Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer?


Abstract
1. Introduction
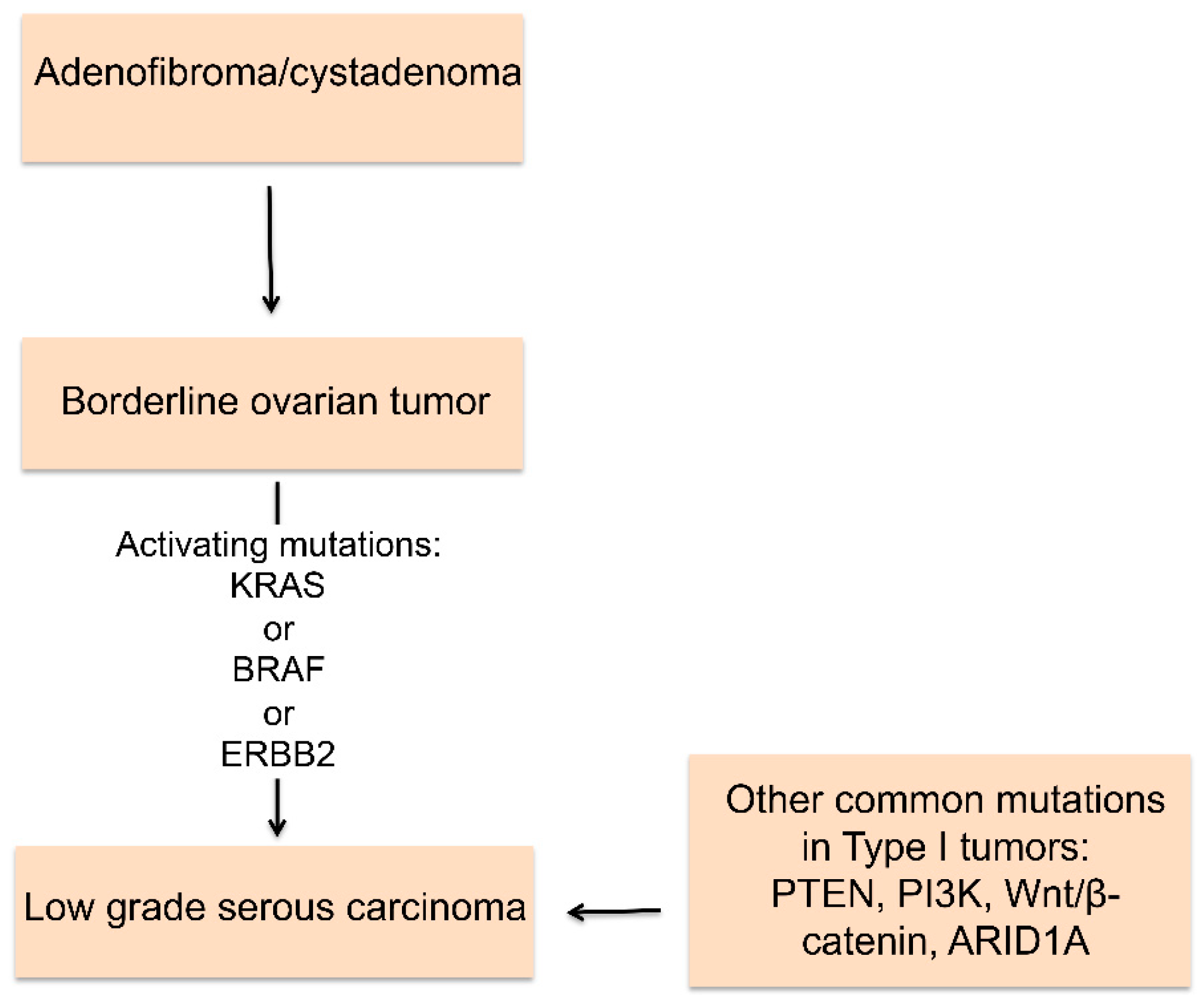
2. Histologic Types of Ovarian Cancer
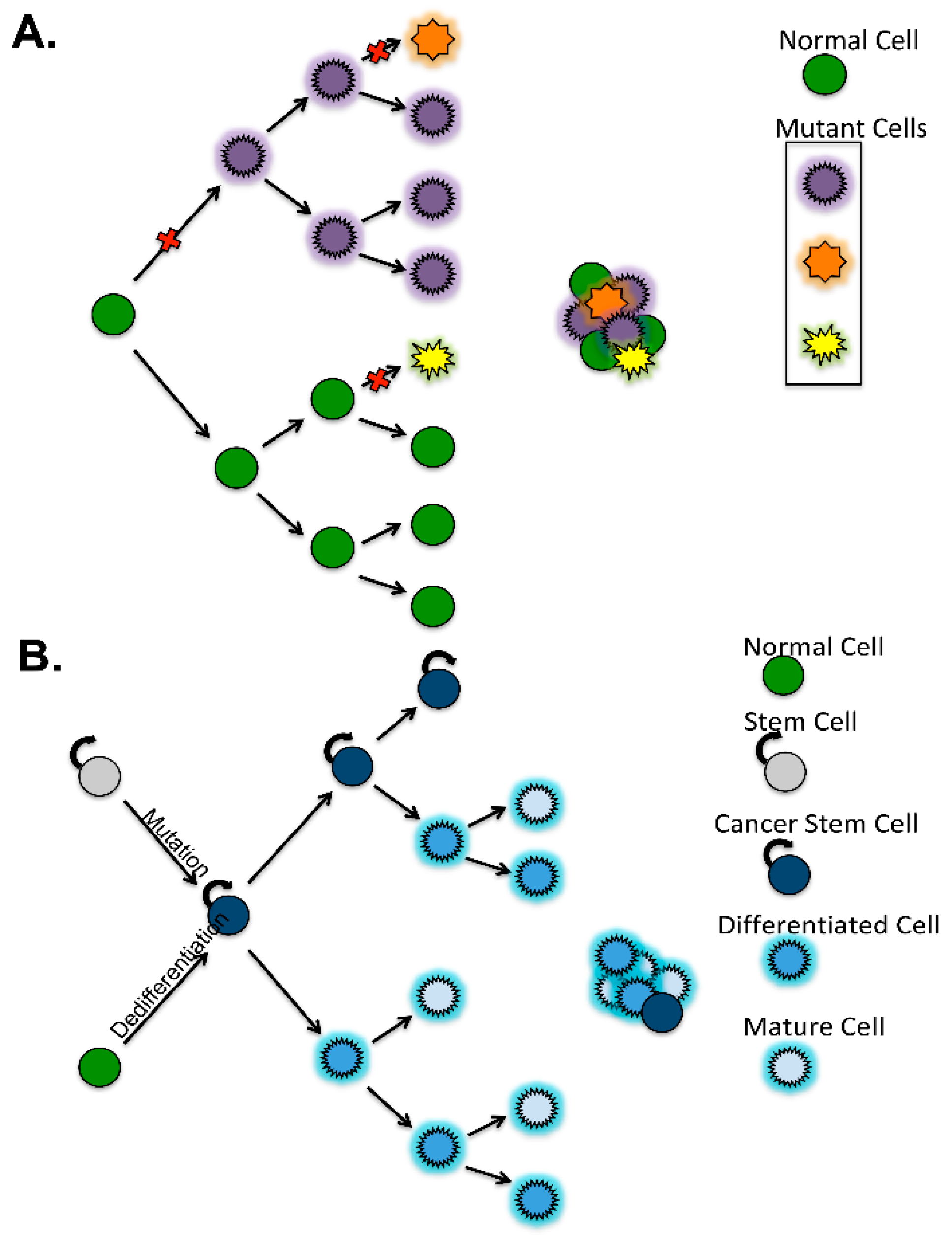
3. Definition of Ovarian Cancer Stem Cells
4. Stem Cell Identification in Ovarian Cancer
4.1. Side Population
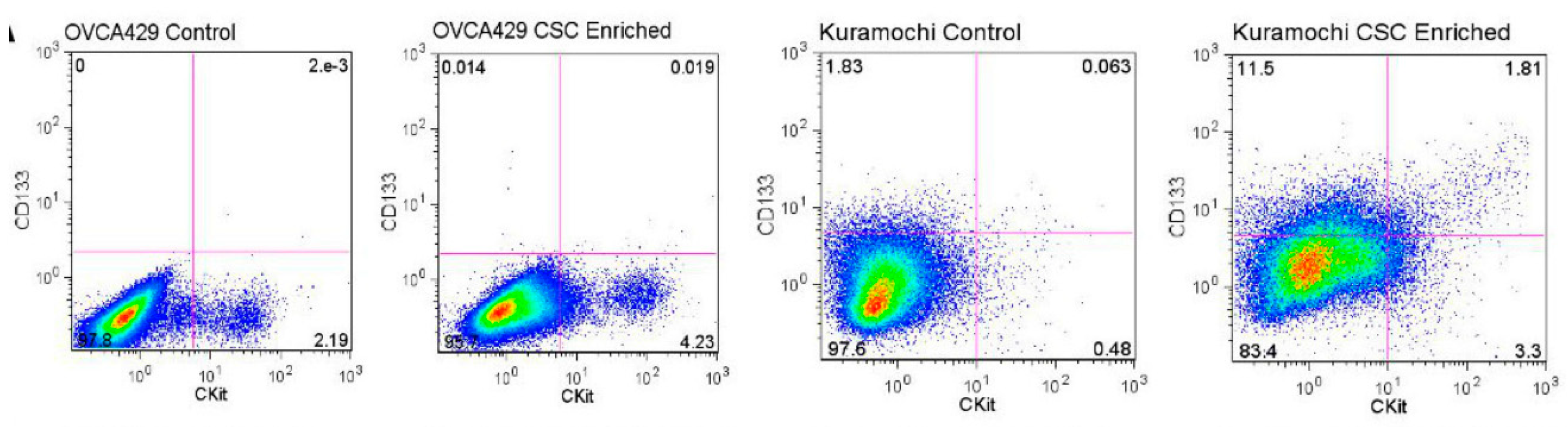
4.2. Cell Surface Markers
4.3. ALDH Activity
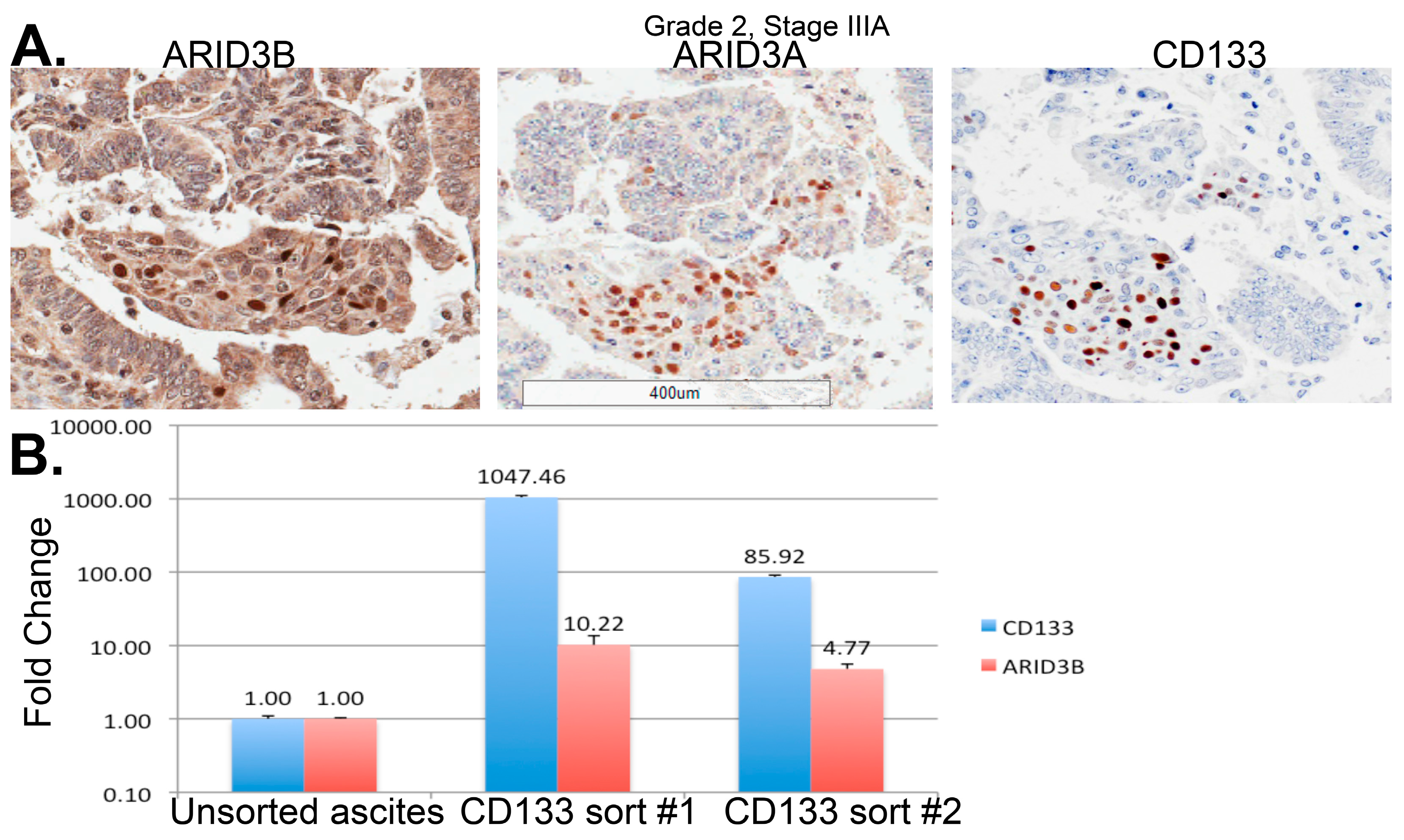
4.4. Transcription Factors
5. Pathways That Promote Stemness and Chemoresistance in HGSOC
5.1. PI3K/PTEN/AKT Signaling
5.2. Jak2/STAT3
5.3. NFκB
5.4. Notch
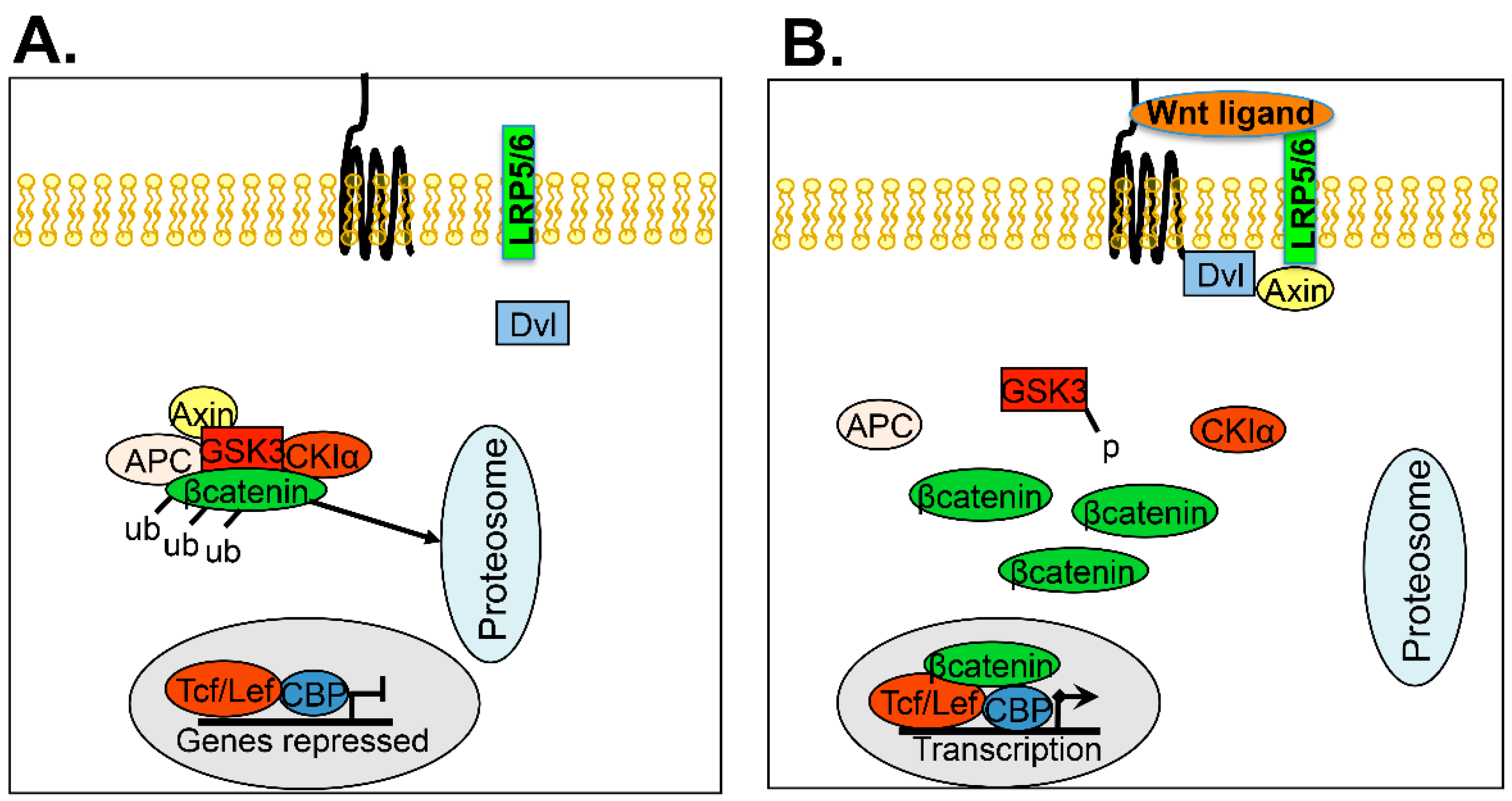
5.5. Wnt
5.6. Hedgehog
5.7. Developing Therapeutics Targeting Ovarian Cancer Stem Cells
6. Future Studies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schorge, J.O.; McCann, C.; Del Carmen, M.G. Surgical debulking of ovarian cancer: What difference does it make? Rev. Obstet. Gynecol. 2010, 3, 111–117. [Google Scholar] [PubMed]
- Lalwani, N.; Prasad, S.R.; Vikram, R.; Shanbhogue, A.K.; Huettner, P.C.; Fasih, N. Histologic, molecular, and cytogenetic features of ovarian cancers: Implications for diagnosis and treatment. Radiographics 2011, 31, 625–646. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, P.T., Jr.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T.; Luesley, D.; Perren, T.; Bannoo, S.; Mascarenhas, M.; et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (chorus): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular characterization of epithelial ovarian cancer: Implications for diagnosis and treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, M.; Matsumura, N.; Konishi, I. Recent concepts of ovarian carcinogenesis: Type I and type II. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Kommoss, S.; Gilks, C.B.; du Bois, A.; Kommoss, F. Ovarian carcinoma diagnosis: The clinical impact of 15 years of change. Br. J. Cancer 2016, 115, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Ottevanger, P.B. Ovarian cancer stem cells more questions than answers. Semin. Cancer Biol. 2017, 44, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F. Treatment goals in ovarian cancer. Int. J. Gynecol. Cancer 2005, 15, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Gilks, C.B.; Prat, J. Ovarian carcinoma pathology and genetics: Recent advances. Hum. Pathol. 