The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer


Abstract
1. Inflammation and EOC
1.1. Signaling Pathways and Transcription Factors
1.2. Innate Immune Response
1.3. Adaptive Immune Response
2. Inflammation as a Risk Factor for EOC
2.1. Ovulation
2.2. Infection
2.3. Other Sources of Inflammation
2.4. NSAIDS and Reduced Risk of EOC
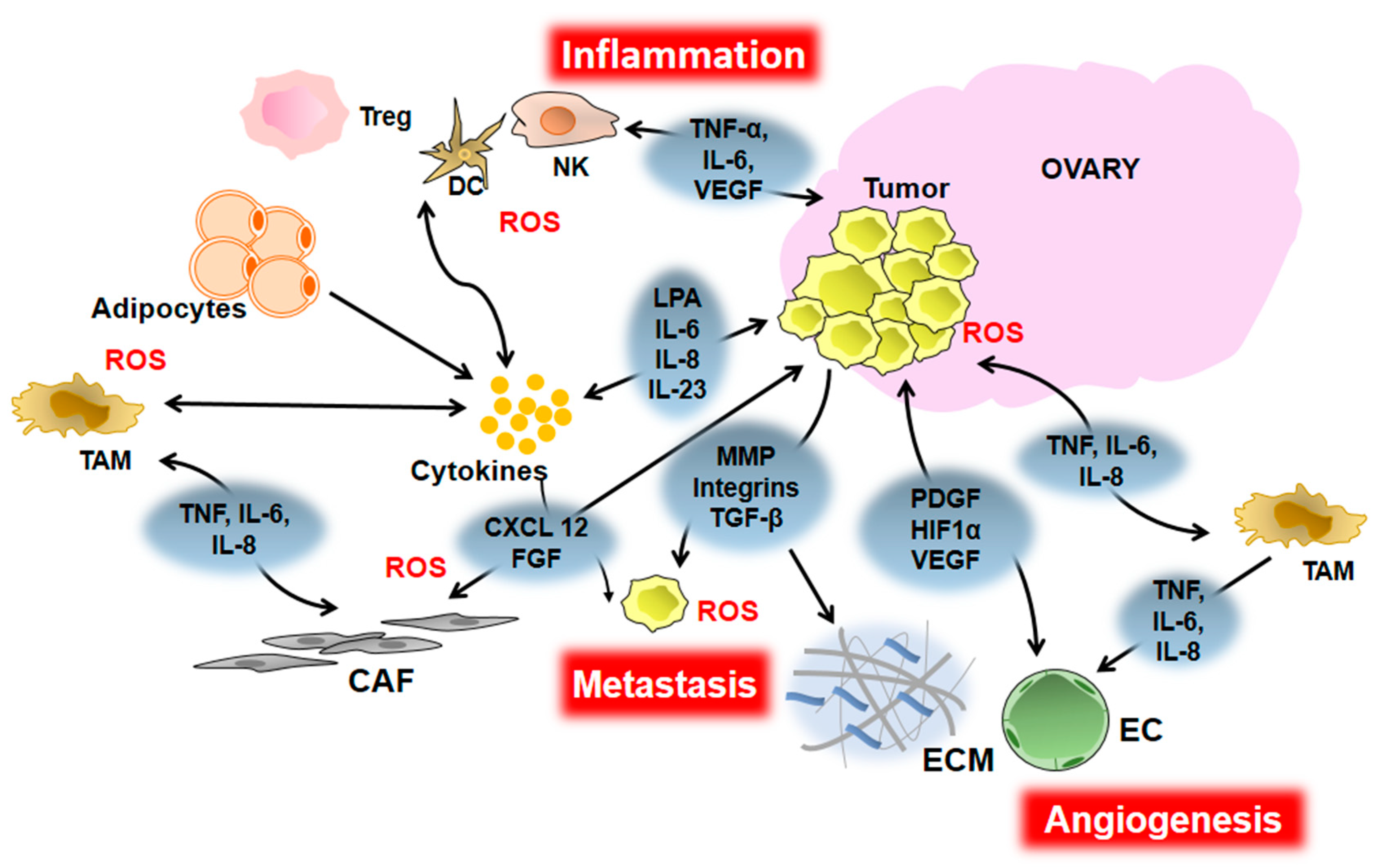
3. Inflammation and EOC Initiation and Progression
3.1. ROS and Oxidative Stress
3.2. TNF-α
3.3. IL-6
3.4. IL-8
3.5. Lyophosphotidic Acid (LPA)
3.6. Prostaglandins and COX-1 and COX-2
4. Inflammation and EOC Angiogenesis
4.1. TNF-α
4.2. IL-6
4.3. IL-8
4.4. LPA
5. Inflammation and EOC Metastasis
5.1. ROS
5.2. TNF-α
5.3. IL-6
5.4. IL-8
5.5. LPA
5.6. TGF-β
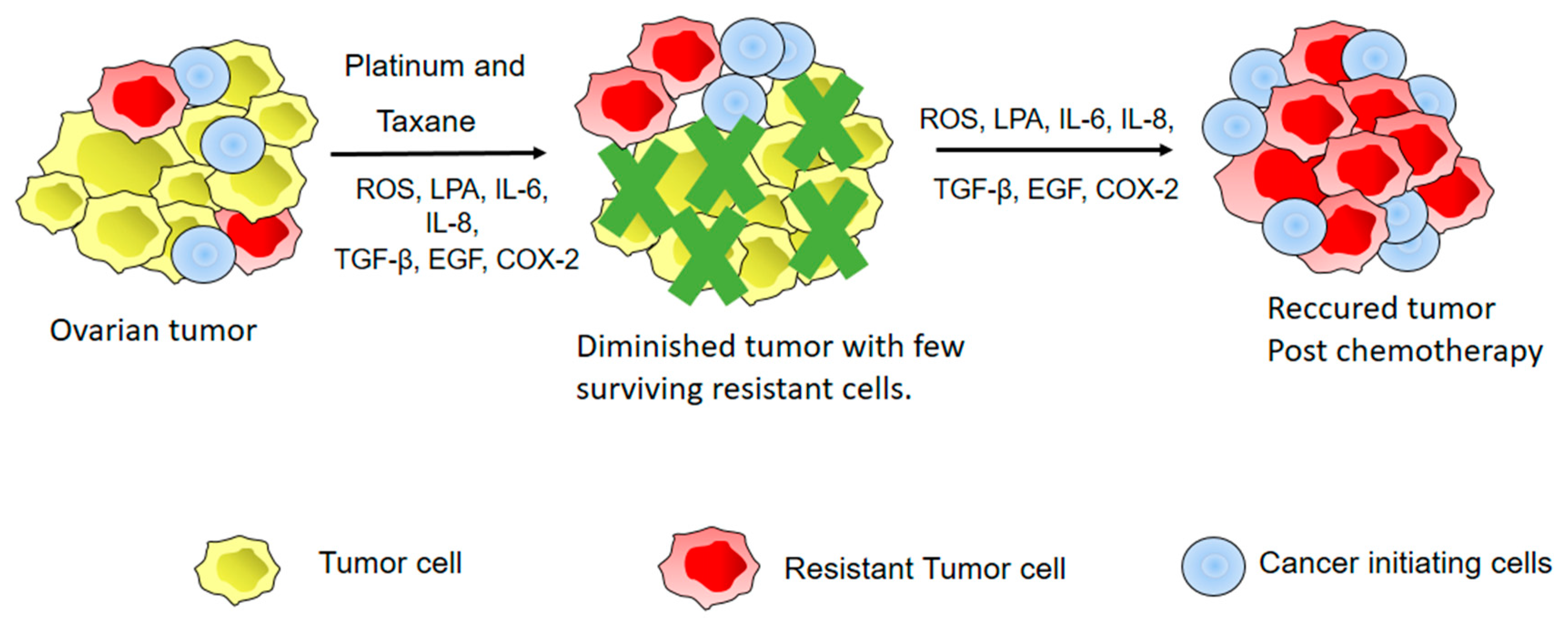
6. Inflammation and EOC Chemoresistance
6.1. ROS
6.2. IL-6
6.3. IL-8
6.4. LPA
6.5. TGF-β and EGF
6.6. COX-2
7. Treatment Strategies Targeting Inflammatory Mediators in EOC
8. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maiuri, A.R.; O’Hagan, H.M. Interplay Between Inflammation and Epigenetic Changes in Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 69–117. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Clendenen, T.V.; Lundin, E.; Zeleniuch-Jacquotte, A.; Koenig, K.L.; Berrino, F.; Lukanova, A.; Lokshin, A.E.; Idahl, A.; Ohlson, N.; Hallmans, G.; et al. Circulating inflammation markers and risk of epithelial ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Wei, L.H.; Kuo, M.L.; Huang, Y.J.; Lai, K.P.; Chen, C.A.; Hsieh, C.Y. Up-regulation of interleukin-6 in human ovarian cancer cell via a Gi/PI3K-Akt/NF-κB pathway by lysophosphatidic acid, an ovarian cancer-activating factor. Carcinogenesis 2005, 26, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Ferrari, D.; Riehemann, K.; Wesselborg, S. Regulation of NF-κ B activation by MAP kinase cascades. Immunobiology 1997, 198, 35–49. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.B.; Rowell, E.; Sekimata, M. Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 2009, 9, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Tone, A.A.; Salvador, S.; Finlayson, S.J.; Tinker, A.V.; Kwon, J.S.; Lee, C.H.; Cohen, T.; Ehlen, T.; Lee, M.; Carey, M.S.; et al. The role of the fallopian tube in ovarian cancer. Clin. Adv. Hematol. Oncol. 2012, 10, 296–306. [Google Scholar] [PubMed]
- Petrovska, M.; Dimitrov, D.G.; Michael, S.D. Quantitative changes in macrophage distribution in normal mouse ovary over the course of the estrous cycle examined with an image analysis system. Am. J. Reprod. Immunol. 1996, 36, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Takaya, R.; Fukaya, T.; Sasano, H.; Suzuki, T.; Tamura, M.; Yajima, A. Macrophages in normal cycling human ovaries; immunohistochemical localization and characterization. Hum. Reprod. 1997, 12, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Van der Hoek, K.H.; Ryan, N.K.; Norman, R.J.; Robker, R.L. Macrophage contributions to ovarian function. Hum. Reprod. Update 2004, 10, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Tingen, C.M.; Kiesewetter, S.E.; Jozefik, J.; Thomas, C.; Tagler, D.; Shea, L.; Woodruff, T.K. A macrophage and theca cell-enriched stromal cell population influences growth and survival of immature murine follicles in vitro. Reproduction 2011, 141, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Kollara, A.; St John, E.; Tone, A.A.; Virtanen, C.; Greenblatt, E.M.; King, W.A.; Brown, T.J. Altered expression of inflammation-associated genes in oviductal cells following follicular fluid exposure: Implications for ovarian carcinogenesis. Exp. Biol. Med. 2014, 239, 24–32. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Hilliard, T.S.; Wu, L.Y.; Jaffe, R.C.; Fazleabas, A.T.; Burdette, J.E. The impact of ovulation on fallopian tube epithelial cells: Evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr. Relat. Cancer 2011, 18, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.A.; Giles, J.R. The hen as a model of ovarian cancer. Nat. Rev. Cancer 2013, 13, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.T.; Wu, Q.J.; Vogtmann, E.; Lin, B.; Wang, Y.L. Age at menarche and risk of ovarian cancer: A meta-analysis of epidemiological studies. Int. J. Cancer 2013, 132, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Chiaffarino, F.; Pelucchi, C.; Parazzini, F.; Negri, E.; Franceschi, S.; Talamini, R.; Conti, E.; Montella, M.; La Vecchia, C. Reproductive and hormonal factors and ovarian cancer. Ann. Oncol. 2001, 12, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Fortner, R.T.; Ose, J.; Merritt, M.A.; Schock, H.; Tjonneland, A.; Hansen, L.; Overvad, K.; Dossus, L.; Clavel-Chapelon, F.; Baglietto, L.; et al. Reproductive and hormone-related risk factors for epithelial ovarian cancer by histologic pathways, invasiveness and histologic subtypes: Results from the EPIC cohort. Int. J. Cancer 2015, 137, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Espey, L.L. Ovulation as an inflammatory reaction—A hypothesis. Biol. Reprod. 1980, 22, 73–106. [Google Scholar] [CrossRef] [PubMed]
- Machelon, V.; Emilie, D. Production of ovarian cytokines and their role in ovulation in the mammalian ovary. Eur. Cytokine Netw. 1997, 8, 137–143. [Google Scholar] [PubMed]
- Chumduri, C.; Gurumurthy, R.K.; Zadora, P.K.; Mi, Y.; Meyer, T.F. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell. Host Microbe 2013, 13, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Ingerslev, K.; Hogdall, E.; Schnack, T.H.; Skovrider-Ruminski, W.; Hogdall, C.; Blaakaer, J. The potential role of infectious agents and pelvic inflammatory disease in ovarian carcinogenesis. Infect. Agent Cancer 2017, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Burney, R.O.; Giudice, L.C. Pathogenesis and pathophysiology of endometriosis. Fertil. Steril. 2012, 98, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; Crosignani, P.; Somigliana, E.; Viganò, P.; Buggio, L.; Bolis, G.; Fedele, L. The ‘incessant menstruation’ hypothesis: A mechanistic ovarian cancer model with implications for prevention. Hum. Reprod. 2011, 26, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Burghaus, S.; Haberle, L.; Schrauder, M.G.; Heusinger, K.; Thiel, F.C.; Hein, A.; Wachter, D.; Strehl, J.; Hartmann, A.; Ekici, A.B.; et al. Endometriosis as a risk factor for ovarian or endometrial cancer—Results of a hospital-based case-control study. BMC Cancer 2015, 15, 751. [Google Scholar] [CrossRef] [PubMed]
