Peroxiredoxin II Regulates Cancer Stem Cells and Stemness-Associated Properties of Cancers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
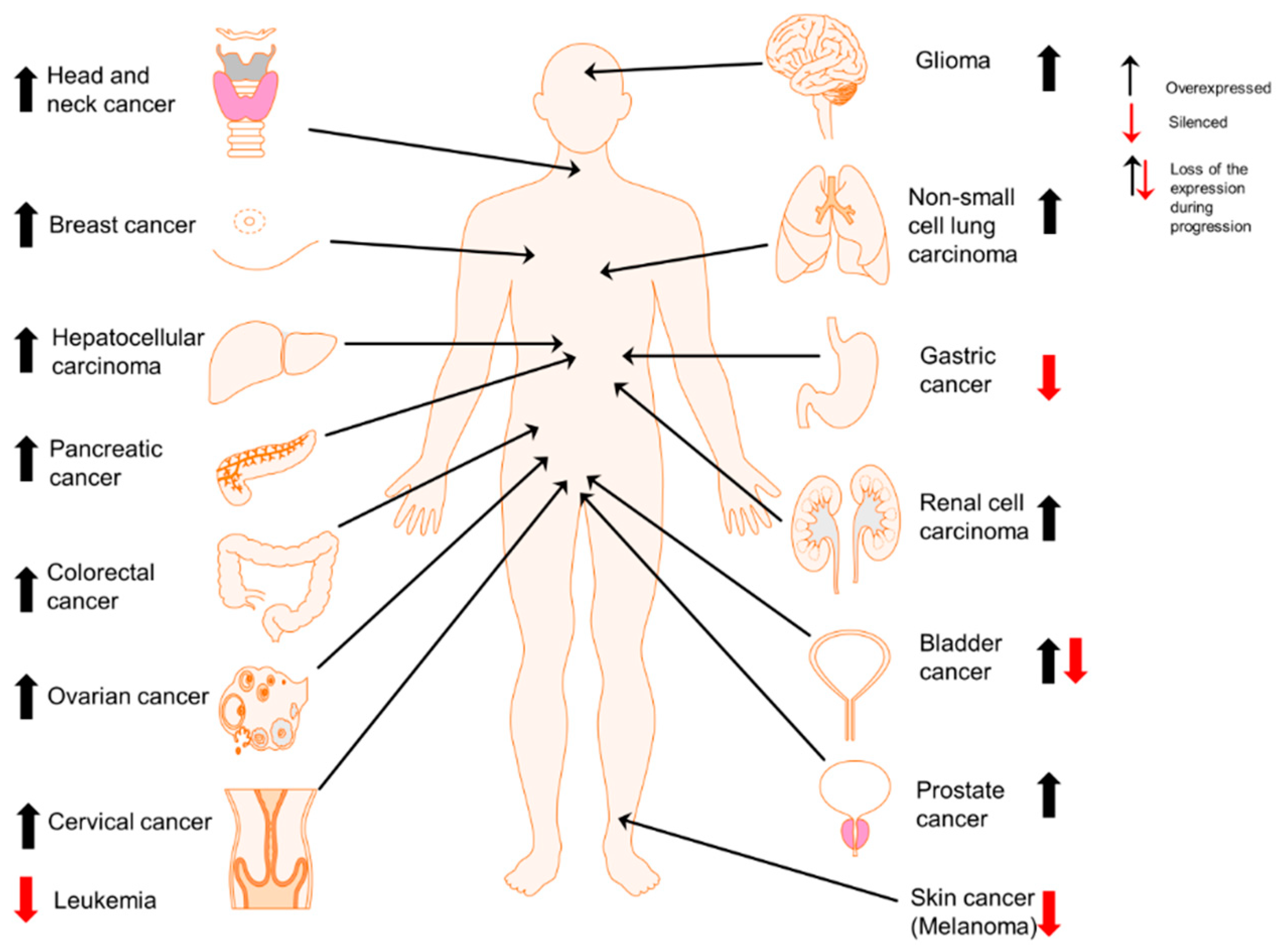
1. Introduction
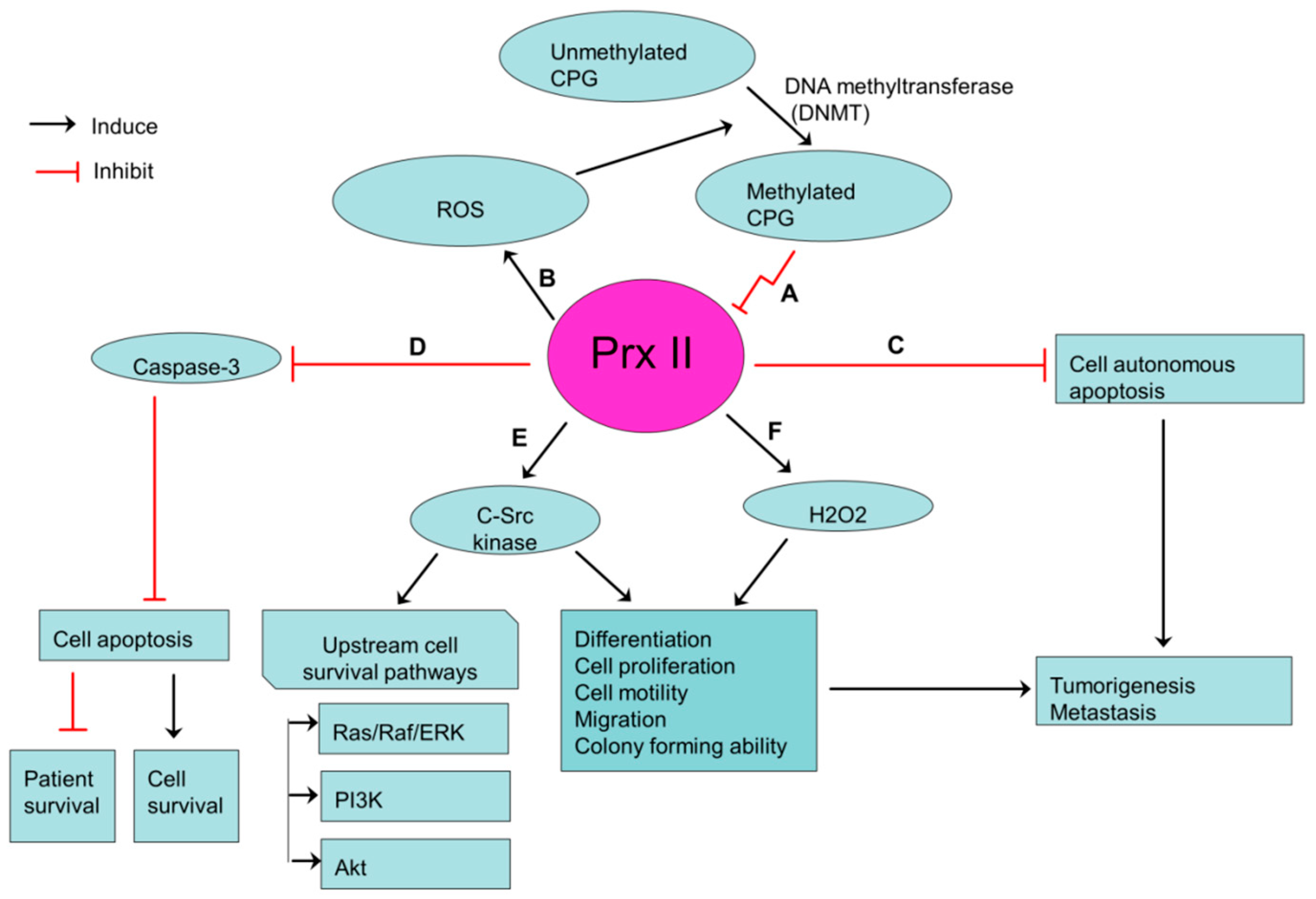
2. Prx II Maintains Cancer Stem Cell Properties of Hepatocellular Carcinoma via VEGF/VEGFR/STAT3 Signaling and Ras/FoxM1 Signaling
3. Inverse Relationship of Prx II Expression and Stemness Characteristics of Gastric Cancers
4. JNK Activation is Induced by Aberrant Prx II Expression in Lung Cancers
5. Prx II Expression Level is Inversely Related to Stemness Characteristics and Cellular Activities of Malignant Melanoma
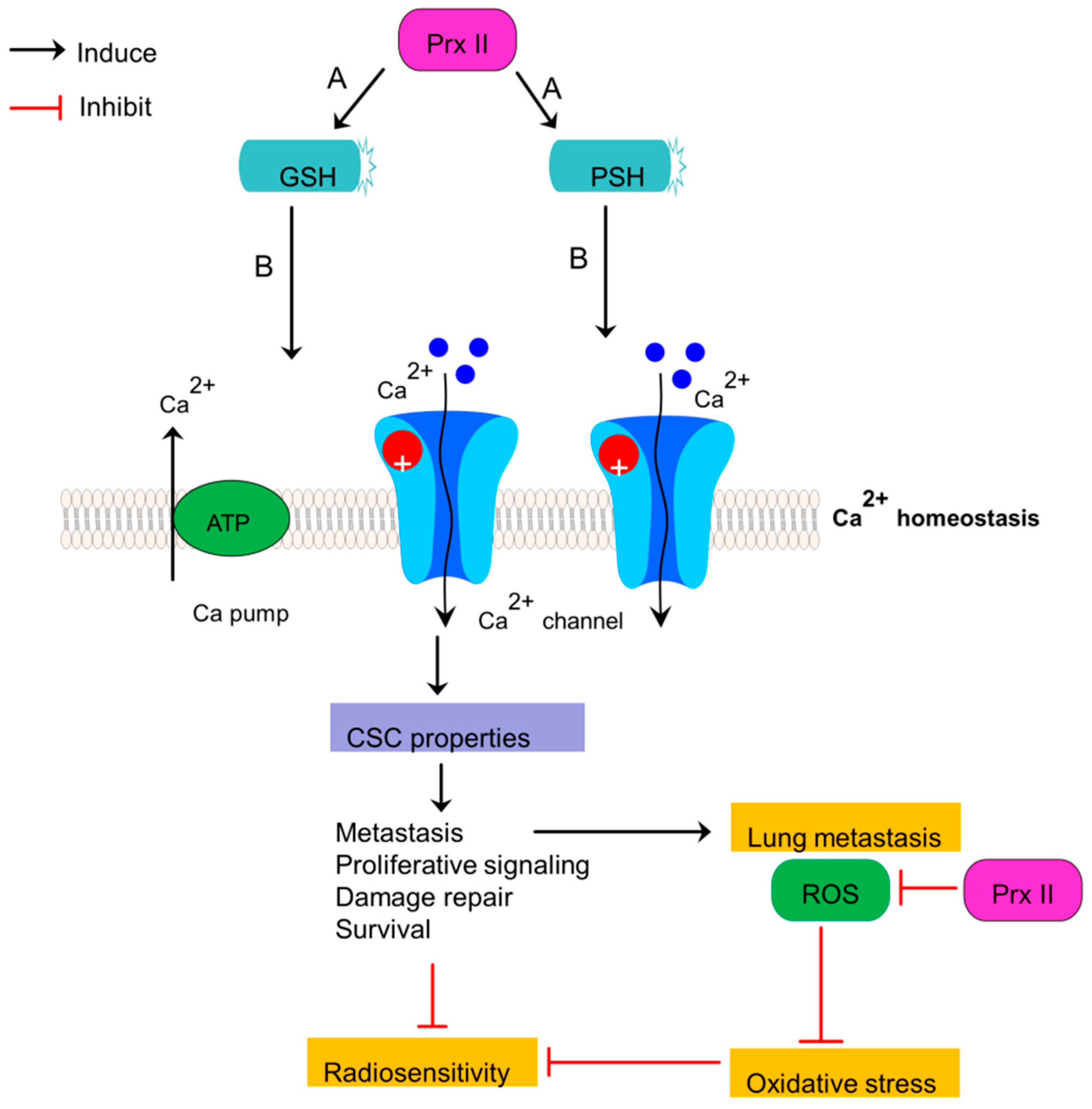
6. Prx II Protects Radioresistant and Metastatic Breast Cancer Stem Cells from Oxidative and Metabolic Stress via Metabolic Adaptation
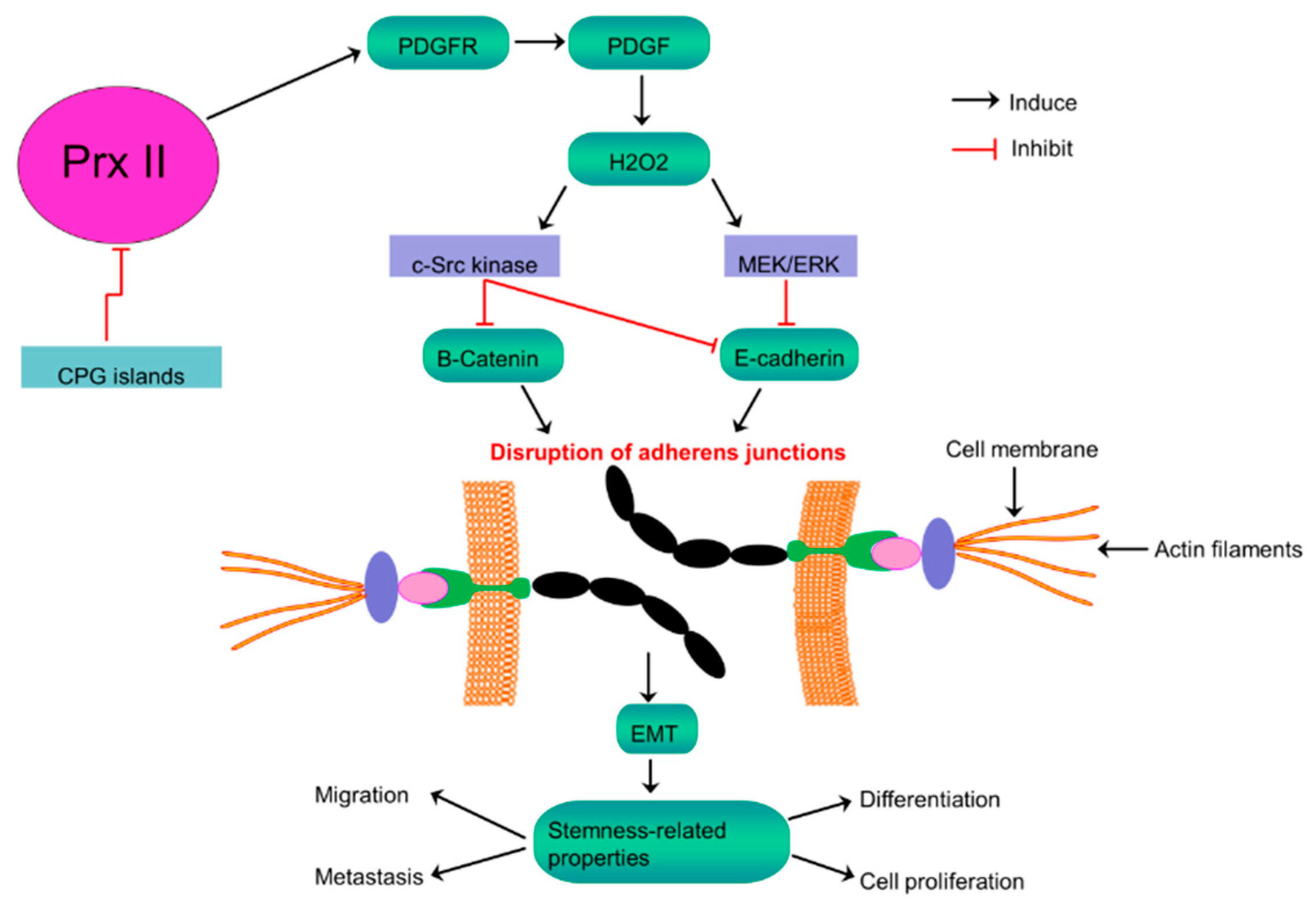
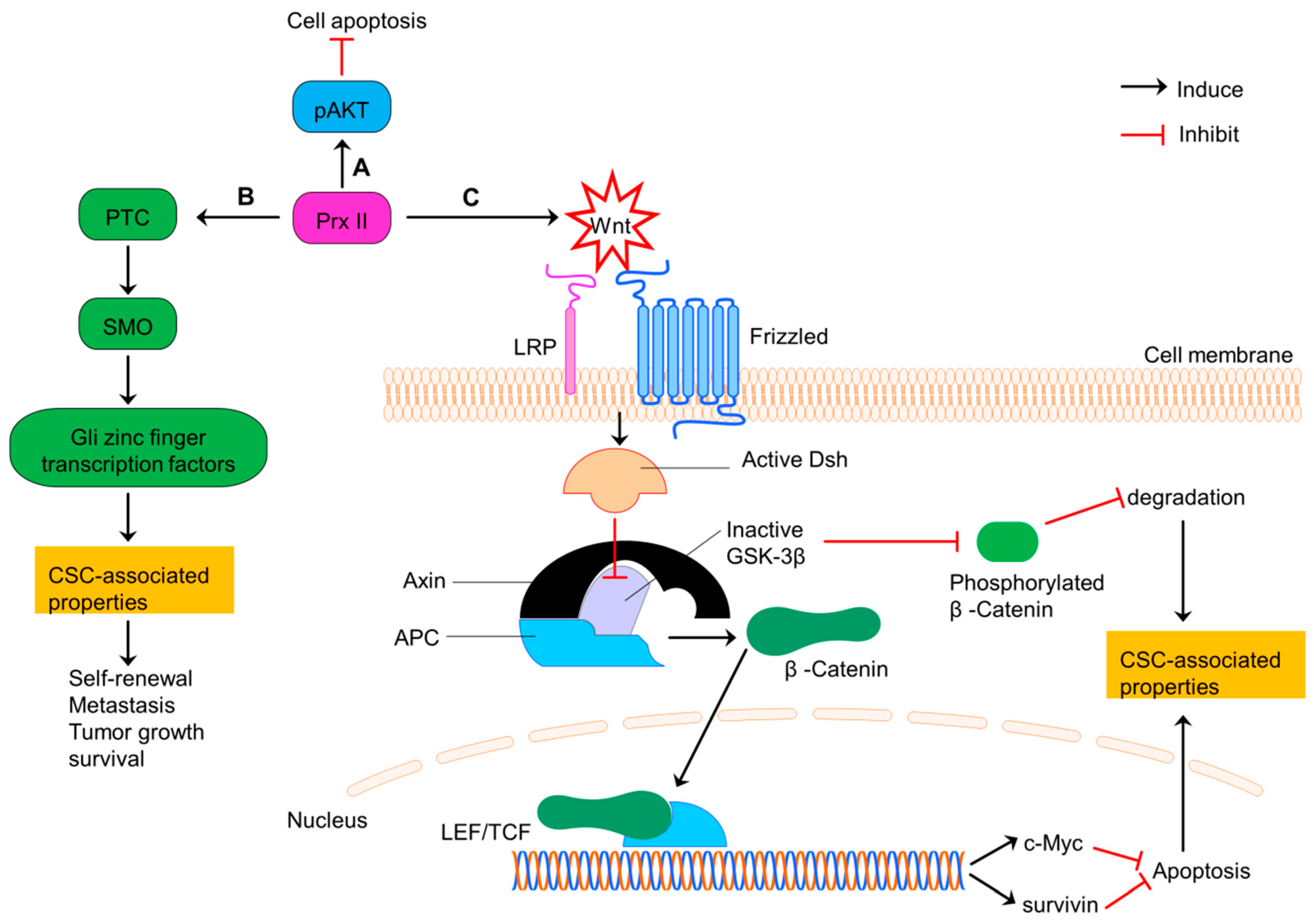
7. Prx II Maintains CSC Phenotype in Colorectal Cancer via Regulating Wnt/β-Catenin and Hedgehog Signaling Pathways
8. Loss of Prx II during Bladder Cancer Progression is Associated to the Induction of Stemness Characteristics
9. Prx II Reduces Radiosensitivity and Oxidative Stress of Glioma
10. Prx II Promotes Prostate Cancer Progression via Distinct Regulation of AR Transactivation
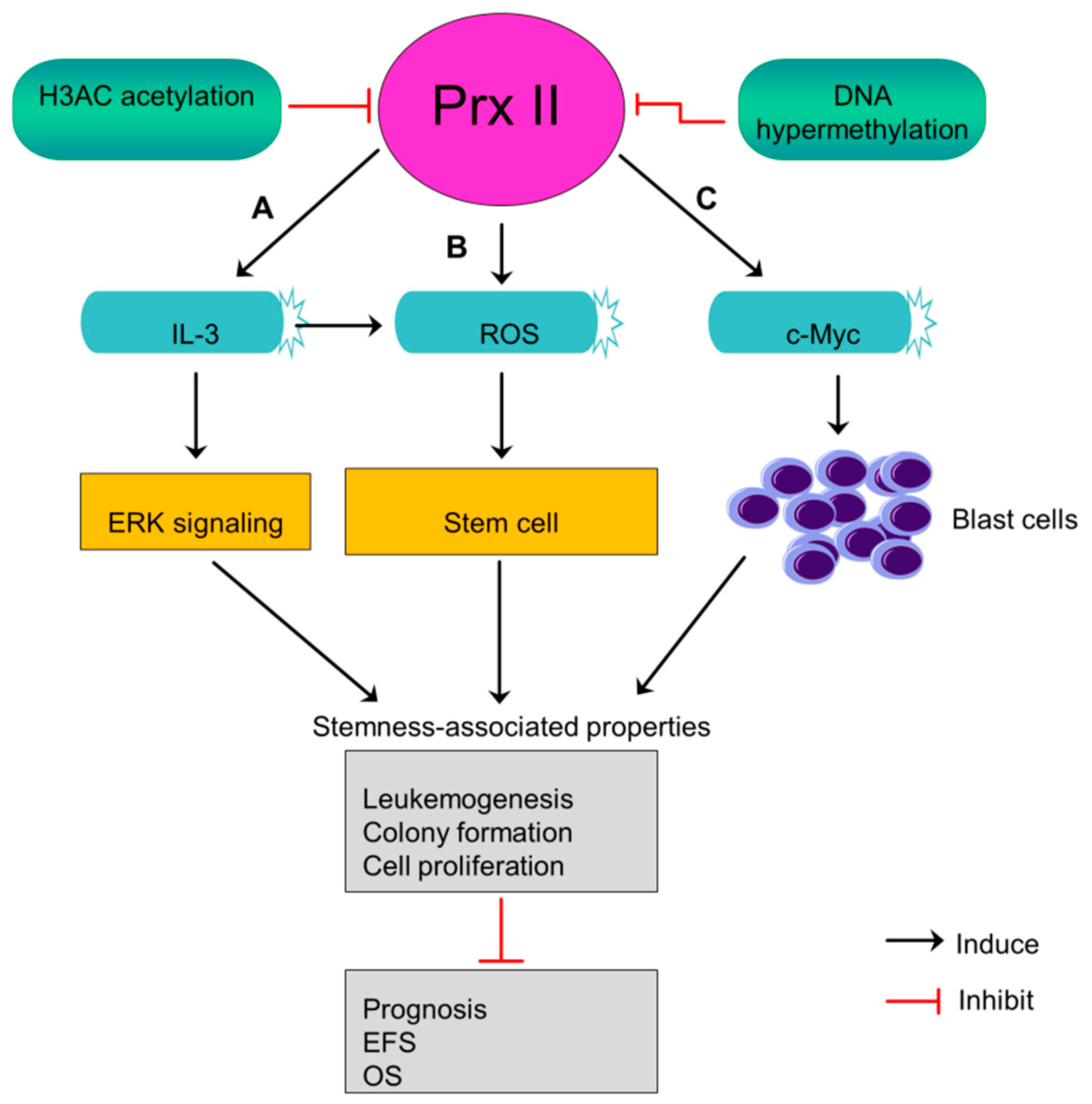
11. Prx II Suppresses Leukemogenesis and the Stem Cell Maintenance of Acute Myeloid Leukemia Blasts
12. Association of Prx II with Stemness Properties in Cancer below Should Be Studied in the Future
12.1. Osteosarcoma
12.2. Head and Neck Cancers
12.3. Renal Cell Carcinoma
12.4. Lymphoma
12.5. Ovarian Cancer
12.6. Pancreatic Cancer
13. Conclusions
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox. Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Immenschuh, S.; Baumgart-Vogt, E. Peroxiredoxins, Oxidative Stress, and Cell Proliferation. Antioxid. Redox. Signal. 2005, 7, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Agrawal-Singh, S.; Isken, F.; Agelopoulos, K.; Klein, H.U.; Thoennissen, N.H.; Koehler, G.; Hascher, A.; Baumer, N.; Berdel, W.E.; Thiede, C.; et al. Genome-wide analysis of histone H3 acetylation patterns in AML identifies PRDX2 as an epigenetically silenced tumor suppressor gene. Blood 2012, 119, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; DeGennaro, M.; Lehmann, R. Redox regulation of cell migration and adhesion. Trends Cell Biol. 2012, 22, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Q.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, H.; Huang, C.; Lei, Y. Cancer drug resistance: Redox resetting renders a way. Oncotarget 2016, 7, 42740–42761. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Magesh, V.; Lee, J.J.; Kim, S.; Knaus, U.G.; Lee, K.J. Ubiquitin C-terminal hydrolase-L1 increases cancer cell invasion by modulating hydrogen peroxide generated via NADPH oxidase 4. Oncotarget 2015, 6, 16287–16303. [Google Scholar] [CrossRef] [PubMed]
- Ow, S.H.; Chua, P.J.; Bay, B.H. Epigenetic regulation of peroxiredoxins: Implications in the pathogenesis of cancer. Exp. Biol. Med. (Maywood) 2017, 242, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Yun, H.M.; Park, K.R.; Hee Park, M.; Myoung Kim, T.; Ho Pak, J.; Jae Lee, S.; Moon, D.C.; Park, C.W.; Song, S.; et al. Lung tumor growth-promoting function of peroxiredoxin 6. Free Radic. Biol. Med. 2013, 61, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y.; Kinnula, V.L. High association of peroxiredoxins with lung cancer. Lung Cancer 2012, 78, 167. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Fu, Z.; Wang, H.; Feng, J.; Wei, J.; Guo, J. Peroxiredoxin 2 is upregulated in colorectal cancer and contributes to colorectal cancer cells’ survival by protecting cells from oxidative stress. Mol. Cell Biochem. 2014, 387, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Riddell, J.R.; Bshara, W.; Moser, M.T.; Spernyak, J.A.; Foster, B.A.; Gollnick, S.O. Peroxiredoxin 1 controls prostate cancer growth through Toll-like receptor 4-dependent regulation of tumor vasculature. Cancer Res. 2011, 71, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Ummanni, R.; Barreto, F.; Venz, S.; Scharf, C.; Barett, C.; Mannsperger, H.A.; Brase, J.C.; Kuner, R.; Schlomm, T.; Sauter, G.; et al. Peroxiredoxins 3 and 4 are overexpressed in prostate cancer tissue and affect the proliferation of prostate cancer cells in vitro. J. Proteome Res. 2012, 11, 2452–2466. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.H.; Lee, D.H.; Kim, Y.; Kim, T.H.; Huh, J.H.; Chung, S.G.; Lee, S.; Lee, C.; Ko, J.J.; An, H.J. Proteomic identification of overexpressed PRDX 1 and its clinical implications in ovarian carcinoma. J. Proteome Res. 2010, 9, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Deighton, R.F.; Le Bihan, T.; Martin, S.F.; Gerth, A.M.J.; McCulloch, M.; Edgar, J.M.; Kerr, L.E.; Whittle, I.R.; McCulloch, J. Interactions among mitochondrial proteins altered in glioblastoma. J. Neurooncol. 2014, 118, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Rhee, S.G.; Chang, T.S.; Jeong, W.; Choi, M.H. 2-Cys peroxiredoxin function in intracellular signal transduction: Therapeutic implications. Trends Mol. Med. 2005, 11, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tamae, D.; LeBon, T.; Shively, J.E.; Yen, Y.; Li, J.J. The role of peroxiredoxin II in radiation-resistant MCF-7 breast cancer cells. Cancer Res. 2005, 65, 10338–10346. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Fu, Z.; Wang, H.; Feng, J.; Wei, J.; Guo, J. Peroxiredoxin 2 knockdown by RNA interference inhibits the growth of colorectal cancer cells by downregulating Wnt/beta-catenin signaling. Cancer Lett. 2014, 343, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Kashiwagi, E.; Takeuchi, A.; Fujimoto, N.; Uchiumi, T.; Naito, S. Peroxiredoxin 2 in the nucleus and cytoplasm distinctly regulates androgen receptor activity in prostate cancer cells. Free Radic. Biol. Med. 2011, 51, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Rho, J.K.; Lee, J.C.; Park, Y.H.; Shin, H.J.; Cho, S.; Kang, Y.K.; Kim, B.Y.; Yoon, D.Y.; Yu, D.Y. An important role for peroxiredoxin II in survival of A549 lung cancer cells resistant to gefitinib. Exp. Mol. Med. 2015, 47, e165. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Bak, Y.; Park, Y.H.; Jang, G.B.; Nam, J.S.; Yoo, J.E.; Park, Y.N.; Bak, I.S.; Kim, J.M.; Yoon, D.Y.; et al. Peroxiredoxin II Is Essential for Maintaining Stemness by Redox Regulation in Liver Cancer Cells. Stem Cells 2016, 34, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Hintsala, H.R.; Soini, Y.; Haapasaari, K.M.; Karihtala, P. Dysregulation of redox-state-regulating enzymes in melanocytic skin tumours and the surrounding microenvironment. Histopathology 2015, 67, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Min, C.; Jun, Y.; Lee, D.J.; Kim, S.H.; Park, J.H.; Cheong, J.H.; Park, Y.J.; Kim, S.Y.; Lee, S.; et al. Silencing of peroxiredoxin II by promoter methylation is necessary for the survival and migration of gastric cancer cells. Exp. Mol. Med. 2018, 50, e443. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Chung, Y.M.; Lee, Y.S.; Kim, H.J.; Kim, J.S.; Chae, H.Z.; Yoo, Y.D. Antisense of human peroxiredoxin II enhances radiation-induced cell death. Clin. Cancer Res. 2000, 6, 4915–4920. [Google Scholar] [PubMed]