2009, 40, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Devouassoux-Shisheboran, M.; Genestie, C. Pathobiology of ovarian carcinomas. Chin. J. Cancer 2015, 34, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P. Morphologic, immunophenotypic, and molecular features of epithelial ovarian cancer. Oncology 2016, 30, 166–176. [Google Scholar] [PubMed]
- Kaldawy, A.; Segev, Y.; Lavie, O.; Auslender, R.; Sopik, V.; Narod, S.A. Low-grade serous ovarian cancer: A review. Gynecol. Oncol. 2016, 143, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Padilla, I.; Malpica, A.L.; Minig, L.; Chiva, L.M.; Gershenson, D.M.; Gonzalez-Martin, A. Ovarian low-grade serous carcinoma: A comprehensive update. Gynecol. Oncol. 2012, 126, 279–285. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. Morphological subtypes of ovarian carcinoma: A review with emphasis on new developments and pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Malpica, A.; Deavers, M.T.; Lu, K.; Bodurka, D.C.; Atkinson, E.N.; Gershenson, D.M.; Silva, E.G. Grading ovarian serous carcinoma using a two-tier system. Am. J. Surg. Pathol. 2004, 28, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. The dualistic model of ovarian carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.L.; Evans, T.; Pearmain, P.; Askew, S.; Singh, K.; Chan, K.K.; Ganesan, R.; Hirschowitz, L. Should all cases of high-grade serous ovarian, tubal, and primary peritoneal carcinomas be reclassified as tubo-ovarian serous carcinoma? Int. J. Gynecol. Cancer 2015, 25, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Soslow, R.A. Histologic subtypes of ovarian carcinoma: An overview. Int. J. Gynecol. Pathol. 2008, 27, 161–174. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.J.; McBride, H.A.; Connolly, L.E.; Deavers, M.T.; Malpica, A.; McCluggage, W.G. High-grade ovarian serous carcinoma exhibits significantly higher p16 expression than low-grade serous carcinoma and serous borderline tumour. Histopathology 2007, 50, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene arid1a in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.N.; Lin, M.C.; Huang, W.C.; Chiang, Y.C.; Kuo, K.T. Loss of arid1a expression and its relationship with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Mod. Pathol. 