- Sayasneh, A.; Tsivos, D.; Crawford, R. Endometriosis and ovarian cancer: A systematic review. ISRN Obstet. Gynecol. 2011, 2011, 140310. [Google Scholar] [CrossRef] [PubMed]
- Rizi, B.S.; Nagrath, D. Linking omentum and ovarian cancer: NO. Oncoscience 2015, 2, 797–798. [Google Scholar] [PubMed]
- Gunderson, C.C.; Ding, K.; Dvorak, J.; Moore, K.N.; McMeekin, D.S.; Benbrook, D.M. The pro-inflammatory effect of obesity on high grade serous ovarian cancer. Gynecol. Oncol. 2016, 143, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.M.; Lee, I.M.; Ridker, P.M.; Buring, J.E.; Hankinson, S.E.; Tworoger, S.S. A prospective study of circulating C-reactive protein, interleukin-6, and tumor necrosis factor alpha receptor 2 levels and risk of ovarian cancer. Am. J. Epidemiol. 2013, 178, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Ose, J.; Schock, H.; Tjonneland, A.; Hansen, L.; Overvad, K.; Dossus, L.; Clavel-Chapelon, F.; Baglietto, L.; Boeing, H.; Trichopolou, A.; et al. Inflammatory Markers and Risk of Epithelial Ovarian Cancer by Tumor Subtypes: The EPIC Cohort. Cancer Epidemiol. Biomark. Prev. 2015, 24, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Duleba, A.J.; Dokras, A. Is PCOS an inflammatory process? Fertil. Steril. 2012, 97, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.C.; Lyall, H.; Petrie, J.R.; Gould, G.W.; Connell, J.M.; Sattar, N. Low grade chronic inflammation in women with polycystic ovarian syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 2453–2455. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Morreale, H.F.; Luque-Ramirez, M.; Gonzalez, F. Circulating inflammatory markers in polycystic ovary syndrome: A systematic review and metaanalysis. Fertil. Steril. 2011, 95, 1048–1058.e1041-1042. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.; Rote, N.S.; Minium, J.; Kirwan, J.P. Evidence of proatherogenic inflammation in polycystic ovary syndrome. Metabolism 2009, 58, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Glintborg, D.; Andersen, M.; Richelsen, B.; Bruun, J.M. Plasma monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1α are increased in patients with polycystic ovary syndrome (PCOS) and associated with adiposity, but unaffected by pioglitazone treatment. Clin. Endocrinol. 2009, 71, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Morreale, H.F.; Botella-Carretero, J.I.; Villuendas, G.; Sancho, J.; San Millan, J.L. Serum interleukin-18 concentrations are increased in the polycystic ovary syndrome: Relationship to insulin resistance and to obesity. J. Clin. Endocrinol. Metab. 2004, 89, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qiao, J.; Li, R.; Li, M.Z. Is interleukin-18 associated with polycystic ovary syndrome? Reprod. Biol. Endocrinol. 2011, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Tarkun, I.; Cetinarslan, B.; Turemen, E.; Canturk, Z.; Biyikli, M. Association between Circulating Tumor Necrosis Factor-α, Interleukin-6, and Insulin Resistance in Normal-Weight Women with Polycystic Ovary Syndrome. Metab. Syndr. Relat. Disord. 2006, 4, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Vgontzas, A.N.; Trakada, G.; Bixler, E.O.; Lin, H.M.; Pejovic, S.; Zoumakis, E.; Chrousos, G.P.; Legro, R.S. Plasma interleukin 6 levels are elevated in polycystic ovary syndrome independently of obesity or sleep apnea. Metabolism 2006, 55, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Dinger, Y.; Akcay, T.; Erdem, T.; Ilker Saygili, E.; Gundogdu, S. DNA damage, DNA susceptibility to oxidation and glutathione level in women with polycystic ovary syndrome. Scand. J. Clin Lab. Investig. 2005, 65, 721–728. [Google Scholar]
- Harris, H.R.; Terry, K.L. Polycystic ovary syndrome and risk of endometrial, ovarian, and breast cancer: A systematic review. Fertil. Res. Pract. 2016, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.S.; Westhoff, C.; Gordon, R.E.; Katz, N. The relationship between perineal cosmetic talc usage and ovarian talc particle burden. Am. J. Obstet. Gynecol. 1996, 174, 1507–1510. [Google Scholar] [CrossRef]
- Henderson, W.J.; Hamilton, T.C.; Griffiths, K. Talc in normal and malignant ovarian tissue. Lancet 1979, 1, 499. [Google Scholar] [CrossRef]
- Muscat, J.E.; Huncharek, M.S. Perineal talc use and ovarian cancer: A critical review. Eur J. Cancer Prev. 2008, 17, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Brasky, T.M.; Moysich, K.B.; Cohn, D.E.; White, E. Non-steroidal anti-inflammatory drugs and endometrial cancer risk in the VITamins And Lifestyle (VITAL) cohort. Gynecol. Oncol. 2013, 128, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Prizment, A.E.; Folsom, A.R.; Anderson, K.E. Nonsteroidal anti-inflammatory drugs and risk for ovarian and endometrial cancers in the Iowa Women’s Health Study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Fairfield, K.M.; Hunter, D.J.; Fuchs, C.S.; Colditz, G.A.; Hankinson, S.E. Aspirin, other NSAIDs, and ovarian cancer risk (United States). Cancer Causes Control. 2002, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Poole, E.M.; White, E.; Visvanathan, K.; Adami, H.O.; Anderson, G.L.; Brasky, T.M.; Brinton, L.A.; Fortner, R.T.; Gaudet, M.; et al. Analgesic Use and Ovarian Cancer Risk: An Analysis in the Ovarian Cancer Cohort Consortium. J. Natl. Cancer Inst. 2018. [Google Scholar] [CrossRef] [PubMed]
- Peres, L.C.; Camacho, F.; Abbott, S.E.; Alberg, A.J.; Bandera, E.V.; Barnholtz-Sloan, J.; Bondy, M.; Cote, M.L.; Crankshaw, S.; Funkhouser, E.; et al. Analgesic medication use and risk of epithelial ovarian cancer in African American women. Br. J. Cancer 2016, 114, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Burford, C.; Barnes, M.N.; Oelschlager, D.K.; Myers, R.B.; Talley, L.I.; Partridge, E.E.; Grizzle, W.E. Effects of nonsteroidal anti-inflammatory agents (NSAIDs) on ovarian carcinoma cell lines: Preclinical evaluation of NSAIDs as chemopreventive agents. Clin. Cancer Res. 2002, 8, 202–209. [Google Scholar] [PubMed]
