The SUMO System and TGFβ Signaling Interplay in Regulation of Epithelial-Mesenchymal Transition: Implications for Cancer Progression



Abstract
1. Introduction
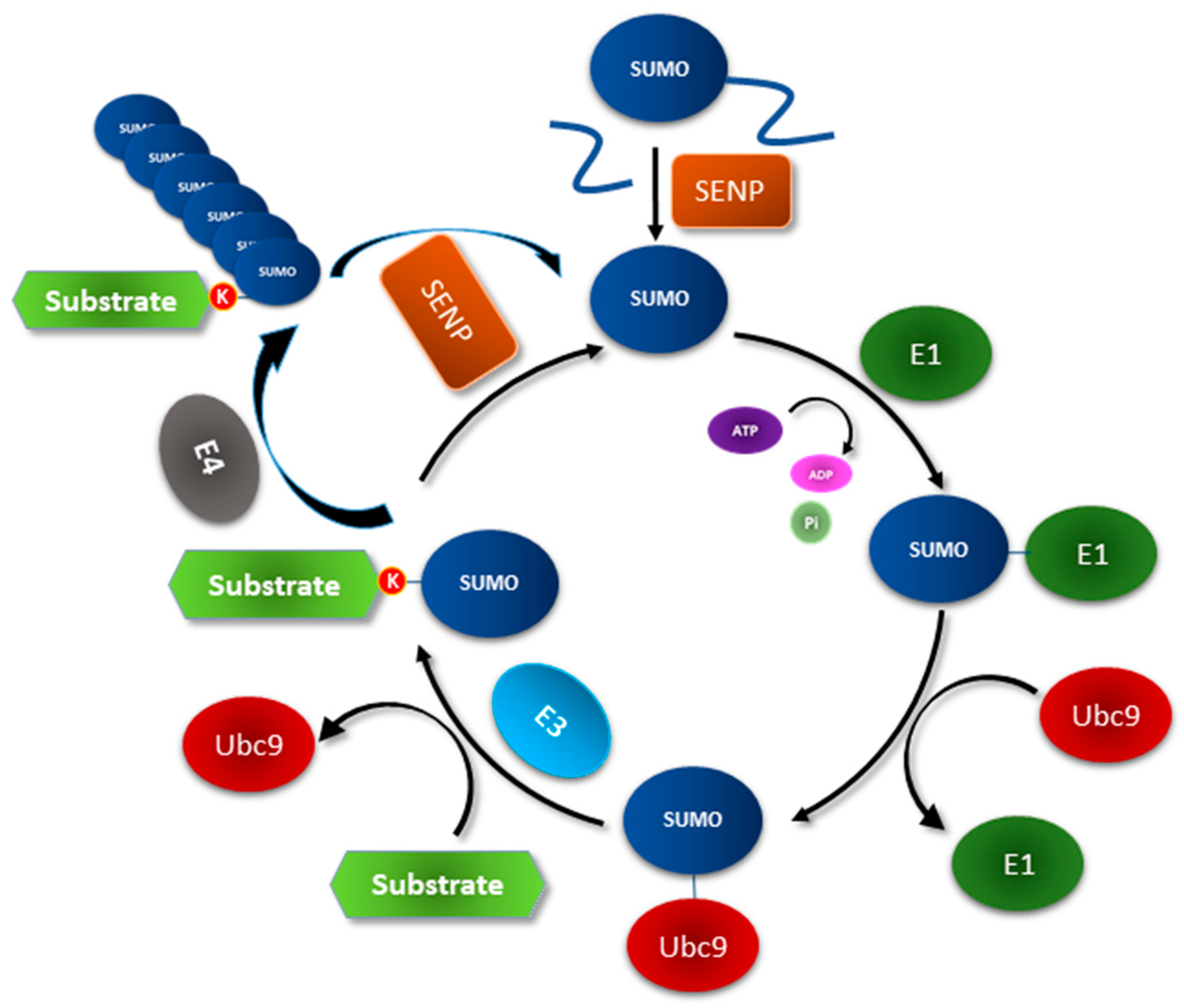
2. The SUMOylation Machinery
3. Epithelial-Mesenchymal Transition (EMT)
4. TGFβ Signaling Pathway
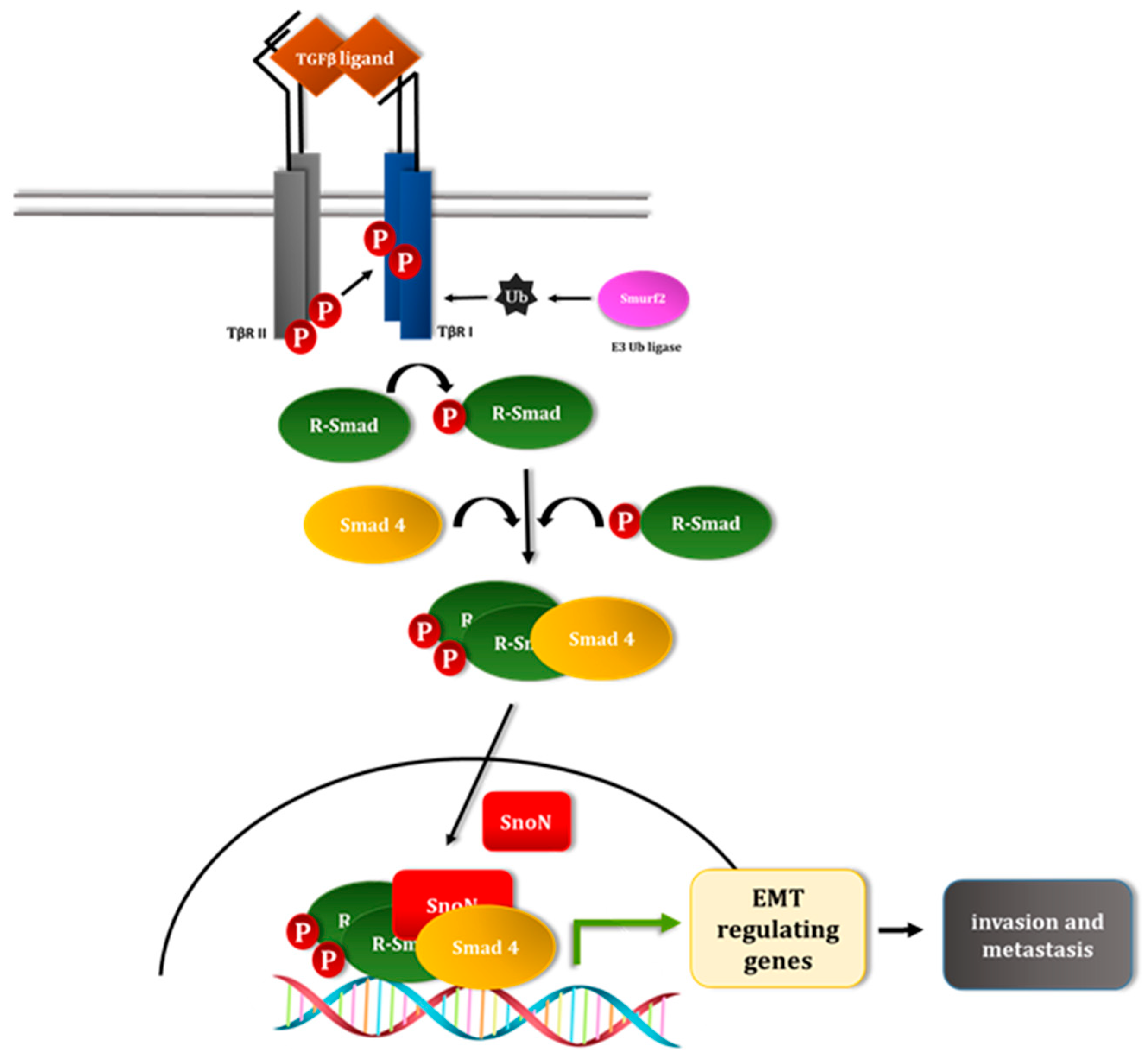
5. SUMOylation of TGFβ Pathway Signal Transducers
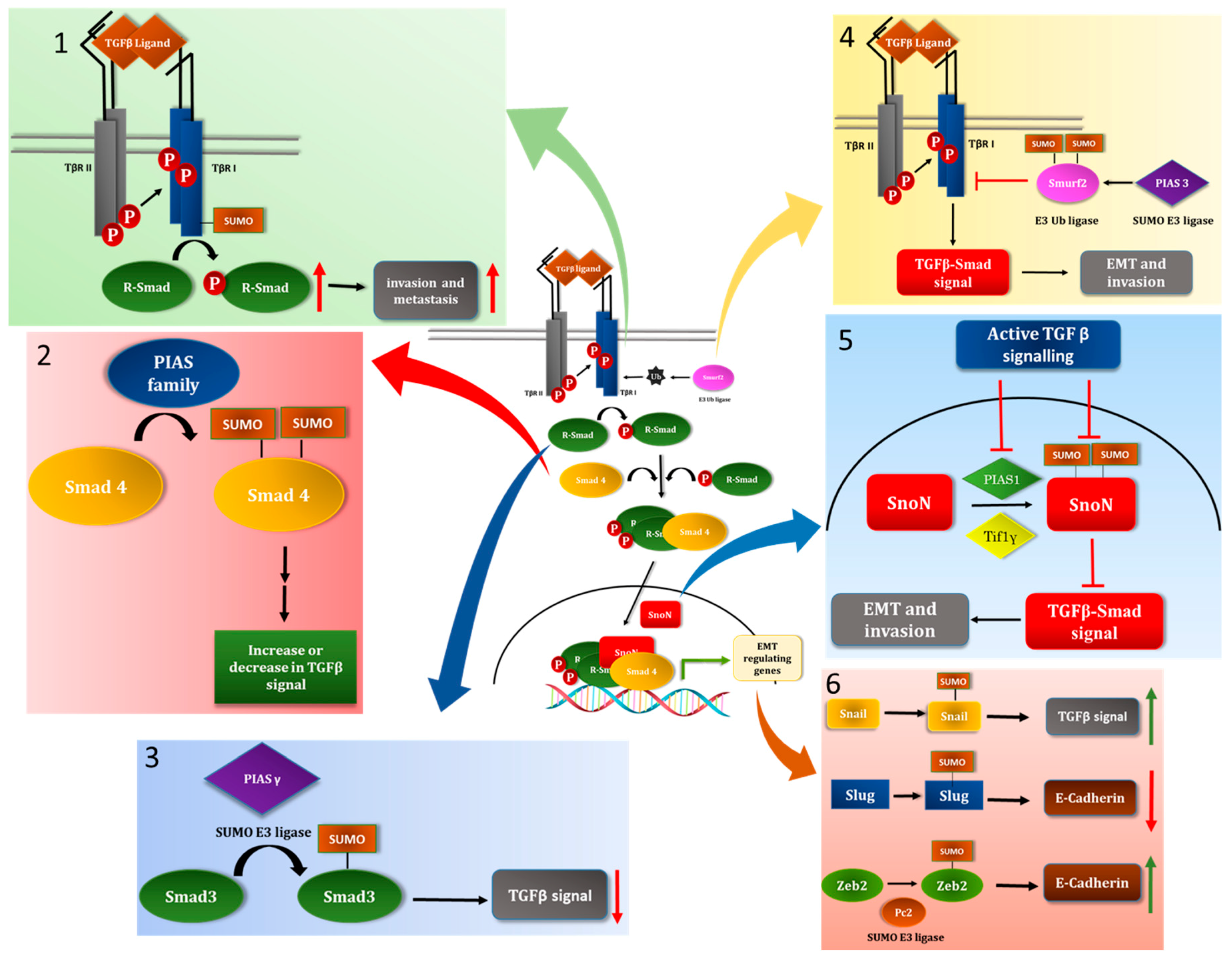
5.1. TGFβ Receptor
5.2. Members of the Sma-Mad (Smad) Family of the Signal Transducers
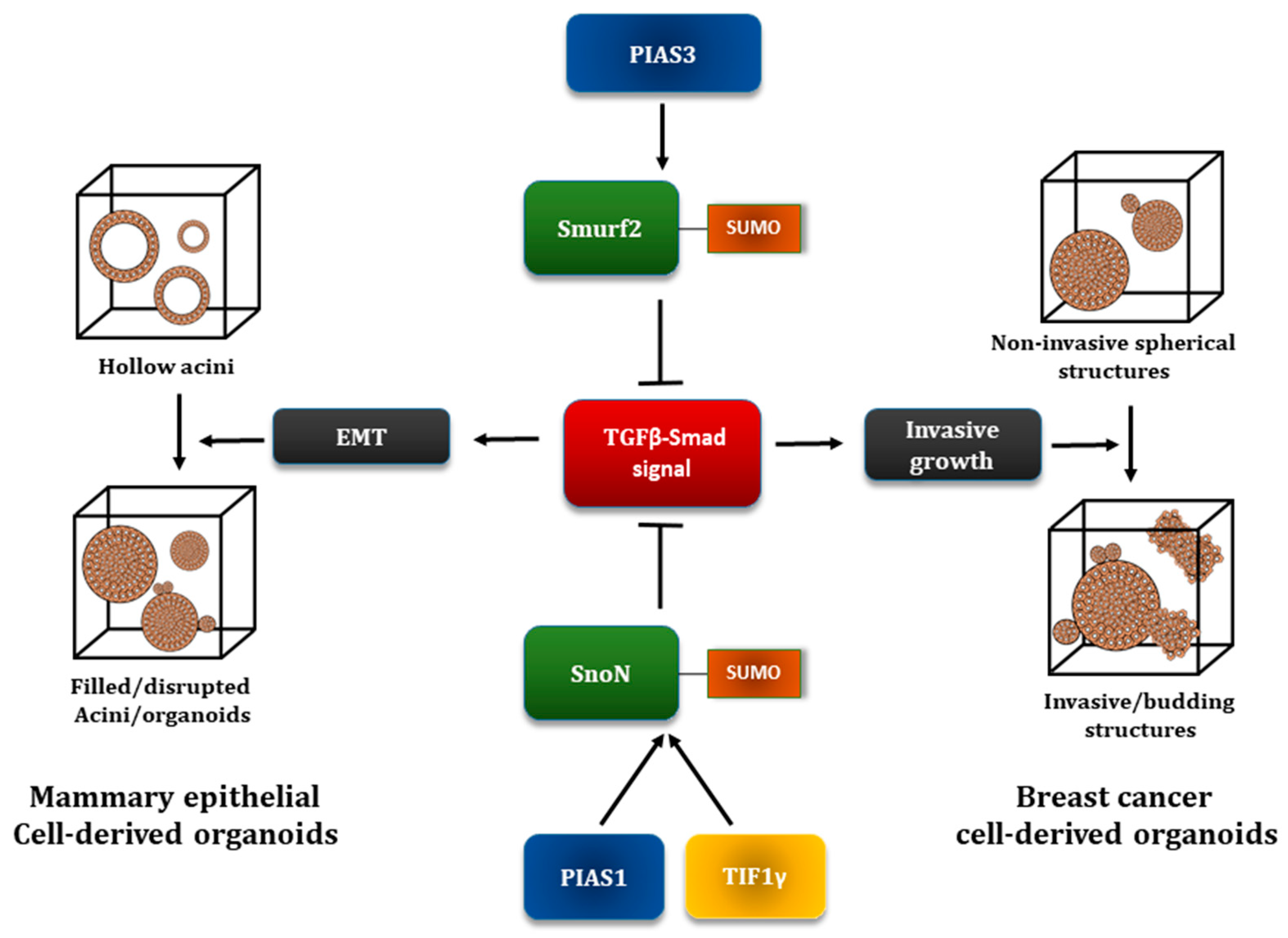
5.3. The E3 Ubiquitin Ligase Smurf2 as a SUMO Substrate
5.4. The Transcriptional Coregulator SnoN as a Target of the SUMO Pathway
6. EMT-TFs as Targets of the SUMO Pathway
6.1. Snail
6.2. Slug
6.3. Zeb2
7. Summary and Future Perspective
7.1. Global Analyses of SUMO System-TGFβ Signaling Interplay
7.2. Therapeutic Targeting of the SUMO Pathway
Author Contributions
Funding
Conflicts of Interest
References
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.; Lindner, A.B. Protein posttranslational modifications: Roles in aging and age-related disease. Oxid. Med. Cell. Longev. 2017, 2017, 5716409. [Google Scholar] [CrossRef] [PubMed]
- Karve, T.M.; Cheema, A.K. Small changes huge impact: The role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids 2011, 2011, 207691. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Benesch, M.; Weis, S.; Haybaeck, J. SUMOylation in carcinogenesis. Cancer Lett. 2012, 316, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, R.J. SUMO protein modification. Biochim. Biophys. Acta 2004, 1695, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Palvimo, J.J. Pias proteins as regulators of small ubiquitin-related modifier (SUMO) modifications and transcription. Biochem. Soc. Trans. 2007, 35, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Bohren, K.M.; Nadkarni, V.; Song, J.H.; Gabbay, K.H.; Owerbach, D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type Ι diabetes mellitus. J. Biol. Chem. 2004, 279, 27233–27238. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Lee, C.C.; Yao, Y.L.; Lai, C.C.; Schmitz, M.L.; Yang, W.M. SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef] [PubMed]
- Seeler, J.S.; Dejean, A. Nuclear and unclear functions of SUMO. Nat. Rev. Mol. Cell Biol. 2003, 4, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.B.; Zanella, C.A.; Henley, J.M.; Cimarosti, H. SUMOylation: Implications for neurodegenerative diseases. Adv. Exp. Med. Biol. 2017, 963, 261–281. [Google Scholar] [PubMed]