2014, 27, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, D.M.; Sun, C.C.; Bodurka, D.; Coleman, R.L.; Lu, K.H.; Sood, A.K.; Deavers, M.; Malpica, A.L.; Kavanagh, J.J. Recurrent low-grade serous ovarian carcinoma is relatively chemoresistant. Gynecol. Oncol. 2009, 114, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Schmeler, K.M.; Sun, C.C.; Bodurka, D.C.; Deavers, M.T.; Malpica, A.; Coleman, R.L.; Ramirez, P.T.; Gershenson, D.M. Neoadjuvant chemotherapy for low-grade serous carcinoma of the ovary or peritoneum. Gynecol. Oncol. 2008, 108, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Santillan, A.; Kim, Y.W.; Zahurak, M.L.; Gardner, G.J.; Giuntoli, R.L., 2nd; Shih, I.M.; Bristow, R.E. Differences of chemoresistance assay between invasive micropapillary/low-grade serous ovarian carcinoma and high-grade serous ovarian carcinoma. Int. J. Gynecol. Cancer 2007, 17, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Fotopoulou, C.; Meyer, T. The molecular fingerprint of high grade serous ovarian cancer reflects its fallopian tube origin. Int. J. Mol. Sci. 2013, 14, 6571–6596. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in BRCA; TP53; PTEN models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Przybycin, C.G.; Kurman, R.J.; Ronnett, B.M.; Shih, I.M.; Vang, R. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am. J. Surg. Pathol. 2010, 34, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Coffey, D.M.; Creighton, C.J.; Yu, Z.; Hawkins, S.M.; Matzuk, M.M. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.J.; Burdette, J.E. Pten loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Coffey, D.M.; Ma, L.; Matzuk, M.M. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 2015, 156, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.K.; Wong, K.K.; Marciano, D.C.; Katsonis, P.; King-Crane, E.R.; Ren, Y.A.; Lichtarge, O.; Richards, J.S. Specific TP53 mutants overrepresented in ovarian cancer impact CNV, TP53 activity, responses to nutlin-3a, and cell survival. Neoplasia 2015, 17, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Merajver, S.D.; Pham, T.M.; Caduff, R.F.; Chen, M.; Poy, E.L.; Cooney, K.A.; Weber, B.L.; Collins, F.S.; Johnston, C.; Frank, T.S. Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat. Genet. 1995, 9, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Berchuck, A.; Heron, K.A.; Carney, M.E.; Lancaster, J.M.; Fraser, E.G.; Vinson, V.L.; Deffenbaugh, A.M.; Miron, A.; Marks, J.R.; Futreal, P.A.; et al. Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin. Cancer Res. 1998, 4, 2433–2437. [Google Scholar] [PubMed]
