Viability Assessment Following Anticancer Treatment Requires Single-Cell Visualization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
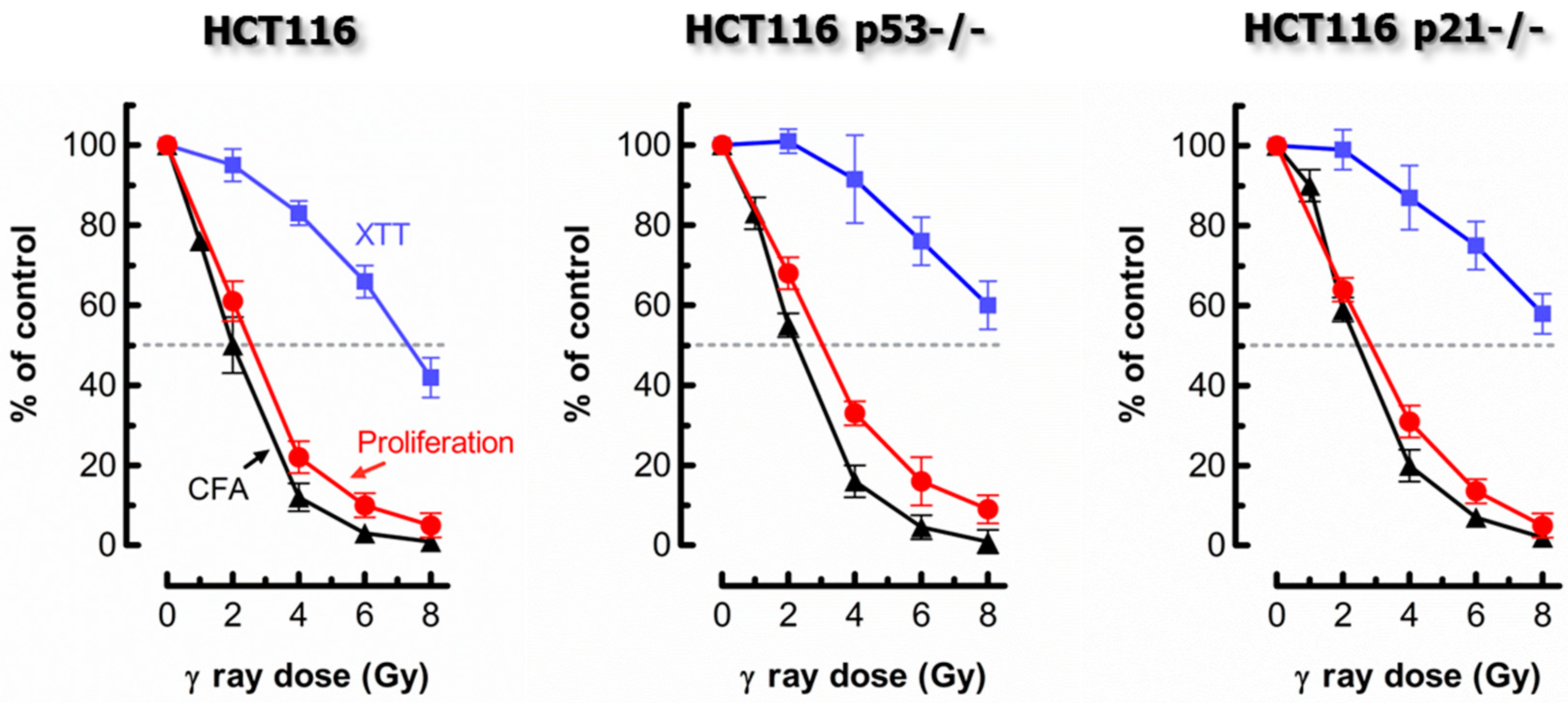
2. Comparing Proliferation Arrest and Colony Formation Assays for Genotoxicity Assessment