- Arango, H.A.; Icely, S.; Roberts, W.S.; Cavanagh, D.; Becker, J.L. Aspirin effects on endometrial cancer cell growth. Obstet. Gynecol. 2001, 97, 423–427. [Google Scholar] [PubMed]
- Gao, J.; Niwa, K.; Sun, W.; Takemura, M.; Lian, Z.; Onogi, K.; Seishima, M.; Mori, H.; Tamaya, T. Non-steroidal anti-inflammatory drugs inhibit cellular proliferation and upregulate cyclooxygenase-2 protein expression in endometrial cancer cells. Cancer Sci. 2004, 95, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Zhang, H.W.; Chen, D.D.; Zhong, L.; Ren, X.D.; St-Tu, R. JTE-522, a selective COX-2 inhibitor, inhibits cell proliferation and induces apoptosis in RL95-2 cells. Acta Pharmacol Sin. 2002, 23, 631–637. [Google Scholar] [PubMed]
- Hsu, C.S.; Li, Y. Aspirin potently inhibits oxidative DNA strand breaks: Implications for cancer chemoprevention. Biochem. Biophys. Res. Commun. 2002, 293, 705–709. [Google Scholar] [PubMed]
- Hales, D.B.; Zhuge, Y.; Lagman, J.A.; Ansenberger, K.; Mahon, C.; Barua, A.; Luborsky, J.L.; Bahr, J.M. Cyclooxygenases expression and distribution in the normal ovary and their role in ovarian cancer in the domestic hen (Gallus domesticus). Endocrine 2008, 33, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Urick, M.E.; Giles, J.R.; Johnson, P.A. VEGF expression and the effect of NSAIDs on ascites cell proliferation in the hen model of ovarian cancer. Gynecol. Oncol. 2008, 110, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Urick, M.E.; Giles, J.R.; Johnson, P.A. Dietary aspirin decreases the stage of ovarian cancer in the hen. Gynecol. Oncol. 2009, 112, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 genes. Biomed. Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, G.; Thompson-Lanza, J.A.; Glassman, A.; Hayes, K.; Patterson, A.; Marquez, R.T.; Auersperg, N.; Yu, Y.; Hahn, W.C.; et al. A genetically defined model for human ovarian cancer. Cancer Res. 2004, 64, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Matte, I.; Rancourt, C.; Piche, A. Prognostic significance of IL-6 and IL-8 ascites levels in ovarian cancer patients. BMC Cancer 2011, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Masoumi-Moghaddam, S.; Amini, A.; Wei, A.Q.; Robertson, G.; Morris, D.L. Intratumoral interleukin-6 predicts ascites formation in patients with epithelial ovarian cancer: A potential tool for close monitoring. J. Ovarian Res. 2015, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Rath, K.S.; Funk, H.M.; Bowling, M.C.; Richards, W.E.; Drew, A.F. Expression of soluble interleukin-6 receptor in malignant ovarian tissue. Am. J. Obstet. Gynecol. 2010, 203, 230.e1–230.e8. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.W.; Chen, M.W.; Hsiao, M.; Wang, S.; Chen, C.A.; Hsiao, S.M.; Chang, J.S.; Lai, T.C.; Rose-John, S.; Kuo, M.L.; et al. IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res. 2011, 71, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Dalal, V.; Kumar, R.; Kumar, S.; Sharma, A.; Kumar, L.; Sharma, J.B.; Roy, K.K.; Singh, N.; Vanamail, P. Biomarker potential of IL-6 and VEGF-A in ascitic fluid of epithelial ovarian cancer patients. Clin. Chim. Acta 2018, 482, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xiang, T.; Zhao, Z.; Lin, K.; Yin, P.; Jiang, L.; Liang, Z.; Zhu, B. Autocrine interleukin-23 promotes self-renewal of CD133+ ovarian cancer stem-like cells. Oncotarget 2016, 7, 76006–76020. [Google Scholar] [CrossRef] [PubMed]
- Maccio, A.; Madeddu, C. Inflammation and ovarian cancer. Cytokine 2012, 58, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Gupta, S.; Sharma, R.K. Role of oxidative stress in female reproduction. Reprod. Biol. Endocrinol. 2005, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Shkolnik, K.; Tadmor, A.; Ben-Dor, S.; Nevo, N.; Galiani, D.; Dekel, N. Reactive oxygen species are indispensable in ovulation. Proc. Natl. Acad. Sci. USA 2011, 108, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Quartuccio, S.M.; Vanderhyden, B.C.; Burdette, J.E. Early transformative changes in normal ovarian surface epithelium induced by oxidative stress require Akt upregulation, DNA damage and epithelial-stromal interaction. Carcinogenesis 2013, 34, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Levanon, K.; Ng, V.; Piao, H.Y.; Zhang, Y.; Chang, M.C.; Roh, M.H.; Kindelberger, D.W.; Hirsch, M.S.; Crum, C.P.; Marto, J.A.; et al. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 2010, 29, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Chu, S.C.; Hsu, C.F.; Chen, P.C.; Ding, D.C.; Chang, M.Y.; Chu, T.Y. Mutagenic, surviving and tumorigenic effects of follicular fluid in the context of p53 loss: Initiation of fimbria carcinogenesis. Carcinogenesis 2015, 36, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Chernov, N.N.; Saprin, A.N.; Kotova, Y.N.; Gavrilova, Y.A.; Chermnykh, N.S.; Shcherbak, N.P. Expression of genes for thioredoxin 1 and thioredoxin 2 in multidrug resistance ovarian carcinoma cells SKVLB. Bull. Exp. Biol. Med. 2007, 144, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Hirota, K.; Nakamura, H.; Masutani, H.; Sasada, T.; Yodoi, J. Thioredoxin and Its Involvement in the Redox Regulation of Transcription Factors, NF-κB and AP-1. In Oxygen Homeostasis and Its Dynamics; Springer: Tokyo, Japan, 1998; pp. 450–456. [Google Scholar]
- Van der Wijst, M.G.; Huisman, C.; Mposhi, A.; Roelfes, G.; Rots, M.G. Targeting Nrf2 in healthy and malignant ovarian epithelial cells: Protection versus promotion. Mol. Oncol. 2015, 9, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, F.; Altinisik, J.; Karateke, A.; Coksuer, H.; Buyru, N. Methylation of tumor suppressor genes in ovarian cancer. Exp. Ther. Med. 2012, 4, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Tsukamoto, T.; Toyoda, T.; Mori, A.; Tanaka, H.; Maekita, T.; Ichinose, M.; Tatematsu, M.; Ushijima, T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010, 70, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Bonham, E.M.; Hannon, B.E.; Amick, T.R.; Baylin, S.B.; O’Hagan, H.M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell. Biol. 2016, 8, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, A.R.; Peng, M.; Sriramkumar, S.; Kamplain, C.M.; DeStefano Shields, C.E.; Sears, C.L.; O’Hagan, H.M. Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis. Cancer Res. 2017, 77, 3467–3478. [Google Scholar] [CrossRef] [PubMed]
