connor, R.J.; et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol. Cancer Ther. 2010, 9, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Aruffo, A.; Stamenkovic, I.; Melnick, M.; Underhill, C.B.; Seed, B. Cd44 is the principal cell surface receptor for hyaluronate. Cell 1990, 61, 1303–1313. [Google Scholar] [CrossRef]
- Marhaba, R.; Zoller, M. Cd44 in cancer progression: Adhesion, migration and growth regulation. J. Mol. Histol. 2004, 35, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Zoller, M. Cd44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.B.; Kohi, S.; Koga, A.; Hirata, K.; Sato, N. Hyaluronan stimulates pancreatic cancer cell motility. Oncotarget 2016, 7, 4829–4840. [Google Scholar] [PubMed]
- Kohi, S.; Sato, N.; Koga, A.; Hirata, K.; Harunari, E.; Igarashi, Y. Hyaluromycin, a novel hyaluronidase inhibitor, attenuates pancreatic cancer cell migration and proliferation. J. Oncol. 2016, 2016, 9063087. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.B.; Sato, N.; Kohi, S.; Koga, A.; Hirata, K. Receptor for hyaluronic acid-mediated motility is associated with poor survival in pancreatic ductal adenocarcinoma. J. Cancer 2015, 6, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Fukushima, N.; Maehara, N.; Matsubayashi, H.; Koopmann, J.; Su, G.H.; Hruban, R.H.; Goggins, M. Sparc/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene 2003, 22, 5021–5030. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A.; Yeo, C.; Iacobuzio-Donahue, C.; Goggins, M. Peritumoral fibroblast sparc expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 2007, 25, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Sinn, M.; Sinn, B.V.; Striefler, J.K.; Lindner, J.L.; Stieler, J.M.; Lohneis, P.; Bischoff, S.; Blaker, H.; Pelzer, U.; Bahra, M.; et al. Sparc expression in resected pancreatic cancer patients treated with gemcitabine: Results from the conko-001 study. Ann. Oncol. 2014, 25, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, J.E.; Oh, P. Antibodies to sparc inhibit albumin binding to sparc, gp60, and microvascular endothelium. Am. J. Physiol. 1992, 263, H1872–H1879. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Stromal depletion goes on trial in pancreatic cancer. J. Natl. Cancer Inst. 2010, 102, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Plaza, C.; Musteanu, M.; Illei, P.; Brachmann, C.B.; Heise, C.; Pierce, D.; Lopez-Casas, P.P.; Menendez, C.; Tabernero, J.; et al. Sparc expression did not predict efficacy of nab-paclitaxel plus gemcitabine or gemcitabine alone for metastatic pancreatic cancer in an exploratory analysis of the phase iii mpact trial. Clin. Cancer Res. 2015, 21, 4811–4818. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Frese, K.K.; Chan, D.S.; Bapiro, T.E.; Howat, W.J.; Richards, F.M.; Ellenrieder, V.; Jodrell, D.I.; Tuveson, D.A. Sparc independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut 2014, 63, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jia, Z.; Gao, Y.; Xie, D.; Wei, D.; Cui, J.; Mishra, L.; Huang, S.; Zhang, Y.; Xie, K. Activation of vitamin d receptor signaling downregulates the expression of nuclear foxm1 protein and suppresses pancreatic cancer cell stemness. Clin. Cancer Res. 2015, 21, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Yeh, C.N.; Hsu, J.T.; Jan, Y.Y.; Chen, L.W.; Kuo, S.F.; Takano, M.; Kittaka, A.; Chen, T.C.; Chen, W.T.; et al. The vitamin d analog, mart-10, represses metastasis potential via downregulation of epithelial-mesenchymal transition in pancreatic cancer cells. Cancer Lett. 2014, 354, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Schwabe, R.F.; Choi, Y.H.; Yang, L.; Paik, Y.H.; Lindquist, J.; Qian, T.; Schoonhoven, R.; Hagedorn, C.H.; Lemasters, J.J.; et al. Nadph oxidase signal transduces angiotensin ii in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Invest. 2003, 112, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Ijichi, H.; Sasaki, T.; Sasahira, N.; Hirano, K.; Kogure, H.; Kawakubo, K.; Yagioka, H.; Yashima, Y.; et al. Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. Br. J. Cancer 2010, 103, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Sasaki, T.; Takahara, N.; Saito, K.; Ishigaki, K.; Hamada, T.; Mizuno, S.; Miyabayashi, K.; Yamamoto, K.; et al. The inhibition of renin-angiotensin system in advanced pancreatic cancer: An exploratory analysis in 349 patients. J. Cancer Res. Clin. Oncol. 2015, 141, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Kudo, M.; Fukuta, N.; Nakatani, T.; Kimura, M.; Park, A.M.; Munakata, H. Involvement of angiotensin ii and reactive oxygen species in pancreatic fibrosis. Pancreatology 2011, 11 (Suppl. 2), 7–13. [Google Scholar] [CrossRef]
- Fendrich, V.; Chen, N.M.; Neef, M.; Waldmann, J.; Buchholz, M.; Feldmann, G.; Slater, E.P.; Maitra, A.; Bartsch, D.K. The angiotensin-i-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut 2010, 59, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase ii and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. Halo 202: Randomized phase ii study of pegph20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase ib study of pegylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]


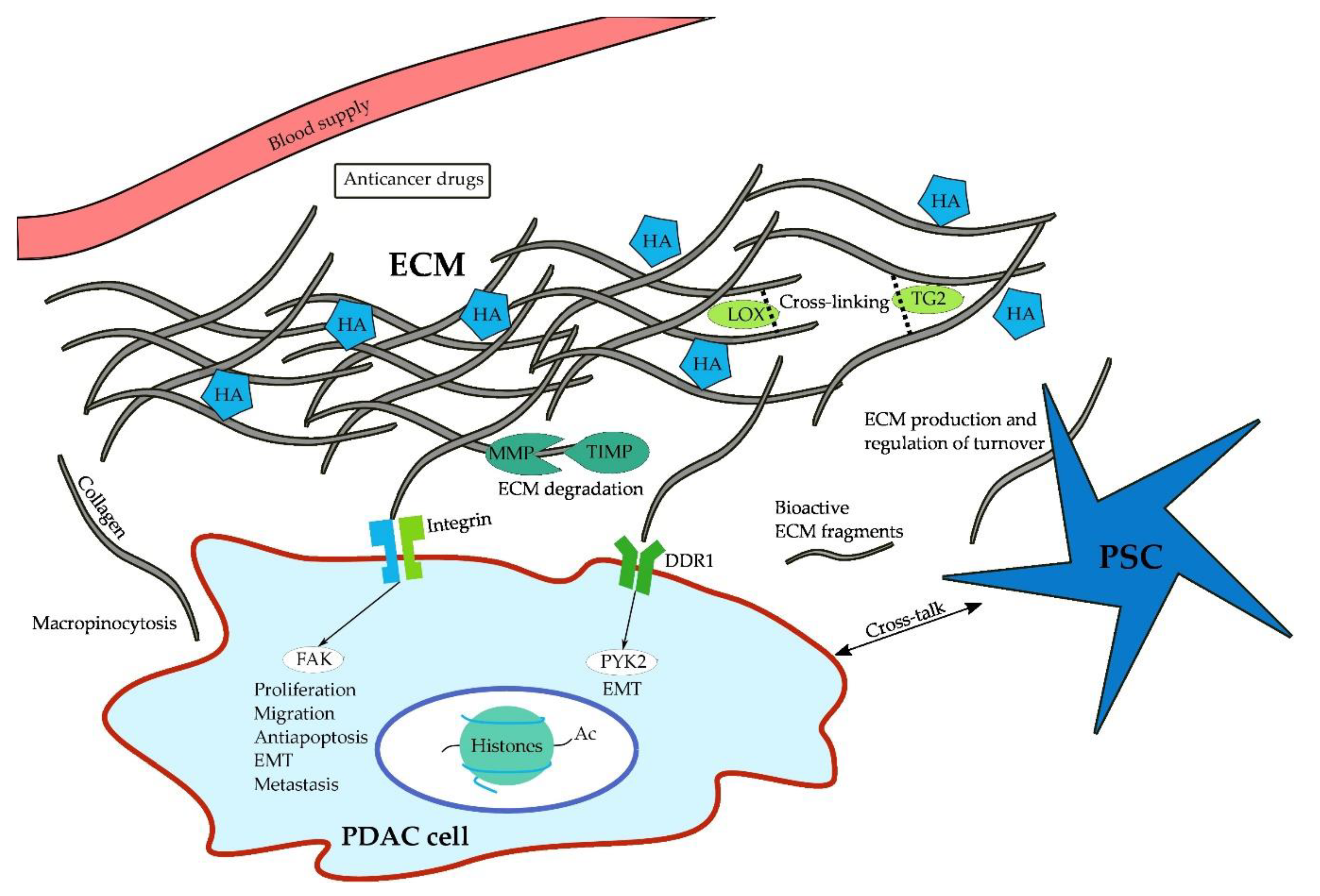{kind=link}
| Type of Collagen | Effect on PDAC Cells | Promotion (+)/Inhibition (−) |
|---|---|---|
| Collagen I | Apoptosis | − |
| EMT | + | |
| FAK pathway | + | |
| Histone acetyltransferases | + | |
| Migration | + | |
| MMP | + | |
| Proliferation | + | |
| Collagen IV | Migration | + |
| Proliferation | + | |
| Collagen V | Adhesion | + |
| Migration | + | |
| Proliferation | + | |