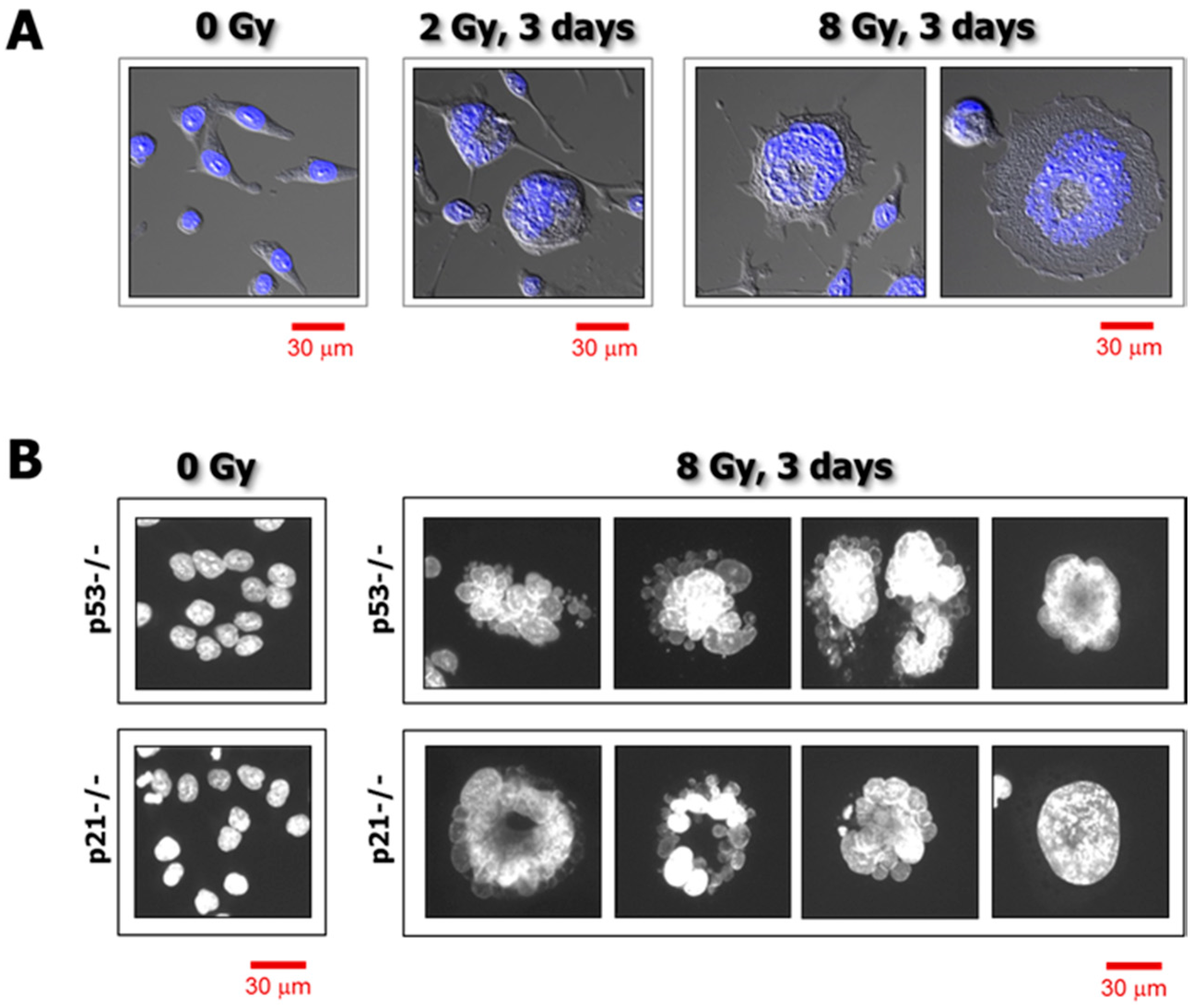
3. Single-Cell Analysis of Cancer Cell Response to Genotoxic Stress
4. Surviving Stress-Induced Apoptotic Signaling
4.1. Apoptotic and Non-Apoptotic Functions of Caspase-3
4.2. Reversibility of Apoptosis in Solid Tumor-Derived Cell Lines