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Fu, Z.X.; Wang, X.H. Peroxiredoxins in colorectal neoplasms. Histol. Histopathol. 2010, 25, 1297–1303. [Google Scholar] [PubMed]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology (Williston Park) 2014, 28, 1101–1107, 1110. [Google Scholar] [PubMed]
- Yeh, D.W.; Chen, Y.S.; Lai, C.Y.; Liu, Y.L.; Lu, C.H.; Lo, J.F.; Chen, L.; Hsu, L.C.; Luo, Y.; Xiang, R.; et al. Downregulation of COMMD1 by miR-205 promotes a positive feedback loop for amplifying inflammatory- and stemness-associated properties of cancer cells. Cell Death Differ. 2016, 23, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Seo, E.J.; Choi, E.J.; Lee, S.I.; Kwon, Y.W.; Jang, I.H.; Kim, S.C.; Kim, K.H.; Suh, D.S.; Seong-Jang, K.; et al. Crucial role of HMGA1 in the self-renewal and drug resistance of ovarian cancer stem cells. Exp. Mol. Med. 2016, 48, e255. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wei, J.; Zhang, S.; Wu, X.; Guo, J.; Liu, M.; Du, K.; Xu, J.; Peng, L.; Lv, Z.; et al. Peroxiredoxin 2 is essential for maintaining cancer stem cell-like phenotype through activation of Hedgehog signaling pathway in colon cancer. Oncotarget 2016, 7, 86816–86828. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kwon, C.H.; Nakano, I. Detoxification of oxidative stress in glioma stem cells: Mechanism, clinical relevance, and therapeutic development. J. Neurosci. Res. 2014, 92, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Cordani, M.; Dalla Pozza, E.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant Mechanisms and ROS-Related MicroRNAs in Cancer Stem Cells. Oxid. Med. Cell Longev. 2015, 2015, 425708. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Okazaki, S.; Saya, H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene 2013, 32, 5191–5198. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Diaz, A.J.; Yen, Y. The role of peroxiredoxin II in chemoresistance of breast cancer cells. Breast Cancer 2014, 6, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Cruz, I.N.; Coley, H.M.; Kramer, H.B.; Madhuri, T.K.; Safuwan, N.A.; Angelino, A.R.; Yang, M. Proteomics Analysis of Ovarian Cancer Cell Lines and Tissues Reveals Drug Resistance-associated Proteins. Cancer Genomics Proteomics 2017, 14, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.T.; Yao, H.R.; Li, Y.Y.; Song, Y.Y.; Su, M.Y. MicroRNA-19b promotes the migration and invasion of ovarian cancer cells by inhibiting the PTEN/AKT signaling pathway. Oncol. Lett. 2018, 16, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, S.; Kuramitsu, Y.; Wang, Y.; Baron, B.; Kitagawa, T.; Akada, J.; Tokuda, K.; Kaino, S.; Maehara, S.; Maehara, Y.; et al. Human pancreatic cancer cells with acquired gemcitabine resistance exhibit significant up-regulation of peroxiredoxin-2 compared to sensitive parental cells. Anticancer Res. 2013, 33, 4821–4826. [Google Scholar] [PubMed]