- Johnson, E.S. Protein modification by SUMO. Annu. Rev. Biophys. Biomol. 2004, 73, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Netherton, S.J.; Bonni, S. Suppression of TGFβ-induced epithelial-mesenchymal transition like phenotype by a PIAS1 regulated SUMOylation pathway in NMuMG epithelial cells. PLoS ONE 2010, 5, e13971. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C. SUMO chains: Polymeric signals. Biochem. Soc. Trans. 2010, 38, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Eisenhardt, N.; Chaugule, V.K.; Koidl, S.; Droescher, M.; Dogan, E.; Rettich, J.; Sutinen, P.; Imanishi, S.Y.; Hofmann, K.; Palvimo, J.J.; et al. A new vertebrate SUMO enzyme family reveals insights into SUMO-chain assembly. Nat. Struct. Mol. Biol. 2015, 22, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Hecker, C.M.; Rabiller, M.; Haglund, K.; Bayer, P.; Dikic, I. Specification of SUMO1-and SUMO2-interacting motifs. J. Biol. Chem. 2006, 281, 16117–16127. [Google Scholar] [CrossRef] [PubMed]
- Rabellino, A.; Andreani, C.; Scaglioni, P.P. The role of pias SUMO E3-ligases in cancer. Cancer Res. 2017, 77, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Gast, A.; Seeler, J.S.; Dejean, A.; Melchior, F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 2002, 108, 109–120. [Google Scholar] [CrossRef]
- Chu, Y.; Yang, X. SUMO E3 ligase activity of trim proteins. Oncogene 2011, 30, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Melhuish, T.A.; Wotton, D. The polycomb protein Pc2 is a SUMO E3. Cell 2003, 113, 127–137. [Google Scholar] [CrossRef]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Gibbons, D.L.; Kurie, J.M. The role of epithelial-mesenchymal transition programming in invasion and metastasis: A clinical perspective. Cancer Manag. Res. 2013, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type i receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining emt during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31C, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Robson, E.J.; Khaled, W.T.; Abell, K.; Watson, C.J. Epithelial-to-mesenchymal transition confers resistance to apoptosis in three murine mammary epithelial cell lines. Differentiation 2006, 74, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, J.J. The dual role of TGFβ in human cancer: From tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Itoh, F.; Goumans, M.J.; Ten Dijke, P. Signaling of transforming growth factor-beta family members through Smad proteins. Eur. J. Biochem. 2000, 267, 6954–6967. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-beta family signaling by inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.W.; Gomez, E.W. Biomechanics of TGFβ-induced epithelial-mesenchymal transition: Implications for fibrosis and cancer. Clin. Trans. Med. 2014, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Saunier, E.F.; Akhurst, R.J.; Derynck, R. The type Ι TGF-beta receptor is covalently modified and regulated by SUMOylation. Nat. Cell Biol. 2008, 10, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Hietakangas, V.; Anckar, J.; Blomster, H.A.; Fujimoto, M.; Palvimo, J.J.; Nakai, A.; Sistonen, L. Pdsm, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA 2006, 103, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Carter, D.; Garrigue-Antar, L.; Reiss, M. Transforming growth factor beta type Ι receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 1998, 58, 4805–4810. [Google Scholar] [PubMed]
- Chen, T.; Yan, W.; Wells, R.G.; Rimm, D.L.; McNiff, J.; Leffell, D.; Reiss, M. Novel inactivating mutations of transforming growth factor-beta type Ι receptor gene in head-and-neck cancer metastases. Int. J. Cancer 2001, 93, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Martin-Malpartida, P.; Massague, J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Burch, M.L.; Zheng, W.; Little, P.J. Smad linker region phosphorylation in the regulation of extracellular matrix synthesis. Cell Mol. Life Sci. 2011, 68, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Chang, C.; Liu, D.; Derynck, R. SUMOylation of Smad4, the common Smad mediator of transforming growth factor-beta family signaling. J. Biol. Chem. 2003, 278, 27853–27863. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, M.; Liang, Y.Y.; Brunicardi, F.C.; Melchior, F.; Feng, X.H. Activation of transforming growth factor-beta signaling by SUMO-1 modification of tumor suppressor Smad4/DPC4. J. Biol. Chem. 2003, 278, 18714–18719. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Wang, G.; He, D.; Liu, F. Repression of Smad4 transcriptional activity by SUMO modification. Biochem. J. 2004, 379, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Shimotohno, K. Transforming growth factor-beta-mediated signaling via the p38 MAP kinase pathway activates Smad-dependent transcription through SUMO-1 modification of Smad4. J. Biol. Chem. 2003, 278, 50833–50842. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, M.; Liang, Y.Y.; Brunicardi, F.C.; Feng, X.H. SUMO-1/Ubc9 promotes nuclear accumulation and metabolic stability of tumor suppressor Smad4. J. Biol. Chem. 2003, 278, 31043–31048. [Google Scholar] [CrossRef] [PubMed]
- Imoto, S.; Sugiyama, K.; Muromoto, R.; Sato, N.; Yamamoto, T.; Matsuda, T. Regulation of transforming growth factor-beta signaling by protein inhibitor of activated stat, piasy through Smad3. J. Biol. Chem. 2003, 278, 34253–34258. [Google Scholar] [CrossRef] [PubMed]