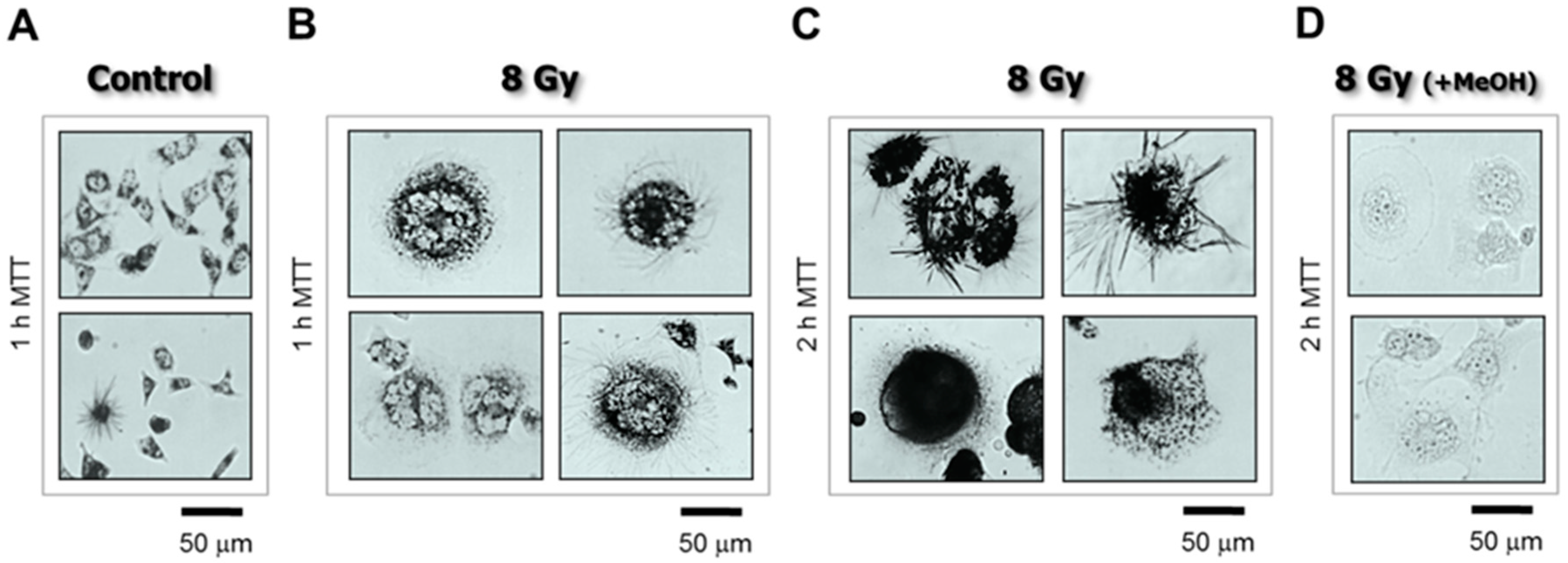
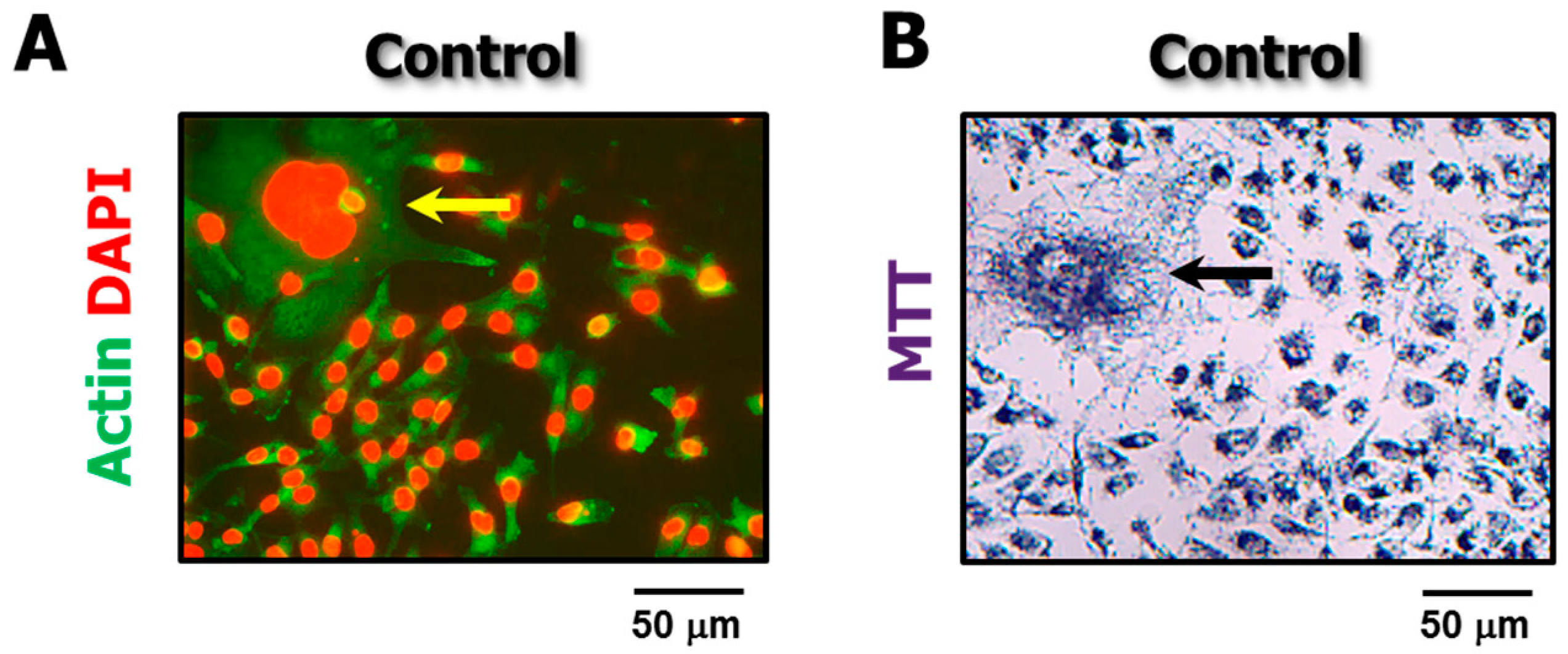
4.3. The Single-Cell MTT Viability Assay: Simple and Sensitive
- Cells were plated in 35 mm dishes (~20,000 cells/2 mL medium/dish) and incubated overnight.