- Memon, A.A.; Chang, J.W.; Oh, B.R.; Yoo, Y.J. Identification of differentially expressed proteins during human urinary bladder cancer progression. Cancer Detect. Prev. 2005, 29, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Kubota, D.; Mukaihara, K.; Yoshida, A.; Tsuda, H.; Kawai, A.; Kondo, T. Proteomics study of open biopsy samples identifies peroxiredoxin 2 as a predictive biomarker of response to induction chemotherapy in osteosarcoma. J. Proteomics 2013, 91, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Su, Y. Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett. 2009, 286, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, J.; Lin, C.; Wang, H.; Jiang, Y.; Wang, J.; Yang, P.; He, F. Peroxiredoxin 2: A potential biomarker for early diagnosis of hepatitis B virus related liver fibrosis identified by proteomic analysis of the plasma. BMC Gastroenterol. 2010, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Han, Q.; Wang, R.; Li, X.; Wang, Q.; Wang, H.; Wang, J.; Ma, Y. PRDX2 protects hepatocellular carcinoma SMMC-7721 cells from oxidative stress. Oncol. Lett. 2016, 12, 2217–2221. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qin, X.; Cui, J.; Dai, Z.; Kang, X.; Yue, H.; Zhang, Y.; Su, J.; Cao, J.; Ou, C.; et al. Proteome analysis of aflatoxin B1-induced hepatocarcinogenesis in tree shrew (Tupaia belangeri chinensis) and functional identification of candidate protein peroxiredoxin II. Proteomics 2008, 8, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Lee, D.J.; Lee, K.W.; Park, Y.S.; Lee, J.Y.; Lee, S.H.; Koh, Y.J.; Koh, G.Y.; Choi, C.; Yu, D.Y.; et al. Peroxiredoxin II is an essential antioxidant enzyme that prevents the oxidative inactivation of VEGF receptor-2 in vascular endothelial cells. Mol. Cell 2011, 44, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Kim, S.U.; Kwon, T.H.; Kim, J.M.; Song, I.S.; Shin, H.J.; Lee, B.K.; Bang, D.H.; Lee, S.J.; Lee, D.S.; et al. Peroxiredoxin II promotes hepatic tumorigenesis through cooperation with Ras/Forkhead box M1 signaling pathway. Oncogene 2016, 35, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, G.H.; Na, D.C.; Ahn, E.Y.; Kim, G.I.; Lee, J.E.; Cho, J.Y.; Yoo, J.E.; Choi, J.S.; Park, Y.N. Human hepatocellular carcinomas with “Stemness”-related marker expression: Keratin 19 expression and a poor prognosis. Hepatology 2011, 54, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.K.; Huang, Y.; He, W.; Yan, Z.W.; Fan, L.; Liu, M.H.; Xiao, W.L.; Sun, H.D.; Chen, G.Q. Adenanthin targets peroxiredoxin I/II to kill hepatocellular carcinoma cells. Cell Death Dis. 2014, 5, e1400. [Google Scholar] [CrossRef] [PubMed]
- Mitsuda, Y.; Morita, K.; Kashiwazaki, G.; Taniguchi, J.; Bando, T.; Obara, M.; Hirata, M.; Kataoka, T.R.; Muto, M.; Kaneda, Y.; et al. RUNX1 positively regulates the ErbB2/HER2 signaling pathway through modulating SOS1 expression in gastric cancer cells. Sci. Rep. 2018, 8, 6423. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, S.; Peat, J.R.; Hore, T.A.; Santos, F.; Dean, W.; Reik, W. Reprogramming DNA methylation in the mammalian life cycle: Building and breaking epigenetic barriers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20110330. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the E-cadherin promoter. Gastroenterology 2008, 135, 2128–2140.e8. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, W.; Okamoto, I.; Yoshida, T.; Okamoto, K.; Takezawa, K.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Arao, T.; Yanagihara, K.; et al. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol. Cancer Ther. 2010, 9, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Yo, Y.D.; Chung, Y.M.; Park, J.K.; Ahn, C.M.; Kim, S.K.; Kim, H.J. Synergistic effect of peroxiredoxin II antisense on cisplatin-induced cell death. Exp. Mol. Med. 2002, 34, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Chandimali, N.; Huynh, D.L.; Zhang, J.J.; Kim, N.; Bak, Y.; Yoon, D.Y.; Yu, D.Y.; Lee, J.C.; Gera, M.; et al. BRM270 inhibits cancer stem cell maintenance via microRNA regulation in chemoresistant A549 lung adenocarcinoma cells. Cell Death Dis. 2018, 9, 244. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.; Park, H.; Chandimali, N.; Huynh, D.L.; Zhang, J.J.; Ghosh, M.; Gera, M.; Kim, N.; Bak, Y.; Yoon, D.Y.; et al. MicroRNA-128 suppresses paclitaxel-resistant lung cancer by inhibiting MUC1-C and BMI-1 in cancer stem cells. Oncotarget 2017, 8, 110540–110551. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, S.T.; Svensk, A.M.; Soini, Y.; Paakko, P.; Hirvikoski, P.; Kang, S.W.; Saily, M.; Kinnula, V.L. Peroxiredoxins, a novel protein family in lung cancer. Int. J. Cancer 2004, 111, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.U.; Park, Y.H.; Kim, J.M.; Sun, H.N.; Song, I.S.; Huang, S.M.; Lee, S.H.; Chae, J.I.; Hong, S.; Sik Choi, S.; et al. Dominant role of peroxiredoxin/JNK axis in stemness regulation during neurogenesis from embryonic stem cells. Stem Cells 2014, 32, 998–1011. [Google Scholar] [CrossRef] [PubMed]
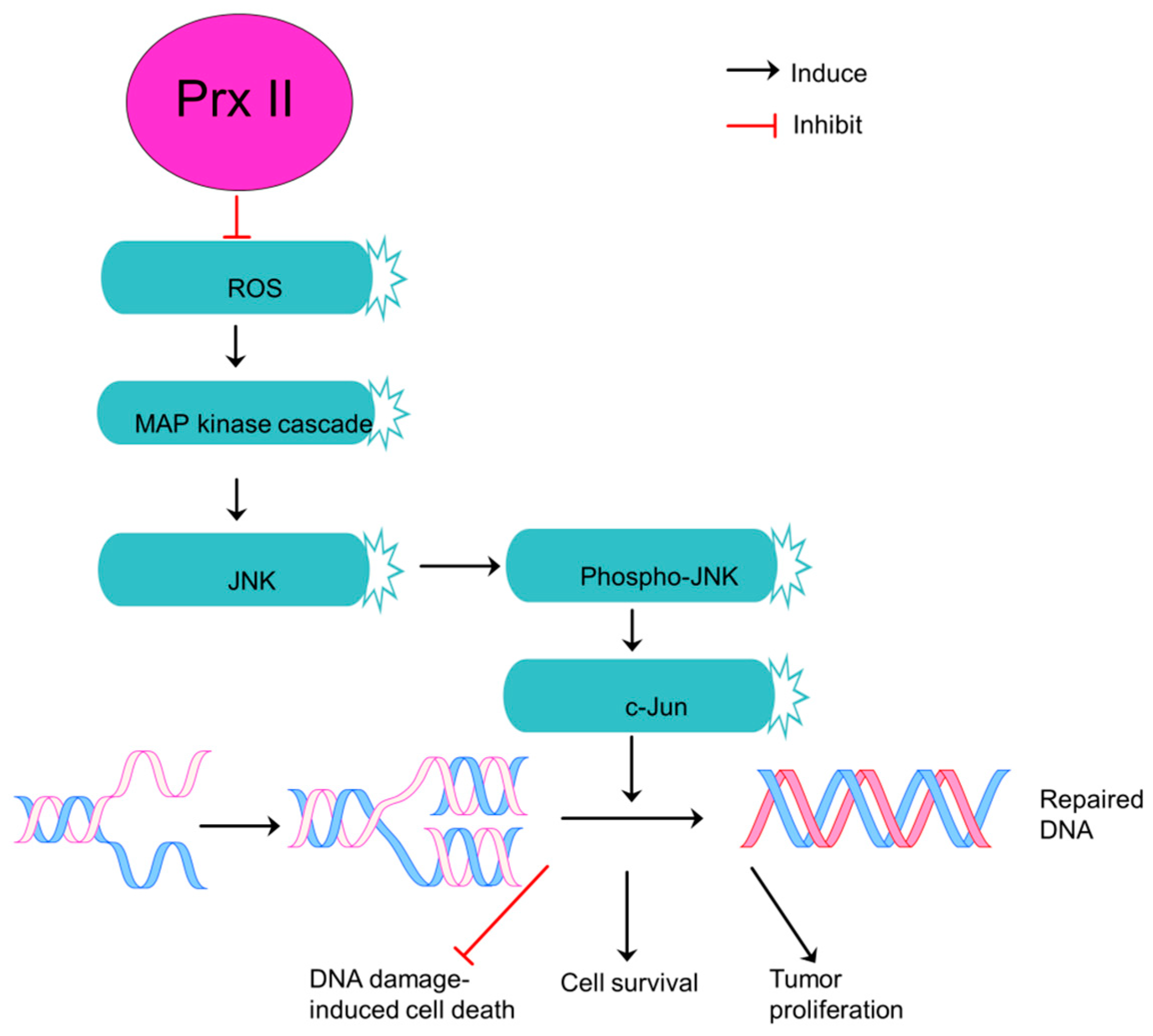
- Lee, K.W.; Lee, D.J.; Lee, J.Y.; Kang, D.H.; Kwon, J.; Kang, S.W. Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J. Biol. Chem. 2011, 286, 8394–8404. [Google Scholar] [CrossRef] [PubMed]
- Kanavy, H.E.; Gerstenblith, M.R. Ultraviolet radiation and melanoma. Semin. Cutan. Med. Surg. 2011, 30, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Furuta, J.; Nobeyama, Y.; Umebayashi, Y.; Otsuka, F.; Kikuchi, K.; Ushijima, T. Silencing of Peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006, 66, 6080–6086. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Demuro, P.P.; Zanini, C.; Santona, A.; Castiglia, D.; D’Atri, S.; Ascierto, P.A.; Napolitano, M.; Cossu, A.; Tadolini, B.; et al. Analysis of candidate genes through a proteomics-based approach in primary cell lines from malignant melanomas and their metastases. Melanoma Res. 2005, 15, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Kang, D.H.; Choi, M.; Choi, Y.J.; Lee, J.Y.; Park, J.H.; Park, Y.J.; Lee, K.W.; Kang, S.W. Peroxiredoxin-2 represses melanoma metastasis by increasing E-Cadherin/beta-Catenin complexes in adherens junctions. Cancer Res. 2013, 73, 4744–4757. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Sung, J.Y.; Kim, O.S.; Kim, Y.J.; Hur, K.C.; Kazlauskas, A.; Rhee, S.G. Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J. Biol. Chem. 2000, 275, 10527–10531. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Lee, I.K.; Kim, G.W.; Kim, B.U.; Han, Y.H.; Yu, D.Y.; Park, H.S.; Kim, K.Y.; Lee, J.S.; Choi, C.; et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature 2005, 435, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Homsi, J.; Cubitt, C.L.; Zhang, S.; Munster, P.N.; Yu, H.; Sullivan, D.M.; Jove, R.; Messina, J.L.; Daud, A.I. Src activation in melanoma and Src inhibitors as therapeutic agents in melanoma. Melanoma Res. 2009, 19, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Roura, S.; Miravet, S.; Piedra, J.; Garcia de Herreros, A.; Dunach, M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J. Biol. Chem. 1999, 274, 36734–36740. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; Simcha, I.; Ben-Yedidia, T.; Blechman, J.; Savagner, P.; Ben-Ze’ev, A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: The roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 2003, 163, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Lilla, C.; Ambrosone, C.B.; Kropp, S.; Helmbold, I.; Schmezer, P.; von Fournier, D.; Haase, W.; Sautter-Bihl, M.L.; Wenz, F.; Chang-Claude, J. Predictive factors for late normal tissue complications following radiotherapy for breast cancer. Breast Cancer Res. Treat. 2007, 106, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Diest, P.J.V.; Perez-Losada, J.; Snijders, A.M. Revisiting the impact of age and molecular subtype on overall survival after radiotherapy in breast cancer patients. Sci. Rep. 2017, 7, 12587. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Duru, N.; Fan, M.; Candas, D.; Menaa, C.; Liu, H.C.; Nantajit, D.; Wen, Y.; Xiao, K.; Eldridge, A.; Chromy, B.A.; et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin. Cancer Res. 2012, 18, 6634–6647. [Google Scholar] [CrossRef] [PubMed]
- Shau, H.; Kim, A. Identification of natural killer enhancing factor as a major antioxidant in human red blood cells. Biochem. Biophys. Res. Commun. 1994, 199, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.J.; Tamae, D.; Yen, Y.; Li, J.; Wang, T. Enhanced radiation response in radioresistant MCF-7 cells by targeting peroxiredoxin II. Breast Cancer 2013, 5, 87–101. [Google Scholar] [PubMed]
- Noh, D.Y.; Ahn, S.J.; Lee, R.A.; Kim, S.W.; Park, I.A.; Chae, H.Z. Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 2001, 21, 2085–2090. [Google Scholar] [PubMed]
- Yuan, C.; Xiu, D.; Tao, M.; Ma, Z.; Jiang, B.; Li, Z.; Li, L.; Wang, L.; Wang, H.; Zhang, T. Data analysis of 36 cases with intraductal papillary mucinous neoplasm of the pancreas for their clinicopathological features, diagnosis, and treatment. Chin. Med. J. (Engl.) 2014, 127, 4087–4091. [Google Scholar] [PubMed]
- Domina, E.A.; Philchenkov, A.; Dubrovska, A. Individual Response to Ionizing Radiation and Personalized Radiotherapy. Crit. Rev. Oncog. 2018, 23, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; Bertoldi, M.; Mohandas, N.; An, X.; Bugatti, A.; Brunati, A.M.; Rusnati, M.; Tibaldi, E.; Siciliano, A.; Turrini, F.; et al. Membrane association of peroxiredoxin-2 in red cells is mediated by the N-terminal cytoplasmic domain of band 3. Free Radic. Biol. Med. 2013, 55, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.B.; Mankad, M.V.; Shriver, S.K.; Mankad, V.N.; Plishker, G.A. Reconstitution of Ca(2+)-dependent K+ transport in erythrocyte membrane vesicles requires a cytoplasmic protein. J. Biol. Chem. 1991, 266, 18964–18968. [Google Scholar] [PubMed]
- Stresing, V.; Baltziskueta, E.; Rubio, N.; Blanco, J.; Arriba, M.C.; Valls, J.; Janier, M.; Clezardin, P.; Sanz-Pamplona, R.; Nieva, C.; et al. Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene 2013, 32, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Gupta, G.P.; Padua, D.; Bos, P.; Nguyen, D.X.; Nuyten, D.; Kreike, B.; Zhang, Y.; Wang, Y.; Ishwaran, H.; et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc. Natl. Acad. Sci. USA 2007, 104, 6740–6745. [Google Scholar] [CrossRef] [PubMed]