- Hennessy, B.T.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D., 2nd; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J. Clin. Oncol. 2010, 28, 3570–3576. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Harrington, P.; Kerr, J.; Russell, P.; DiCioccio, R.A.; Scott, I.V.; Jacobs, I.; Chenevix-Trench, G.; Ponder, B.A.; Gayther, S.A. Somatic and germline mutations of the BRCA2 gene in sporadic ovarian cancer. Cancer Res. 1996, 56, 3622–3625. [Google Scholar] [PubMed]
- Hilton, J.L.; Geisler, J.P.; Rathe, J.A.; Hattermann-Zogg, M.A.; DeYoung, B.; Buller, R.E. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J. Natl. Cancer Inst. 2002, 94, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.P.; Hatterman-Zogg, M.A.; Rathe, J.A.; Buller, R.E. Frequency of BRCA1 dysfunction in ovarian cancer. J. Natl. Cancer Inst. 2002, 94, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Rzepecka, I.K.; Szafron, L.; Stys, A.; Bujko, M.; Plisiecka-Halasa, J.; Madry, R.; Osuch, B.; Markowska, J.; Bidzinski, M.; Kupryjanczyk, J. High frequency of allelic loss at the BRCA1 locus in ovarian cancers: Clinicopathologic and molecular associations. Cancer Genet. 2012, 205, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni-Datar, K.; Orsulic, S.; Foster, R.; Rueda, B.R. Ovarian tumor initiating cell populations persist following paclitaxel and carboplatin chemotherapy treatment in vivo. Cancer Lett. 2013, 339, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2013, 338, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug resistance driven by cancer stem cells and their niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Ojeda, D.; Rueda, B.R.; Buckanovich, R.J. Ovarian cancer stem cell markers: Prognostic and therapeutic implications. Cancer Lett. 2012, 322, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Cancerous ovarian stem cells: Obscure targets for therapy but relevant to chemoresistance. J. Cell. Biochem. 