- Chandhoke, A.S.; Karve, K.; Dadakhujaev, S.; Netherton, S.; Deng, L.; Bonni, S. The ubiquitin ligase Smurf2 suppresses TGFβ-induced epithelial-mesenchymal transition in a SUMOylation-regulated manner. Cell Death Differ. 2016, 23, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Chandhoke, A.S.; Chanda, A.; Karve, K.; Deng, L.; Bonni, S. The PIAS3-Smurf2 SUMOylation pathway suppresses breast cancer organoid invasiveness. Oncotarget 2017, 8, 21001–21014. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, Y.; Dadakhujaev, S.; Chandhoke, A.S.; Huynh, M.A.; Oldenborg, A.; Ikeuchi, M.; Deng, L.; Bennett, E.J.; Harper, J.W.; Bonni, A.; et al. TIF1γ protein regulates epithelial-mesenchymal transition by operating as a small ubiquitin-like modifier (SUMO) E3 ligase for the transcriptional regulator SnoN1. J. Biol. Chem. 2014, 289, 25067–25078. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Chan, A.; Deng, L.; Kornaga, E.N.; Enwere, E.K.; Morris, D.G.; Bonni, S. Identification of the SUMO E3 ligase PIAS1 as a potential survival biomarker in breast cancer. PLoS ONE 2017, 12, e0177639. [Google Scholar] [CrossRef] [PubMed]
- Gudey, S.K.; Sundar, R.; Heldin, C.H.; Bergh, A.; Landstrom, M. Pro-invasive properties of Snail1 are regulated by SUMOylation in response to TGFβ stimulation in cancer. Oncotarget 2017, 8, 97703–97726. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates E-cadherin repression via p19Arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated SUMOylation of Smad-interacting protein 1 attenuates transcriptional repression of e-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gao, C.; Huang, W.; Yang, M.; Chen, G.; Jiang, L.; Gou, F.; Feng, H.; Ai, N.; Xu, Y. High glucose induces SUMOylation of Smad4 via SUMO2/3 in mesangial cells. BioMed Res. Int. 2014, 2014, 782625. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Ma, Y.L.; Liu, Y.C.; Lee, E.H.Y. Smad4 SUMOylation is essential for memory formation through upregulation of the skeletal myopathy gene tpm2. BMC Biol. 2017, 15, 112. [Google Scholar] [CrossRef] [PubMed]
- Xiu, D.; Wang, Z.; Cui, L.; Jiang, J.; Yang, H.; Liu, G. SUMOylation of Smad 4 ameliorates the oxidative stress-induced apoptosis in osteoblasts. Cytokine 2018, 102, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Wang, G.; Matsuura, I.; He, D.; Liu, F. Activation of Smad transcriptional activity by protein inhibitor of activated STAT3 (PIAS3). Proc. Natl. Acad. Sci. USA 2004, 101, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Matsuura, I.; He, D.; Wang, G.; Shuai, K.; Liu, F. Repression of Smad transcriptional activity by piasy, an inhibitor of activated stat. Proc. Natl. Acad. Sci. USA 2003, 100, 9791–9796. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kavsak, P.; Abdollah, S.; Wrana, J.L.; Thomsen, G.H. A Smad ubiquitin ligase targets the bmp pathway and affects embryonic pattern formation. Nature 1999, 400, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, M.; Feng, X.H. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J. Biol. chem. 2000, 275, 36818–36822. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Imamura, T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008, 99, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Bonni, S.; Wang, H.R.; Causing, C.G.; Kavsak, P.; Stroschein, S.L.; Luo, K.; Wrana, J.L. TGF-beta induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat. Cell Biol. 2001, 3, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Dadakhujaev, S.; Salazar-Arcila, C.; Netherton, S.J.; Chandhoke, A.S.; Singla, A.K.; Jirik, F.R.; Bonni, S. A novel role for the SUMO E3 ligase PIAS1 in cancer metastasis. Oncoscience 2014, 1, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Banerjee, S. Differential pias3 expression in human malignancy. Oncol. Rep. 2004, 11, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Bonni, S.; Bonni, A. SnoN signaling in proliferating cells and postmitotic neurons. FEBS Lett. 2012, 586, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Deheuninck, J.; Luo, K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009, 19, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Stroschein, S.L.; Wang, W.; Zhou, S.; Zhou, Q.; Luo, K. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science 1999, 286, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, Y.; Stegmuller, J.; Netherton, S.; Huynh, M.A.; Masu, M.; Frank, D.; Bonni, S.; Bonni, A. A SnoN-Ccd1 pathway promotes axonal morphogenesis in the mammalian brain. J. Neurosci. 2009, 29, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Sarker, K.P.; Wilson, S.M.; Bonni, S. SnoN is a cell type-specific mediator of transforming growth factor-beta responses. J. Biol. Chem. 2005, 280, 13037–13046. [Google Scholar] [CrossRef] [PubMed]
- Wallden, K.; Nyman, T.; Hallberg, B.M. SnoN stabilizes the Smad3/Smad4 protein complex. Sci. Rep. 2017, 7, 46370. [Google Scholar] [CrossRef] [PubMed]
- Mizuide, M.; Hara, T.; Furuya, T.; Takeda, M.; Kusanagi, K.; Inada, Y.; Mori, M.; Imamura, T.; Miyazawa, K.; Miyazono, K. Two short segments of Smad3 are important for specific interaction of Smad3 with c-Ski and SnoN. J. Biol. Chem. 2003, 278, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Khan, M.M.; Kaul, S.C.; Dong, H.D.; Wadhwa, R.; Colmenares, C.; Kohno, I.; Ishii, S. Ski is a component of the histone deacetylase complex required for transcriptional repression by mad and thyroid hormone receptor. Genes Dev. 1999, 13, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Xu, C.; Zheng, X.; Yuan, H.; Liu, M.; Qiu, Y.; Chen, J. SnoN