- Iles, K.E.; Forman, H.J. Macrophage signaling and respiratory burst. Immunol. Res. 2002, 26, 95–105. [Google Scholar] [CrossRef]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, Y.; Wang, X.; Zhang, L.; Liu, H. An Optimal Mean Based Block Robust Feature Extraction Method to Identify Colorectal Cancer Genes with Integrated Data. Sci. Rep. 2017, 7, 8584. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, S.; Wang, R.; Wu, X.; Zeng, L.; Fu, Z. Knockdown of PRDX2 sensitizes colon cancer cells to 5-FU by suppressing the PI3K/AKT signaling pathway. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wang, R.; Shang, J.; Xiong, Y.; Fu, Z. Peroxiredoxin 2 is associated with colorectal cancer progression and poor survival of patients. Oncotarget 2017, 8, 15057–15070. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Lee, D.J.; Lee, S.; Lee, S.Y.; Jun, Y.; Kim, Y.; Kim, Y.; Lee, J.S.; Lee, D.K.; Lee, S.; et al. Interaction of tankyrase and peroxiredoxin II is indispensable for the survival of colorectal cancer cells. Nat. Commun. 2017, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Lee, J.H.S.; Kang, S.W. Survival of APC-mutant colorectal cancer cells requires interaction between tankyrase and a thiol peroxidase, peroxiredoxin II. BMB Rep. 2017, 50, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Cerda, M.B.; Lloyd, R.; Batalla, M.; Giannoni, F.; Casal, M.; Policastro, L. Silencing peroxiredoxin-2 sensitizes human colorectal cancer cells to ionizing radiation and oxaliplatin. Cancer Lett. 2017, 388, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.S.; Shipley, W.U.; Feldman, A.S. Bladder cancer. Lancet 2009, 374, 239–249. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chen, C.L.; Chen, H.W.; Chung, T.; Wu, C.C.; Chen, C.D.; Hsu, C.W.; Chen, M.C.; Tsui, K.H.; Chang, P.L.; et al. Discovery of novel bladder cancer biomarkers by comparative urine proteomics using iTRAQ technology. J. Proteome Res. 2010, 9, 5803–5815. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y.; Haapasaari, K.M.; Vaarala, M.H.; Turpeenniemi-Hujanen, T.; Karja, V.; Karihtala, P. 8-hydroxydeguanosine and nitrotyrosine are prognostic factors in urinary bladder carcinoma. Int. J. Clin. Exp. Pathol. 2011, 4, 267–275. [Google Scholar] [PubMed]
- Zhou, Q.; Liu, J.; Quan, J.; Liu, W.; Tan, H.; Li, W. microRNAs as potential biomarkers for the diagnosis of glioma: A systematic review and meta-analysis. Cancer Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Smith-Pearson, P.S.; Kooshki, M.; Spitz, D.R.; Poole, L.B.; Zhao, W.; Robbins, M.E. Decreasing peroxiredoxin II expression decreases glutathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H2O2. Free Radic. Biol. Med. 2008, 45, 1178–1789. [Google Scholar] [CrossRef] [PubMed]
- Jarvela, S.; Rantala, I.; Rodriguez, A.; Kallio, H.; Parkkila, S.; Kinnula, V.L.; Soini, Y.; Haapasalo, H. Specific expression profile and prognostic significance of peroxiredoxins in grade II-IV astrocytic brain tumors. BMC Cancer 2010, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Maestroni, U.; Morandin, F.; Ferretti, S.; Dinale, F.; Ziglioli, F. Recurrence of prostate cancer after HIFU. Proposal of a novel predictive index. Acta Biomed. 2018, 89, 220–226. [Google Scholar] [PubMed]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef] [PubMed]
- Eden, S.; Hashimshony, T.; Keshet, I.; Cedar, H.; Thorne, A.W. DNA methylation models histone acetylation. Nature 1998, 394, 842. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, X.; Yang, Y.; He, Z.; Qu, X.; Zhang, Y. Long noncoding RNAs in the progression, metastasis, and prognosis of osteosarcoma. Cell Death Dis. 2016, 7, e2389. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, K.; Tochigi, N.; Saito, S.; Shimoda, T.; Morioka, H.; Toyama, Y.; Hosono, A.; Suehara, Y.; Beppu, Y.; Kawai, A.; et al. Peroxiredoxin 2 as a chemotherapy responsiveness biomarker candidate in osteosarcoma revealed by proteomics. Proteomics Clin. Appl. 2010, 4, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, J.; Guo, L.; Nie, J.; Zhu, C.; Ma, X. The value of narrow band imaging in diagnosis of head and neck cancer: A meta-analysis. Sci. Rep. 2018, 8, 515. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y.; Kallio, J.P.; Hirvikoski, P.; Helin, H.; Kellokumpu-Lehtinen, P.; Kang, S.W.; Tammela, T.L.; Peltoniemi, M.; Martikainen, P.M.; Kinnula, V.L. Oxidative/nitrosative stress and peroxiredoxin 2 are associated with grade and prognosis of human renal carcinoma. APMIS 2006, 114, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Szaumkessel, M.; Richter, J.; Ammerpohl, O.; Hansmann, M.L.; Kuppers, R.; Siebert, R.; Giefing, M. The PRDX2 gene is transcriptionally silenced and de novo methylated in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Blood 2014, 123, 3672–3674. [Google Scholar] [CrossRef] [PubMed]
- Trzeciecka, A.; Klossowski, S.; Bajor, M.; Zagozdzon, R.; Gaj, P.; Muchowicz, A.; Malinowska, A.; Czerwoniec, A.; Barankiewicz, J.; Domagala, A.; et al. Dimeric peroxiredoxins are druggable targets in human Burkitt lymphoma. Oncotarget 2016, 7, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Pylvas, M.; Puistola, U.; Kauppila, S.; Soini, Y.; Karihtala, P. Oxidative stress-induced antioxidant enzyme expression is an early phenomenon in ovarian carcinogenesis. Eur. J. Cancer 2010, 46, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Sova, H.; Kangas, J.; Puistola, U.; Santala, M.; Liakka, A.; Karihtala, P. Down-regulation of 8-hydroxydeoxyguanosine and peroxiredoxin II in the pathogenesis of endometriosis-associated ovarian cancer. Anticancer Res. 2012, 32, 3037–3044. [Google Scholar] [PubMed]
- Chandimali, N.; Huynh, D.L.; Jin, W.Y.; Kwon, T. Combination Effects of Hispidin and Gemcitabine via Inhibition of Stemness in Pancreatic Cancer Stem Cells. Anticancer Res. 2018, 38, 3967–3975. [Google Scholar] [CrossRef] [PubMed]
- Kinnula, V.L.; Lehtonen, S.; Sormunen, R.; Kaarteenaho-Wiik, R.; Kang, S.W.; Rhee, S.G.; Soini, Y. Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J. Pathol. 2002, 196, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Kontostathi, G.; Zoidakis, J.; Makridakis, M.; Lygirou, V.; Mermelekas, G.; Papadopoulos, T.; Vougas, K.; Vlamis-Gardikas, A.; Drakakis, P.; Loutradis, D.; et al. Cervical Cancer Cell Line Secretome Highlights the Roles of Transforming Growth Factor-Beta-Induced Protein ig-h3, Peroxiredoxin-2, and NRF2 on Cervical Carcinogenesis. BioMed Res. Int. 2017, 2017, 4180703. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandimali, N.; Jeong, D.K.; Kwon, T. Peroxiredoxin II Regulates Cancer Stem Cells and Stemness-Associated Properties of Cancers. Cancers 2018, 10, 305. https://doi.org/10.3390/cancers10090305
Chandimali N, Jeong DK, Kwon T. Peroxiredoxin II Regulates Cancer Stem Cells and Stemness-Associated Properties of Cancers. Cancers. 2018; 10(9):305. https://doi.org/10.3390/cancers10090305
Chicago/Turabian StyleChandimali, Nisansala, Dong Kee Jeong, and Taeho Kwon. 2018. "Peroxiredoxin II Regulates Cancer Stem Cells and Stemness-Associated Properties of Cancers" Cancers 10, no. 9: 305. https://doi.org/10.3390/cancers10090305
APA StyleChandimali, N., Jeong, D. K., & Kwon, T. (2018). Peroxiredoxin II Regulates Cancer Stem Cells and Stemness-Associated Properties of Cancers. Cancers, 10(9), 305. https://doi.org/10.3390/cancers10090305