2013, 114, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Borovski, T.; De Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Starbuck, K.; Al-Alem, L.; Eavarone, D.A.; Hernandez, S.F.; Bellio, C.; Prendergast, J.M.; Stein, J.; Dransfield, D.T.; Zarrella, B.; Growdon, W.B.; et al. Treatment of ovarian cancer by targeting the tumor stem cell-associated carbohydrate antigen, sialyl-thomsen-nouveau. Oncotarget 2018, 9, 23289–23305. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.M.; Joseph, S.; Sudhahar, C.G.; Cowden Dahl, K.D. Enrichment for chemoresistant ovarian cancer stem cells from human cell lines. J. Vis. Exp. 2014, 51891. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lai, D.; Liu, T.; Cheng, W.; Guo, L. Cancer stem-like cells can be isolated with drug selection in human ovarian cancer cell line SKOV3. Acta Biochim. Biophys. Sin. 2010, 42, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.; Samyesudhas, S.J.; Carrasco, M.; Li, J.; Joseph, S.; Dahl, R.; Cowden Dahl, K.D. Arid3b increases ovarian tumor burden and is associated with a cancer stem cell gene signature. Oncotarget 2014, 5, 8355–8366. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Agro, L.; O’Brien, C. In vitro and in vivo limiting dilution assay for colorectal cancer. Bio-protocol 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.K. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol. Aspects Med. 2014, 39, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Q.; Choi, Y.P.; Kang, S.; Youn, J.H.; Cho, N.H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gu, C.; Huang, K.; Zhang, Z.; Ye, M.; Fan, W.; Han, W.; Meng, Y. The prognostic value and clinicopathological significance of CD44 expression in ovarian cancer: A meta-analysis. Arch. Gynecol. Obstet. 2016, 294, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Elzarkaa, A.A.; Sabaa, B.E.; Abdelkhalik, D.; Mansour, H.; Melis, M.; Shaalan, W.; Farouk, M.; Malik, E.; Soliman, A.A. Clinical relevance of CD44 surface expression in advanced stage serous epithelial ovarian cancer: A prospective study. J. Cancer Res. Clin. Oncol. 2016, 142, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, C.; Rouzier, R.; Geyl, C.; Cortez, A.; Castela, M.; Lis, R.; Darai, E.; Touboul, C. Predictive markers of chemoresistance in advanced stages epithelial ovarian carcinoma. Gynecol. Oncol. 2015, 136, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Peyrollier, K.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction activates stem cell marker nanog, stat-3-mediated mdr1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 2008, 283, 17635–17651. [Google Scholar] [CrossRef] [PubMed]