suppresses TGF-beta-induced epithelial-mesenchymal transition and invasion of bladder cancer in a tif1gamma-dependent manner. Oncol. Rep. 2016, 36, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- De Herreros, A.G.; Peiro, S.; Nassour, M.; Savagner, P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J. Mammary Gland Biol. 2010, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The role of Snail in emt and tumorigenesis. Curr. Cancer Drug Target 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by gsk-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. Traf6 ubiquitinates TGFβ type Ι receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; Zang, S.; Li, H.; Zimmerman, L.J.; Champer, J.; Tsuyada, A.; Chow, A.; Zhou, W.; Yu, Y.; Gao, H.; et al. Nuclear translocation of type Ι transforming growth factor beta receptor confers a novel function in RNA processing. Mol. Cell Biol. 2012, 32, 2183–2195. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, W.; Kuo, M.L.; Tago, K.; Williams, R.T.; Sherr, C.J. Ubiquitination of, and SUMOylation by, the Arf tumor suppressor. Israel Med. Assoc. J. 2006, 8, 249–251. [Google Scholar]
- Neo, S.H.; Itahana, Y.; Alagu, J.; Kitagawa, M.; Guo, A.K.; Lee, S.H.; Tang, K.; Itahana, K. TRIM28 is an E3 ligase for Arf-mediated NPM1/B23 SUMOylation that represses centrosome amplification. Mol. Cell. Biol. 2015, 35, 2851–2863. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Munoz-Antonia, T.; Cress, W.D. TRIM28 contributes to EMT via regulation of E-cadherin and N-cadherin in lung cancer cell lines. PLoS ONE 2014, 9, e101040. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Cheng, J.; Zhou, B.; Zhu, L.; Khan, M.A.; He, T.; Zhou, S.; He, J.; Lu, X.; Chen, H.; et al. Tripartite motif containing 28 (TRIM28) promotes breast cancer metastasis by stabilizing TWIST1 protein. Sci. Rep. 2016, 6, 29822. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Shi, Y.; Sawada, J.; Sui, G.; Affar el, B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Mendler, L.; Braun, T.; Muller, S. The ubiquitin-like SUMO system and heart function: From development to disease. Circ. Res. 2016, 118, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2009, 2, ra24. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Thompson, J.W.; Wang, Z.; Wang, L.; Sheng, H.; Foster, M.W.; Moseley, M.A.; Paschen, W. Analysis of oxygen/glucose-deprivation-induced changes in SUMO3 conjugation using silac-based quantitative proteomics. J. Proteome Res. 2012, 11, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Bossis, G.; Melchior, F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell 2006, 21, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lam, L.S.; Lam, L.H.; Chau, S.F.; Ng, T.B.; Au, S.W. Molecular basis of the redox regulation of SUMO proteases: A protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008, 22, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Eifler, K.; Vertegaal, A.C. Mapping the SUMOylated landscape. FEBS J. 2015, 282, 3669–3680. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Barysch, S.V.; Karaca, S.; Dittner, C.; Hsiao, H.H.; Berriel Diaz, M.; Herzig, S.; Urlaub, H.; Melchior, F. Detecting endogenous SUMO targets in mammalian cells and tissues. Nat. Struct. Mol. Biol. 2013, 20, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Tatham, M.H.; Plechanovova, A.; Matic, I.; Garg, A.K.; Hay, R.T. Purification and identification of endogenous polySUMO conjugates. EMBO Rep. 2011, 12, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; D’Souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Tammsalu, T.; Matic, I.; Jaffray, E.G.; Ibrahim, A.F.M.; Tatham, M.H.; Hay, R.T. Proteome-wide identification of SUMO2 modification sites. Sci. Signal. 2014, 7, rs2. [Google Scholar] [CrossRef] [PubMed]
- Petushkova, N.A.; Zgoda, V.G.; Pyatnitskiy, M.A.; Larina, O.V.; Teryaeva, N.B.; Potapov, A.A.; Lisitsa, A.V. Post-translational modifications of FDA-approved plasma biomarkers in glioblastoma samples. PLoS ONE 2017, 12, e0177427. [Google Scholar] [CrossRef] [PubMed]
- Noberini, R.; Uggetti, A.; Pruneri, G.; Minucci, S.; Bonaldi, T. Pathology tissue-quantitative mass spectrometry analysis to profile histone post-translational modification patterns in patient samples. Mol. Cell. Proteom. 2016, 15, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Bossis, G.; Sarry, J.E.; Kifagi, C.; Ristic, M.; Saland, E.; Vergez, F.; Salem, T.; Boutzen, H.; Baik, H.; Brockly, F.; et al. The ROS/SUMO axis contributes to the response of acute myeloid leukemia cells to chemotherapeutic drugs. Cell Rep. 2014, 7, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Chen, B.; He, L.; Tang, Y.; Jiang, Z.; Yin, G.; Wang, J.; Jiang, X. Anacardic acid (6-pentadecylsalicylic acid) induces apoptosis of prostate cancer cells through inhibition of androgen receptor and activation of p53 signaling. Chin. J. Cancer Res. 2012, 24, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.J.; Muluhngwi, P.; Alizadeh-Rad, N.; Green, M.A.; Rouchka, E.C.; Waigel, S.J.; Klinge, C.M. Genome-wide miRNA response to anacardic acid in breast cancer cells. PLoS ONE 2017, 12, e0184471. [Google Scholar] [CrossRef] [PubMed]
- Xiu, Y.L.; Zhao, Y.; Gou, W.F.; Chen, S.; Takano, Y.; Zheng, H.C. Anacardic acid enhances the proliferation of human ovarian cancer cells. PLoS ONE 2014, 9, e99361. [Google Scholar] [CrossRef] [PubMed]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef] [PubMed]