- The cells were then either exposed to γ radiation or sham-irradiated (controls) and incubated for 3 days.
- The medium was replaced with fresh medium containing MTT (final concentration, 0.5 mg/mL) and the cells were returned to the incubator.
- After incubation with MTT for 1–2 h, the cells were viewed under a microscope (bright-field; 20× objective) and their images acquired.
5. Conclusions
Funding
Conflicts of Interest
References
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of polyploid/multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated giant cancer cells produced in response to ionizing radiation retain viability and replicate their genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Impact of premature senescence on radiosensitivity measured by high throughput cell-based assays. Int. J. Mol. Sci. 2017, 18, 1460. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Do multiwell plate high throughput assays measure loss of cell viability following exposure to genotoxic agents? Int. J. Mol. Sci. 2017, 18, 1679. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Waters, R. DNA damage and its repair in human normal or xeroderma pigmentosum fibroblasts treated with 4-nitroquinoline 1-oxide or its 3-methyl derivative. Carcinogenesis 1982, 2, 1359–1362. [Google Scholar] [CrossRef]
- Flusberg, D.A.; Sorger, P.K. Surviving apoptosis: Life-death signaling in single cells. Trends Cell Biol. 2015, 25, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the cell cycle: A complex landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Garijo, A. When dying is not the end: Apoptotic caspases as drivers of proliferation. Semin. Cell Dev. Biol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Niepel, M.; Spencer, S.L.; Sorger, P.K. Non-genetic cell-to-cell variability and the consequences for pharmacology. Curr. Opin. Chem. Biol. 2009, 13, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Loewer, A. Recent progress and open challenges in modeling p53 dynamics in single cells. Curr. Opin. Syst. Biol. 2017, 3, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Albeck, J.G.; Burke, J.M.; Aldridge, B.B.; Zhang, M.; Lauffenburger, D.A.; Sorger, P.K. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol. Cell 2008, 30, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Feinerman, O.; Veiga, J.; Dorfman, J.R.; Germain, R.N.; Altan-Bonnet, G. Variability and robustness in T cell activation from regulated heterogeneity in protein levels. Science 2008, 321, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. Cancer cells display profound intra and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Tang, Y.; Shi, J.; Loy, C.T.; Amendt, C.; Wilm, C.; Zenke, F.T.; Mitchison, T.J. Quantitative live imaging of cancer and normal cells treated with kinesin-5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol. Cancer Ther. 2008, 7, 3480–3489. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Nongenetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Irish, J.M.; Hovland, R.; Krutzik, P.O.; Perez, O.D.; Bruserud, O.; Gjertsen, B.T.; Nolan, G.P. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 2004, 118, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; Frenkel-Morgenstern, M.; Issaeva, I.; Sigal, A.; Milo, R.; Cohen-Saidon, C.; Liron, Y.; Kam, Z.; et al. Dynamic proteomics of individual cancer cells in response to a drug. Science 2008, 322, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Geva-Zatorsky, N.; Rosenfeld, N.; Itzkovitz, S.; Milo, R.; Sigal, A.; Dekel, E.; Yarnitzky, T.; Liron, Y.; Polak, P.; Lahav, G.; et al. Oscillations and variability in the p53 system. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 2018, 173, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Engle, J.T.; Griffin, E.A.; Miller, J.P.; Chu, W.; Zhou, D.; Mach, R.H. Imaging caspase-3 activation as a marker of apoptosis-targeted treatment response in cancer. Mol. Imaging Biol. 2015, 17, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E.; Cioccari, L.; Jakob, S.; Vallan, C.; Schaffner, T.; Zimmermann, A.; Brunner, T. Active caspase 3 and DNA fragmentation as markers for apoptotic cell death in primary and metastatic liver tumours. Pathology 2007, 39, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Jonges, L.E.; Nagelkerke, J.F.; Ensink, N.G.; van der Velde, E.A.; Tollenaar, R.A.; Fleuren, G.J.; van de Velde, C.J.; Morreau, H.; Kuppen, P.J. Caspase-3 activity as a prognostic factor in colorectal carcinoma. Lab. Investig. 2001, 81, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.Y. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.Y. Caspase-3 promotes genetic instability and carcinogenesis. Mol. Cell 2015, 58, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yu, Y.; He, S.; Cheng, J.; Gong, Y.; Zhang, Z.; Yang, X.; Xu, B.; Liu, X.; Li, C.Y.; et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017, 385, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low levels of caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016, 7, e2087. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. The growing complexity of cancer cell response to DNA-damaging agents: Caspase 3 mediates cell death or survival? Int. J. Mol. Sci. 2016, 17, 708. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: Impact on chemotherapy outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed]
- Esmatabadi, M.J.D.; Bakhshinejad, B.; Motalegh, F.M.; Babashah, S.; Sadeghizadeh, M. Therapeutic resistance and cancer recurrence mechanisms: Unfolding the story of tumour coming back. J. Biosci. 2016, 41, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Yuen, K.L.; Tang, H.M.; Fung, M.C. Reversibility of apoptosis in cancer cells. Br. J. Cancer 2009, 100, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Fung, M.C.; Hardwick, J.M. In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci. Rep. 2015, 5, 9015. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Hardwick, J.M.; Fung, M.C. Strategies for tracking anastasis, a cell survival phenomenon that reverses apoptosis. J. Vis. Exp. JoVE 2015, 96, e51964. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Talbot, Jr. C.C.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda MD, USA, 2004. [Google Scholar]
- Riss, T.; O’Brien, M.; Moravec, R. Selecting cell-based assays for drug discovery screening. Cell Notes 2005, 13, 16–21. [Google Scholar]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [PubMed]
- Liu, Y.; Peterson, D.A.; Kimura, H.; Schubert, D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem. 1997, 69, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. Characterisation of the cellular reduction of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M. Vital dyes and virtual deaths. Cell Death Differ. 2013, 20, 963. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, K.; Oehling, V.; Dusny, C.; Schmid, A. Beyond the bulk: Disclosing the life of single microbial cells. FEMS Microbiol. Rev. 2017, 41, 751–780. [Google Scholar] [CrossRef] [PubMed]
- Rantalainen, M. Application of single-cell sequencing in human cancer. Brief. Funct. Genom. 2017, 17, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Tellez-Gabriel, M.; Ory, B.; Lamoureux, F.; Heymann, M.F.; Heymann, D. Tumour heterogeneity: The key advantages of single-cell analysis. Int. J. Mol. Sci. 2016, 17, 2142. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed]
- Saadatpour, A.; Lai, S.; Guo, G.; Yuan, G.H. Single-cell analysis in cancer genomics. Trends Genet. 2015, 31, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20. [Google Scholar]
- Thomas, F.; Ujvari, B.; Renaud, F.; Vincent, M. Cancer adaptations: Atavism, de novo selection, or something in between? Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, F.; Xing, K.; He, X. The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nat. Commun. 2015, 6, 6367. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M. Resistance to cancer chemotherapy as an atavism? Bioessays 2016, 38, 1065. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Carcinogenesis: Recent insights in protist stem cell biology lead to a better understanding of atavistic mechanisms implied in cancer development. MOJ Tumor Res. 2018, 1, 18–29. [Google Scholar]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzayans, R.; Andrais, B.; Murray, D. Viability Assessment Following Anticancer Treatment Requires Single-Cell Visualization. Cancers 2018, 10, 255. https://doi.org/10.3390/cancers10080255
Mirzayans R, Andrais B, Murray D. Viability Assessment Following Anticancer Treatment Requires Single-Cell Visualization. Cancers. 2018; 10(8):255. https://doi.org/10.3390/cancers10080255
Chicago/Turabian StyleMirzayans, Razmik, Bonnie Andrais, and David Murray. 2018. "Viability Assessment Following Anticancer Treatment Requires Single-Cell Visualization" Cancers 10, no. 8: 255. https://doi.org/10.3390/cancers10080255
APA StyleMirzayans, R., Andrais, B., & Murray, D. (2018). Viability Assessment Following Anticancer Treatment Requires Single-Cell Visualization. Cancers, 10(8), 255. https://doi.org/10.3390/cancers10080255