- Sapoznik, S.; Bahar-Shany, K.; Brand, H.; Pinto, Y.; Gabay, O.; Glick-Saar, E.; Dor, C.; Zadok, O.; Barshack, I.; Zundelevich, A.; et al. Activation-Induced Cytidine Deaminase Links Ovulation-Induced Inflammation and Serous Carcinogenesis. Neoplasia 2016, 18, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Babic, A.; Beck, A.H.; Terry, K. TNF-α expression, risk factors, and inflammatory exposures in ovarian cancer: Evidence for an inflammatory pathway of ovarian carcinogenesis? Hum. Pathol. 2016, 54, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.M.; Carson, L.F.; Weinberg, B.; Haney, A.F.; Twiggs, L.B.; Ramakrishnan, S. Serum and ascitic fluid levels of interleukin-1, interleukin-6, and tumor necrosis factor-α in patients with ovarian epithelial cancer. Cancer 1993, 72, 2433–2440. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Grimshaw, M.J.; Kulbe, H.; Wilson, J.L.; Wilbanks, G.D.; Burke, F.; Balkwill, F.R. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol. Cancer Ther. 2006, 5, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Thompson, R.; Wilson, J.L.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill, F. The inflammatory cytokine tumor necrosis factor-α generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007, 67, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Charles, K.A.; Kulbe, H.; Soper, R.; Escorcio-Correia, M.; Lawrence, T.; Schultheis, A.; Chakravarty, P.; Thompson, R.G.; Kollias, G.; Smyth, J.F.; et al. The tumor-promoting actions of TNF-α involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J. Clin. Investig. 2009, 119, 3011–3023. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Chung, C.; Kaldi, K.; Watson, J.M.; Knox, R.M.; Martinez-Maza, O. Serum interleukin-6 levels correlate with disease status in patients with epithelial ovarian cancer. Am. J. Obstet. Gynecol. 1991, 164, 1038–1042; discussion 1042–1033. [Google Scholar] [CrossRef]
- Scambia, G.; Testa, U.; Benedetti Panici, P.; Foti, E.; Martucci, R.; Gadducci, A.; Perillo, A.; Facchini, V.; Peschle, C.; Mancuso, S. Prognostic significance of interleukin 6 serum levels in patients with ovarian cancer. Br. J. Cancer 1995, 71, 354. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFkB pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Robinson, S.C.; Thompson, R.G.; Charles, K.; Kulbe, H.; Balkwill, F.R. Ovarian cancer cell-derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol. Cancer Ther. 2007, 6, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Moon, H.G.; Lee, D.S.; Yoo, Y.B. Tissue transglutaminase-interleukin-6 axis facilitates peritoneal tumor spreading and metastasis of human ovarian cancer cells. Lab. Anim. Res. 2015, 31, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Guo, X.; Jin, X.; Sun, W.; Zhang, X.; Xu, R.C. Interleukin-6 signaling regulates anchorage-independent growth, proliferation, adhesion and invasion in human ovarian cancer cells. Cytokine 2012, 59, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Maccio, A.; Lai, P.; Santona, M.C.; Pagliara, L.; Melis, G.B.; Mantovani, G. High serum levels of soluble IL-2 receptor, cytokines, and C reactive protein correlate with impairment of T cell response in patients with advanced epithelial ovarian cancer. Gynecol. Oncol. 1998, 69, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, A.; Medina, L.; Piura, B.; Segal, S.; Huleihel, M. Regulation of ovarian carcinoma SKOV-3 cell proliferation and secretion of MMPs by autocrine IL-6. Anticancer Res. 2007, 27, 267–272. [Google Scholar] [PubMed]
- Runesson, E.; Bostrom, E.K.; Janson, P.O.; Brannstrom, M. The human preovulatory follicle is a source of the chemotactic cytokine interleukin-8. Mol. Hum. Reprod 1996, 2, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Brannstrom, M.; Mayrhofer, G.; Robertson, S.A. Localization of leukocyte subsets in the rat ovary during the periovulatory period. Biol. Reprod. 1993, 48, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Kassim, S.K.; El-Salahy, E.M.; Fayed, S.T.; Helal, S.A.; Helal, T.; Azzam Eel, D.; Khalifa, A. Vascular endothelial growth factor and interleukin-8 are associated with poor prognosis in epithelial ovarian cancer patients. Clin. Biochem. 2004, 37, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, K.; Runesson, E.; Sundfeldt, K.; Haeger, M.; Hedin, L.; Janson, P.O.; Brannstrom, M. The chemotactic cytokine interleukin-8—A cyst fluid marker for malignant epithelial ovarian cancer? Gynecol. Oncol. 1998, 71, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.; Schmidt, D.; Bohme, M.; Schmidt, U.; Weise, W.; Morenz, J. Cytokine level in malignant ascites and peripheral blood of patients with advanced ovarian carcinoma. Geburtshilfe Frauenheilkd. 1996, 56, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, R.C.; Zhang, X.L.; Niu, X.L.; Qu, Y.; Li, L.Z.; Meng, X.Y. Interleukin-8 secretion by ovarian cancer cells increases anchorage-independent growth, proliferation, angiogenic potential, adhesion and invasion. Cytokine 2012, 59, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Hakozaki, M.; Nyunoya, Y.; Kobayashi, Y.; Matsushita, K.; Takenouchi, T.; Mikata, A.; Mukaida, N.; Matsushima, K. Chemokine gene transfection into tumour cells reduced tumorigenicity in nude mice in association with neutrophilic infiltration. Br. J. Cancer 1995, 72, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Hellendall, R.P.; Wang, Y.; Haskill, J.S.; Mukaida, N.; Matsushima, K.; Ting, J.P. IL-8 reduced tumorigenicity of human ovarian cancer in vivo due to neutrophil infiltration. J. Immunol. 2000, 164, 2769–2775. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Miyake, M.; Nishioka, Y.; Yamano, S.; Aono, T.; Fukuzawa, K. Production of lysophosphatidic acids by lysophospholipase D in human follicular fluids of In vitro fertilization patients. Biol. Reprod. 