- Klemba, A.; Purzycka-Olewiecka, J.K.; Wcislo, G.; Czarnecka, A.M.; Lewicki, S.; Lesyng, B.; Szczylik, C.; Kieda, C. Surface markers of cancer stem-like cells of ovarian cancer and their clinical relevance. Contemp. Oncol. 2018, 22, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zeng, J.; Liang, B.; Zhao, Z.; Sun, L.; Cao, D.; Yang, J.; Shen, K. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp. Mol. Pathol. 2011, 91, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Vanderhyden, B.C. AKT mediates the pro-survival effects of kit in ovarian cancer cells and is a determinant of sensitivity to imatinib mesylate. Gynecol. Oncol. 2007, 105, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Ruscito, I.; Cacsire Castillo-Tong, D.; Vergote, I.; Ignat, I.; Stanske, M.; Vanderstichele, A.; Ganapathi, R.N.; Glajzer, J.; Kulbe, H.; Trillsch, F.; et al. Exploring the clonal evolution of CD133/aldehyde-dehydrogenase-1 (ALDH1)-positive cancer stem-like cells from primary to recurrent high-grade serous ovarian cancer (HGSOC). A study of the ovarian cancer therapy-innovative models prolong survival (OCTIPS) consortium. Eur. J. Cancer 2017, 79, 214–225. [Google Scholar] [PubMed]
- Liang, J.; Yang, B.; Cao, Q.; Wu, X. Association of vasculogenic mimicry formation and CD133 expression with poor prognosis in ovarian cancer. Gynecol. Obstet. Investig. 2016, 81, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, A.; Song, H.; Tao, J.; Yang, H.; Zuo, M. Prognostic value of cancer stem cell marker CD133 in ovarian cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 3080–3088. [Google Scholar] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.; Bobbs, A.; Sattler, R.; Kurkewich, J.L.; Dausinas, P.B.; Nallathamby, P.; Cowden Dahl, K.D. CD133 promotes adhesion to the ovarian cancer metastatic niche. Cancer Growth Metastasis 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N., Jr.; Goodman, B.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther. 2010, 9, 3186–3199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yo, Y.T.; Lee, H.Y.; Liao, Y.P.; Chao, T.K.; Su, P.H.; Lai, H.C. Aldh1-bright epithelial ovarian cancer cells are associated with CD44 expression, drug resistance, and poor clinical outcome. Am. J. Pathol. 