| Viability | + | |
| Collagen XV | Migration | − |
| HA Degrading Enzymes | |||
| PEGPH20 | Phase | Stage | Design |
| NCT02910882 | II | LAPC | PEGPH20 + GEM + Radiation |
| NCT01959139 | I/II | Metastatic | PEGPH20 + FOLFIRINOX vs. FOLFIRINOX |
| NCT03193190 | I/II | Metastatic | GEM/nab/mFOLFOX6 vs. Atezolizumab + Cobimetinib vs. Atezolizumab + PEGPH20 vs. Atezolizumab + BL-8040 |
| NCT03481920 | I | LAPC/Metastatic | PEGPH20 + Avelumab (single arm) |
| NCT01839487 | II | Metastatic | PEGPH20+ GEM/nab vs GEM/nab |
| Angiotensin inhibitors | |||
| Losartan | Phase | Stage | Design |
| NCT01821729 | II | LAPC | Losartan + FOLFIRINOX + Proton Beam Radiation (single arm) |
| Vitamin D receptor agonists | |||
| Paricalcitol | Phase | Stage | Design |
| NCT03520790 | I/II | Metastatic | GEM/nab + Placebo vs. GEM/nab + Paricalcitol |
| NCT03415854 | II | Metastatic | Paricalcitol + Cisplatin + GEM/nab (single arm) |
| NCT02930902 | I | Resectable | Pembrolizumab + Paricalcitol vs. Pembrolizumab + Paricalcitol+ GEM/nab |
| NCT03331562 | II | Metastatic | Pembrolizumab + Paricalcitol vs. Pembrolizumab +Placebo |
| NCT03300921 | I | Resectable | Pembrolizumab + Paricalcitol vs. Pembrolizumab + Placebo |
| NCT03519308 | I | Resectable | Nivolumab + GEM/nab + Paricalcitol vs. Nivolumab + GEM/nab |
| Retinoic acid receptor agonists | |||
| ATRA | Phase | Stage | Design |
| NCT03307148 | I | LAPC/Metastatic | ATRA + GEM/nab (single arm) |
| Macropinocytosis inhibitors | |||
| Hydroxychloroquine | Phase | Stage | Design |
| NCT01978184 | II | Resectable | Hydroxychloroquine + GEM/nab vs. GEM/nab |
| NCT03344172 | II | Resectable | GEM/nab + Hydroxychloroquine + Avelumab vs. GEM/nab + Hydroxychloroquine |
| NCT01506973 | I/II | Metastatic | Hydroxychloroquine + GEM (single arm) |
| NCT01494155 | II | Resectable | Hydroxychloroquine + Capecitabine + Radiation (single arm) |
| NCT01128296 | I/II | Resectable | Hydroxychloroquine + GEM (single arm) |
| FAK inhibitors | |||
| Defactinib | Phase | Stage | Design |
| NCT02758587 | I/II | LAPC/Metastatic | Defactinib + Pembrolizumab (single arm) |
| NCT02546531 | I | LAPC/Metastatic | Defactinib + Pembrolizumab + GEM (single arm) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. https://doi.org/10.3390/cancers10090316
Weniger M, Honselmann KC, Liss AS. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers. 2018; 10(9):316. https://doi.org/10.3390/cancers10090316
Chicago/Turabian StyleWeniger, Maximilian, Kim C. Honselmann, and Andrew S. Liss. 2018. "The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship" Cancers 10, no. 9: 316. https://doi.org/10.3390/cancers10090316
APA StyleWeniger, M., Honselmann, K. C., & Liss, A. S. (2018). The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers, 10(9), 316. https://doi.org/10.3390/cancers10090316