1999, 61, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.U.; Chou, C.H.; Lee, H.; Ho, C.H.; Lin, C.W.; Yang, Y.S. Lysophosphatidic acid up-regulates expression of interleukin-8 and -6 in granulosa-lutein cells through its receptors and nuclear factor-κB dependent pathways: Implications for angiogenesis of corpus luteum and ovarian hyperstimulation syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Gaudette, D.; Furui, T.; Mao, M.; Estrella, V.; Eder, A.; Pustilnik, T.; Sasagawa, T.; Lapushin, R.; Yu, S.; et al. Lysophospholipid growth factors in the initiation, progression, metastases, and management of ovarian cancer. Ann. N. Y. Acad. Sci. 2000, 905, 188–208. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.J.; Schwartz, B.; Washington, M.; Kennedy, A.; Webster, K.; Belinson, J.; Xu, Y. Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: Comparison of the lysophospholipid contents in malignant vs nonmalignant ascitic fluids. Anal. Biochem. 2001, 290, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Eder, A.; Fang, X.; Hasegawa, Y.; Mao, M.; Lu, Y.; Tanyi, J.; Tabassam, F.H.; Wiener, J.; Lapushin, R.; et al. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat. Res. 2002, 107, 259–283. [Google Scholar] [PubMed]
- Westermann, A.M.; Havik, E.; Postma, F.R.; Beijnen, J.H.; Dalesio, O.; Moolenaar, W.H.; Rodenhuis, S. Malignant effusions contain lysophosphatidic acid (LPA)-like activity. Ann. Oncol. 1998, 9, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gaudette, D.C.; Boynton, J.D.; Frankel, A.; Fang, X.J.; Sharma, A.; Hurteau, J.; Casey, G.; Goodbody, A.; Mellors, A.; et al. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin. Cancer Res. 1995, 1, 1223–1232. [Google Scholar] [PubMed]
- Symowicz, J.; Adley, B.P.; Woo, M.M.; Auersperg, N.; Hudson, L.G.; Stack, M.S. Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells. Cancer Res. 2005, 65, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.; Bast, R.C.; Liu, S.; Xu, H.J.; Hu, S.X.; LaPushin, R.; Claret, F.X.; Aggarwal, B.B.; Lu, Y.; et al. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. J. Biol. Chem. 2004, 279, 9653–9661. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.A.; Rogers, L.C.; Klomsiri, C.; Poole, L.B.; Daniel, L.W. Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic. Biol. Med. 2010, 49, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xiao Yj, Y.J.; Baudhuin, L.M.; Hong, G.; Xu, Y. Role of ether-linked lysophosphatidic acids in ovarian cancer cells. J. Lipid Res. 2002, 43, 463–476. [Google Scholar] [PubMed]
- Ali-Fehmi, R.; Morris, R.T.; Bandyopadhyay, S.; Che, M.; Schimp, V.; Malone, J.M., Jr.; Munkarah, A.R. Expression of cyclooxygenase-2 in advanced stage ovarian serous carcinoma: Correlation with tumor cell proliferation, apoptosis, angiogenesis, and survival. Am. J. Obstet. Gynecol. 2005, 192, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Rask, K.; Zhu, Y.; Wang, W.; Hedin, L.; Sundfeldt, K. Ovarian epithelial cancer: A role for PGE2-synthesis and signalling in malignant transformation and progression. Mol. Cancer 2006, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, P.K.; Metsa-Ketela, T. Prostaglandin and thromboxane production in ovarian cancer tissue. Gynecol. Obstet. Investig. 1984, 18, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Muthuswamy, R.; Odunsi, K.; Edwards, R.P.; Kalinski, P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Tranguch, S.; Trofimova, I.N.; Dinulescu, D.M.; Jacks, T.; Nikitin, A.Y.; Connolly, D.C.; Dey, S.K. Cyclooxygenase-1 is overexpressed in multiple genetically engineered mouse models of epithelial ovarian cancer. Cancer Res. 2006, 66, 2527–2531. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, J.H.; Zhang, J.; Tang, Y.X.; Wan, L. Effects of cyclooxygenase inhibitors in combination with taxol on expression of cyclin D1 and Ki-67 in a xenograft model of ovarian carcinoma. Int. J. Mol. Sci. 2012, 13, 9741–9753. [Google Scholar] [CrossRef] [PubMed]
- Paweletz, N.; Knierim, M. Tumor-related angiogenesis. Crit. Rev. Oncol. Hematol. 1989, 9, 197–242. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Giannoudis, A.; Corke, K.P.; Gill, D.; Wells, M.; Ziegler-Heitbrock, L.; Lewis, C.E. Hypoxia-induced gene expression in human macrophages: Implications for ischemic tissues and hypoxia-regulated gene therapy. Am. J. Pathol. 2003, 163, 1233–1243. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Lewis, C.E. Hypoxia regulates macrophage functions in inflammation. J. Immunol. 2005, 175, 6257–6263. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Wheeler-Jones, C.; Abu-Ghazaleh, R.; Cospedal, R.; Houliston, R.A.; Martin, J.; Zachary, I. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen-activated protein kinase. FEBS Lett. 1997, 420, 28–32. [Google Scholar] [CrossRef]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 1998, 273, 13313–13316. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Liu, Q.; Mao, F.; Wu, J.; Lei, T. TNF-α-induced VEGF and MMP-9 expression promotes hemorrhagic transformation in pituitary adenomas. Int. J. Mol. Sci. 2011, 12, 4165–4179. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Brooks, J.; Willard, M.; Liang, K.; Yoon, Y.; Kang, S.; Shim, H. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem. Biophys. Res. Commun. 2007, 359, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lange, A.; Mottram, P.; Alvarez, X.; Cheng, P.; Hogan, M.; Moons, L.; Wei, S.; Zou, L.; Machelon, V.; et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005, 65, 465–472. [Google Scholar] [PubMed]
- Motro, B.; Itin, A.; Sachs, L.; Keshet, E. Pattern of interleukin 6 gene expression in vivo suggests a role for this cytokine in angiogenesis. Proc. Natl. Acad. Sci. USA 1990, 87, 3092–3096. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Langley, R.R.; Fidler, I.J. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005, 65, 10794–10800. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Scheller, J.; Elson, G.; Jones, S.A. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J. Leukocyte Biol. 2006, 80, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.E.; Hori, Y.; Fan, T.P. Interleukin-8 stimulates angiogenesis in rats. Inflammation 1993, 17, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Polverini, P.J.; Kunkel, S.L.; Harlow, L.A.; DiPietro, L.A.; Elner, V.M.; Elner, S.G.; Strieter, R.M. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 1992, 258, 1798–1801. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. J. Clin. Investig. 1999, 103, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.S.; Chan, L.K.; Wong, E.C.; Hui, C.W.; Sneddon, K.; Cheung, T.H.; Yim, S.F.; Lee, J.H.; Yeung, C.S.; Chung, T.K.; et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFα-TGFα-EGFR. Oncogene 2017, 36, 3576–3587. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.J.; Wilson, J.L.; Scott, K.; Stamp, G.; Wilbanks, G.D.; Fricker, S.; Bridger, G.; Balkwill, F.R. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002, 62, 5930–5938. [Google Scholar] [PubMed]