2012, 180, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiu, J.; Ye, C.; Yang, D.; Gao, L.; Su, Y.; Tang, X.; Xu, N.; Zhang, D.; Xiong, L.; et al. Ror1 expression correlated with poor clinical outcome in human ovarian cancer. Sci. Rep. 2014, 4, 5811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, B.; Lai, H.; Liu, G.; Ghia, E.M.; Widhopf, G.F., 2nd; Zhang, Z.; Wu, C.C.; Chen, L.; Wu, R.; et al. Ovarian cancer stem cells express ror1, which can be targeted for anti-cancer-stem-cell therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 17266–17271. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ji, X.; Zhang, F.; Li, L.; Ma, L. Embryonic stem cell markers. Molecules 2012, 17, 6196–6236. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hou, Y.; Huang, Z.; Cai, J.; Wang, Z. Sox2 is required to maintain cancer stem cells in ovarian cancer. Cancer Sci. 2017, 108, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Di, J.; Duiveman-de Boer, T.; Zusterzeel, P.L.; Figdor, C.G.; Massuger, L.F.; Torensma, R. The stem cell markers OCT4A, nanog and C-MYC are expressed in ascites cells and tumor tissue of ovarian cancer patients. Cell. Oncol. 2013, 36, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.X.; Luo, X.; Xu, M.; Feng, X.; Wang, J. Let-7d increases ovarian cancer cell sensitivity to a genistein analog by targeting c-MYC. Oncotarget 2017, 8, 74836–74845. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter BCRP1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Garson, K.; Vanderhyden, B.C. Epithelial ovarian cancer stem cells: Underlying complexity of a simple paradigm. Reproduction 2015, 149, R59–R70. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; McArthur, C.; Jaffe, R.B. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br. J. Cancer 2010, 102, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Seino, K.; Hosonuma, S.; Ohara, T.; Itamochi, H.; Isonishi, S.; Kita, T.; Wada, H.; Kojo, S.; Kiguchi, K. Side population is increased in paclitaxel-resistant ovarian cancer cell lines regardless of resistance to cisplatin. Gynecol. Oncol. 