- Bogachek, M.V.; De Andrade, J.P.; Weigel, R.J. Regulation of epithelial-mesenchymal transition through SUMOylation of transcription factors. Cancer Res. 2015, 75, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y. Advances in the development of SUMO specific protease (SENP) inhibitors. Comput. Struct. Biotechnol. 2015, 13, 204–211. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SUMO Substrate | Effect on Transcriptional Responses | Effect on Biological Responses | References |
|---|---|---|---|
| TGFβ Receptor I | Not reported | Promotes TGFβ-induced invasion and lung metastasis of Ras-transformed fibroblasts. | [44] |
| Smad 3 and 4 | Positive or negative in a cell and context dependent manner | Not reported | [50,51,52,53,54] |
| Smurf2 | Not reported | Supresses TGFβ-induced EMT and invasive growth in non-transformed and transformed mammary cells respectively. | [56,57] |
| SnoN | Supresses TGFβ-induced gene expression in multiple cell types | Supresses TGFβ-induced EMT and invasive growth in non-transformed and transformed mammary cells, respectively. Similar effect may occur in bladder cancer. | [14,58,59,84] |
| Snail | Promotes c-Jun-Snail complex induced gene expression in different cancer cells. | Promotes TGFβ-induced migration and invasion of prostate and breast cancer cells. | [60] |
| Slug | Not reported | Promotes TGFβ-induced migration and invasion of prostate cancer cells. | [61] |
| Zeb2 | Suppresses ability to bind to E-cadherin promoter. | Supresses EMT but effect on migration and invasion of tumor cells needs further analyses. | [62] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanda, A.; Sarkar, A.; Bonni, S. The SUMO System and TGFβ Signaling Interplay in Regulation of Epithelial-Mesenchymal Transition: Implications for Cancer Progression. Cancers 2018, 10, 264. https://doi.org/10.3390/cancers10080264
Chanda A, Sarkar A, Bonni S. The SUMO System and TGFβ Signaling Interplay in Regulation of Epithelial-Mesenchymal Transition: Implications for Cancer Progression. Cancers. 2018; 10(8):264. https://doi.org/10.3390/cancers10080264
Chicago/Turabian StyleChanda, Ayan, Anusi Sarkar, and Shirin Bonni. 2018. "The SUMO System and TGFβ Signaling Interplay in Regulation of Epithelial-Mesenchymal Transition: Implications for Cancer Progression" Cancers 10, no. 8: 264. https://doi.org/10.3390/cancers10080264
APA StyleChanda, A., Sarkar, A., & Bonni, S. (2018). The SUMO System and TGFβ Signaling Interplay in Regulation of Epithelial-Mesenchymal Transition: Implications for Cancer Progression. Cancers, 10(8), 264. https://doi.org/10.3390/cancers10080264