- Kulbe, H.; Hagemann, T.; Szlosarek, P.W.; Balkwill, F.R.; Wilson, J.L. The inflammatory cytokine tumor necrosis factor-α regulates chemokine receptor expression on ovarian cancer cells. Cancer Res. 2005, 65, 10355–10362. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Gwak, H.; Kim, H.S.; Kim, B.; Dhanasekaran, D.N.; Song, Y.S. Malignant ascites enhances migratory and invasive properties of ovarian cancer cells with membrane bound IL-6R in vitro. Oncotarget 2016, 7, 83148–83159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, X.L.; Qu, Y.; Wu, J.; Zhu, Y.Q.; Sun, W.J.; Li, L.Z. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 2010, 295, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Yung, M.M.; Tang, H.W.; Cai, P.C.; Leung, T.H.; Ngu, S.F.; Chan, K.K.; Xu, D.; Yang, H.; Ngan, H.Y.; Chan, D.W. GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [PubMed]
- Cuello, M.; Ettenberg, S.A.; Nau, M.M.; Lipkowitz, S. Synergistic induction of apoptosis by the combination of trail and chemotherapy in chemoresistant ovarian cancer cells. Gynecol. Oncol. 2001, 81, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, J.; Oyesanya, R.A.; Lee, Z.; Mukherjee, A.; Fang, X. Sp-1 and c-Myc mediate lysophosphatidic acid-induced expression of vascular endothelial growth factor in ovarian cancer cells via a hypoxia-inducible factor-1-independent mechanism. Clin. Cancer Res. 2009, 15, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Pustilnik, T.B.; Estrella, V.; Wiener, J.R.; Mao, M.; Eder, A.; Watt, M.A.; Bast, R.C., Jr.; Mills, G.B. Lysophosphatidic acid induces urokinase secretion by ovarian cancer cells. Clin. Cancer Res. 1999, 5, 3704–3710. [Google Scholar] [PubMed]
- Duffy, M.J. Proteases as prognostic markers in cancer. Clin. Cancer Res. 1996, 2, 613–618. [Google Scholar] [PubMed]
- Masferrer, J.L.; Leahy, K.M.; Koki, A.T.; Zweifel, B.S.; Settle, S.L.; Woerner, B.M.; Edwards, D.A.; Flickinger, A.G.; Moore, R.J.; Seibert, K. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000, 60, 1306–1311. [Google Scholar] [PubMed]
- Liu, X.H.; Yao, S.; Kirschenbaum, A.; Levine, A.C. NS398, a selective cyclooxygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expression in LNCaP cells. Cancer Res. 1998, 58, 4245–4249. [Google Scholar] [PubMed]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.; Mills, G.B. Peptide and lipid growth factors decrease cis-diamminedichloroplatinum-induced cell death in human ovarian cancer cells. Clin. Cancer Res. 1996, 2, 1307–1313. [Google Scholar] [PubMed]
- Gao, Y.; Shan, N.; Zhao, C.; Wang, Y.; Xu, F.; Li, J.; Yu, X.; Gao, L.; Yi, Z. LY2109761 enhances cisplatin antitumor activity in ovarian cancer cells. Int J. Clin. Exp. Pathol. 2015, 8, 4923–4932. [Google Scholar] [PubMed]
- Hu, Y.L.; Tee, M.K.; Goetzl, E.J.; Auersperg, N.; Mills, G.B.; Ferrara, N.; Jaffe, R.B. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J. Natl. Cancer Inst. 2001, 93, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Yang-Hartwich, Y.; Gurrea-Soteras, M.; Sumi, N.; Joo, W.D.; Holmberg, J.C.; Craveiro, V.; Alvero, A.B.; Mor, G. Ovulation and extra-ovarian origin of ovarian cancer. Sci. Rep. 2014, 4, 6116. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Cain, R.J.; Ridley, A.J. Phosphoinositide 3-kinases in cell migration. Biol. Cell 2009, 101, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Devreotes, P.; Horwitz, A.R. Signaling networks that regulate cell migration. Cold Spring Harb Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Smith, T.M.; Isaacsohn, I.; Mercer, C.A.; Zhou, M.; Van Rooijen, N.; Husseinzadeh, N.; McFarland-Mancini, M.M.; Drew, A.F. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res. 2007, 67, 5708–5716. [Google Scholar] [CrossRef] [PubMed]
- Wilkosz, S.; Ireland, G.; Khwaja, N.; Walker, M.; Butt, R.; de Giorgio-Miller, A.; Herrick, S.E. A comparative study of the structure of human and murine greater omentum. Anat. Embryol. 2005, 209, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Pulliam, N.; Ozes, A.; Buechlein, A.; Ding, N.; Keer, H.; Rusch, D.; O’Hagan, H.; Stack, M.S.; Nephew, K.P. Epigenetic Targeting of Adipocytes Inhibits High-Grade Serous Ovarian Cancer Cell Migration and Invasion. Mol. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Meza-Perez, S.; Randall, T.D. Immunological Functions of the Omentum. Trends Immunol. 2017, 38, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.A.; Rybalko, V.Y.; Bigelow, C.E.; Lugade, A.A.; Foster, T.H.; Frelinger, J.G.; Lord, E.M. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am. J. Pathol. 2006, 169, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.; Krishnan, V.; Schoof, M.; Rodriguez, I.; Theriault, B.; Chekmareva, M.; Rinker-Schaeffer, C. Milky spots promote ovarian cancer metastatic colonization of peritoneal adipose in experimental models. Am. J. Pathol. 2013, 183, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Wang, Y.; Ma, J.; Shen, H.; Wang, C.; Sun, Y.; Howell, S.B.; Lin, X. Reactive oxygen species promote ovarian cancer progression via the HIF-1α/LOX/E-cadherin pathway. Oncol. Rep. 2014, 32, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Zhang, S.; Fulton, A.M. Sublethal oxidative stress inhibits tumor cell adhesion and enhances experimental metastasis of murine mammary carcinoma. Clin. Exp. Metast. 1995, 13, 16–22. [Google Scholar] [CrossRef]
- Haskill, S.; Becker, S.; Fowler, W.; Walton, L. Mononuclear-cell infiltration in ovarian cancer. I. Inflammatory-cell infiltrates from tumour and ascites material. Br. J. Cancer 1982, 45, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Griffin, J.D.; Rambaldi, A.; Chen, Z.G.; Mantovani, A. Induction of monocyte migration by recombinant macrophage colony-stimulating factor. J. Immunol. 1988, 141, 575–579. [Google Scholar] [PubMed]
- Ramakrishnan, S.; Xu, F.J.; Brandt, S.J.; Niedel, J.E.; Bast, R.C., Jr.; Brown, E.L. Constitutive production of macrophage colony-stimulating factor by human ovarian and breast cancer cell lines. J. Clin. Investig. 1989, 83, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Plante, M.; Rubin, S.C.; Wong, G.Y.; Federici, M.G.; Finstad, C.L.; Gastl, G.A. Interleukin-6 level in serum and ascites as a prognostic factor in patients with epithelial ovarian cancer. Cancer 1994, 73, 1882–1888. [Google Scholar] [CrossRef]
- Scambia, G.; Testa, U.; Panici, P.B.; Martucci, R.; Foti, E.; Petrini, M.; Amoroso, M.; Masciullo, V.; Peschle, C.; Mancuso, S. Interleukin-6 serum levels in patients with gynecological tumors. Int. J. Cancer 1994, 57, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Tempfer, C.; Zeisler, H.; Sliutz, G.; Haeusler, G.; Hanzal, E.; Kainz, C. Serum evaluation of interleukin 6 in ovarian cancer patients. Gynecol. Oncol. 1997, 66, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Isobe, A.; Sawada, K.; Kinose, Y.; Ohyagi-Hara, C.; Nakatsuka, E.; Makino, H.; Ogura, T.; Mizuno, T.; Suzuki, N.; Morii, E.; et al. Interleukin 6 receptor is an independent prognostic factor and a potential therapeutic target of ovarian cancer. PLoS ONE 2015, 10, e0118080. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.A.; Liu, Y.; Ellerbroek, S.M.; Stack, M.S. Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and MMP-dependent invasion in ovarian cancer cells. Cancer Res. 2001, 61, 3194–3199. [Google Scholar] [PubMed]
- Reiser, C.O.; Lanz, T.; Hofmann, F.; Hofer, G.; Rupprecht, H.D.; Goppelt-Struebe, M. Lysophosphatidic acid-mediated signal-transduction pathways involved in the induction of the early-response genes prostaglandin G/H synthase-2 and Egr-1: A critical role for the mitogen-activated protein kinase p38 and for Rho proteins. Biochem. J. 1998, 330 (Pt 3), 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, W.H.; Kruijer, W.; Tilly, B.C.; Verlaan, I.; Bierman, A.J.; de Laat, S.W. Growth factor-like action of phosphatidic acid. Nature 1986, 323, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.M.; Hong, G.; Morrison, B.H.; Wu, W.; Baudhuin, L.M.; Xiao, Y.J.; Mok, S.C.; Xu, Y. Lysophospholipids increase interleukin-8 expression in ovarian cancer cells. Gynecol. Oncol. 2001, 81, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, Y.; Chen, H. LPA receptor 1 mediates LPA-induced ovarian cancer metastasis: An in vitro and in vivo study. BMC Cancer 2016, 16, 846. [Google Scholar] [CrossRef] [PubMed]
- Loizzi, V.; Del Vecchio, V.; Gargano, G.; De Liso, M.; Kardashi, A.; Naglieri, E.; Resta, L.; Cicinelli, E.; Cormio, G. Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer. Int. J. Mol. Sci. 2017, 18, 1967. [Google Scholar] [CrossRef] [PubMed]
- Le Page, C.; Puiffe, M.L.; Meunier, L.; Zietarska, M.; de Ladurantaye, M.; Tonin, P.N.; Provencher, D.; Mes-Masson, A.M. BMP-2 signaling in ovarian cancer and its association with poor prognosis. J. Ovarian Res. 2009, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F. Treatment goals in ovarian cancer. Int. J. Gynecol. Cancer 2005, 15 (Suppl. 1), 3–11. [Google Scholar] [CrossRef] [PubMed]
- Koti, M.; Siu, A.; Clement, I.; Bidarimath, M.; Turashvili, G.; Edwards, A.; Rahimi, K.; Mes-Masson, A.M.; Squire, J.A. A distinct pre-existing inflammatory tumour microenvironment is associated with chemotherapy resistance in high-grade serous epithelial ovarian cancer. Br. J. Cancer 2015, 112, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev. Oncol Hematol 2007, 63, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [PubMed]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Cusmai, A.; Iodice, G.; Dammacco, F. Autocrine interleukin-6 production and highly malignant multiple myeloma: Relation with resistance to drug-induced apoptosis. Blood 2001, 97, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Mehrabi, S.; Yao, X.; Millingen, S.; Aikhionbare, F.O. Reactive Oxygen Species and Serous Epithelial Ovarian Adenocarcinoma. Cancer Res. J. 2016, 4, 106–114. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Jung, J.W.; Jung, J.; Han, Y.; Suh, D.H.; Kim, H.S.; Dhanasekaran, D.N.; Song, Y.S. PGC1α induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget 2017, 8, 60299–60311. [Google Scholar] [PubMed]
- Duan, Z.; Feller, A.J.; Penson, R.T.; Chabner, B.A.; Seiden, M.V. Discovery of differentially expressed genes associated with paclitaxel resistance using cDNA array technology: Analysis of interleukin (IL) 6, IL-8, and monocyte chemotactic protein 1 in the paclitaxel-resistant phenotype. Clin. Cancer Res. 1999, 5, 3445–3453. [Google Scholar] [PubMed]
- Duan, Z.; Foster, R.; Bell, D.A.; Mahoney, J.; Wolak, K.; Vaidya, A.; Hampel, C.; Lee, H.; Seiden, M.V. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin. Cancer Res. 2006, 12, 5055–5063. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, T.; Robertson, N.M.; Abdollahi, A.; Litwack, G. Identification of interleukin 8 as an inhibitor of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in the ovarian carcinoma cell line OVCAR3. Cancer Res. 2003, 63, 4521–4526. [Google Scholar] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.E.; Baldwin, R.L.; Yamada, S.D.; Korc, M.; Karlan, B.Y. Altered expression of transforming growth factor-beta ligands and receptors in primary and recurrent ovarian carcinoma. Cancer 1999, 85, 658–668. [Google Scholar] [CrossRef]
- Li, M.; Balch, C.; Montgomery, J.S.; Jeong, M.; Chung, J.H.; Yan, P.; Huang, T.H.; Kim, S.; Nephew, K.P. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med. Genom. 2009, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; Kassar, M.; Yu, Z.; Bamias, A.; Weinberger, P.M.; Markakis, S.; Kowalski, D.; Camp, R.L.; Rimm, D.L.; Dimopoulos, M.A. Effect of epidermal growth factor receptor expression level on survival in patients with epithelial ovarian cancer. Clin. Cancer Res. 2005, 11, 8637–8643. [Google Scholar] [CrossRef] [PubMed]
- Crew, A.J.; Langdon, S.P.; Miller, E.P.; Miller, W.R. Mitogenic effects of epidermal growth factor and transforming growth factor-α on EGF-receptor positive human ovarian carcinoma cell lines. Eur. J. Cancer 1992, 28, 337–341. [Google Scholar] [CrossRef]
- Ferrandina, G.; Ranelletti, F.O.; Martinelli, E.; Paglia, A.; Zannoni, G.F.; Scambia, G. Cyclo-oxygenase-2 (Cox-2) expression and resistance to platinum versus platinum/paclitaxel containing chemotherapy in advanced ovarian cancer. BMC Cancer 2006, 6, 182. [Google Scholar] [CrossRef] [PubMed]
- Ferrandina, G.; Lauriola, L.; Zannoni, G.F.; Fagotti, A.; Fanfani, F.; Legge, F.; Maggiano, N.; Gessi, M.; Mancuso, S.; Ranelletti, F.O.; et al. Increased cyclooxygenase-2 (COX-2) expression is associated with chemotherapy resistance and outcome in ovarian cancer patients. Ann. Oncol. 2002, 13, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2012, 31, 14. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.R.; Charles, K.A.; Hoare, S.A.; Rye, R.L.; Jodrell, D.I.; Aird, R.E.; Vora, R.; Prabhakar, U.; Nakada, M.; Corringham, R.E.; et al. A clinical study assessing the tolerability and biological effects of infliximab, a TNF-α inhibitor, in patients with advanced cancer. Ann. Oncol. 2008, 19, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nemeth, J.; O’Brien, C.; Susa, M.; Liu, X.; Zhang, Z.; Choy, E.; Mankin, H.; Hornicek, F.; Duan, Z. Effects of siltuximab on the IL-6-induced signaling pathway in ovarian cancer. Clin. Cancer Res. 2010, 16, 5759–5769. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Grande, F.; Garofalo, A.; Neamati, N. Discovery of a novel orally active small-molecule gp130 inhibitor for the treatment of ovarian cancer. Mol. Cancer Ther. 2013, 12, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Santegoets, S.J.; Reyners, A.K.; Goedemans, R.; Wouters, M.C.; Kenter, G.G.; van Erkel, A.R.; van Poelgeest, M.I.; Nijman, H.W.; van der Hoeven, J.J.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Alsina-Sanchis, E.; Figueras, A.; Lahiguera, A.; Gil-Martin, M.; Pardo, B.; Piulats, J.M.; Marti, L.; Ponce, J.; Matias-Guiu, X.; Vidal, A.; et al. TGFbeta Controls Ovarian Cancer Cell Proliferation. Int. J. Mol. Sci. 2017, 18, 1658. [Google Scholar] [CrossRef] [PubMed]
- Secord, A.A.; Blessing, J.A.; Armstrong, D.K.; Rodgers, W.H.; Miner, Z.; Barnes, M.N.; Lewandowski, G.; Mannel, R.S.; Gynecologic Oncology, G. Phase II trial of cetuximab and carboplatin in relapsed platinum-sensitive ovarian cancer and evaluation of epidermal growth factor receptor expression: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 108, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Chekerov, R.; Klare, P.; Krabisch, P.; Potenberg, J.; Heinrich, G.; Mueller, L.; Kurbacher, C.M.; Grischke, E.-M.; Braicu, E.I.; Wimberger, P.; et al. Panitumumab in platinum-sensitive epithelial ovarian cancer patients with KRAS wild-type: The PROVE-study, a phase II randomized multicenter study of the North-Eastern German Society of Gynaecologic Oncology. J. Clin. Oncol. 2017, 35 (Suppl. 1), 5558. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Inflammatory Mediators | Secreting Cell Type | Stages in Tumor Progression | |||
|---|---|---|---|---|---|
| Initiation and Progression | Angiogenesis | Metastasis | Chemoresistance | ||
| TNF-α ligands, TNFRI, TNFRII | OC cells, infiltrating monocytes, macrophages | ↑ autocrine production of TNF-α and IL-6, M-CSF, CXCL2, CCL2 [93,94] and AIDS mRNA level [90] | ↑ VEGF, VEGF↑ CXCL12 and promotes angiogenesis [139,140,141] | ↑ TGF-α secretion by stromal fibroblasts which promote peritoneal metastasis [148] Enhances migration of OC cells towards CXCL12 [149,150] | |
| IL-6 | Ovarian epithelial cells, OC cells, M2 macrophages, mesothelial cells, TAMS, ascites | ↑Proliferation by promoting G1 to S transition and MAPK-ERK- Akt activation and STAT3 activation [101,102] ↓IL-2, resulting in immune suppression [103] | Induces STAT3 and MAPK phosphorylation which enhances migration of endothelial cells [143] sIL-6R-IL-6 facilitates angiogenesis in cells lacking IL-6 receptor [144] | Stimulates production of MMPs in OCs which ↑ invasion and migration [101,104] ↑ IL-6 in ascites enhances invasion via JAK-STAT signaling [151] | ↓ Caspase- 3 cleavage and makes OC cells resistant to cisplatin and paclitaxel [152] ↑Expression of MDR1, GSTpi, Bcl-2, Bcl-xL, and XIAP [152] |
| IL-8 | Pre-ovulatory follicles, OC cells, ascites | ↑ Proliferation by ↑ cyclin B1 and cyclin D1 via pAkt [110] | ↑ Expression of VEGF, MMP-2, MMP-9 promoting angiogenesis [110] | Activates TAK1/ NF-κB via CXCR2 [153] | Blocks TRAIL induced apoptosis to promote resistance [154] |
| LPA | Follicular fluid, corpus luteum, OC cells, ascites | ↑ IL-6 and IL-8 via NF-κB and AP-1 [113,114,122] ↑COX-2 AND MMP2 [115,120,121] ↑ phosphorylation of Akt and ERK resulting in increased cell cycle [123,124] | ↑ Expression of VEGF via Myc and Sp-1 [155] | ↑ urokinase, which results in degradation of basemembrane protein to promote metastasis [156,157] | |
| Prostaglandins, COX-1 and COX-2 | Ovary, FT, uterus, MDSCs | ↑ CXCR4 and SDF1 in MDSCs resulting in immune suppression [128] | ↑ Bcl-2 and blood vessel formation [158,159] | ↑ Bcl-2, thus inhibiting apoptosis in lung, colon, breast and prostate cancers [158,159] | |
| TGF-β and EGF | OC cells, CAFs | TGF-β ↑ VCAN, which activates NF-κB and ↑MM-9 [160] | ↑ EGF protects cells from cisplatin-induced apoptosis [161]. Inhibiting TGF-β sensitizes resistant cells [162] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savant, S.S.; Sriramkumar, S.; O’Hagan, H.M. The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer. Cancers 2018, 10, 251. https://doi.org/10.3390/cancers10080251
Savant SS, Sriramkumar S, O’Hagan HM. The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer. Cancers. 2018; 10(8):251. https://doi.org/10.3390/cancers10080251
Chicago/Turabian StyleSavant, Sudha S., Shruthi Sriramkumar, and Heather M. O’Hagan. 2018. "The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer" Cancers 10, no. 8: 251. https://doi.org/10.3390/cancers10080251
APA StyleSavant, S. S., Sriramkumar, S., & O’Hagan, H. M. (2018). The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer. Cancers, 10(8), 251. https://doi.org/10.3390/cancers10080251