2011, 121, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and mullerian inhibiting substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Seshacharyulu, P.; Lakshmanan, I.; Vaz, A.P.; Chugh, S.; Sheinin, Y.M.; Mahapatra, S.; Batra, S.K.; Ponnusamy, M.P. HPAF1/PD2 interacts with OCT3/4 to promote self-renewal of ovarian cancer stem cells. Oncotarget 2017, 8, 14806–14820. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Maihle, N.J.; Huang, Y. Pluripotency factors LIN28 and OCT4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene 2010, 29, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Varas-Godoy, M.; Rice, G.; Illanes, S.E. The crosstalk between ovarian cancer stem cell niche and the tumor microenvironment. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Vathipadiekal, V.; Saxena, D.; Mok, S.C.; Hauschka, P.V.; Ozbun, L.; Birrer, M.J. Identification of a potential ovarian cancer stem cell gene expression profile from advanced stage papillary serous ovarian cancer. PLoS ONE 2012, 7, e29079. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, M.P.; Batra, S.K. Ovarian cancer: Emerging concept on cancer stem cells. J. Ovarian Res. 2008, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Cowden Dahl, K.D.; Dahl, R.; Kruichak, J.N.; Hudson, L.G. The epidermal growth factor receptor responsive MIR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia 2009, 11, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.; Bobbs, A.; Cowden Dahl, K. Indiana University School of Medicine-South Bend, University of Notre Dame. Unpublished data. 2016. [Google Scholar]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Wang, H.Q.; Skele, K.L.; De Rienzo, A.; Klein-Szanto, A.J.; Godwin, A.K.; Testa, J.R. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 2004, 23, 5853–5857. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Xu, L.; Tang, H.; Yang, Q.; Yi, X.; Fang, Y.; Zhu, Y.; Wang, Z. The role of the PTEN/PI3K/AKT pathway on prognosis in epithelial ovarian cancer: A meta-analysis. Oncologist 2014, 19, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Dong, Z.; Chen, Y.; Yang, L.; Lai, D. Enrichment of ovarian cancer stem-like cells is associated with epithelial to mesenchymal transition through an miRNA-activated AKT pathway. Cell Prolif. 2013, 46, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.J.; Kwon, Y.W.; Jang, I.H.; Kim, D.K.; Lee, S.I.; Choi, E.J.; Kim, K.H.; Suh, D.S.; Lee, J.H.; Choi, K.U.; et al. Autotaxin regulates maintenance of ovarian cancer stem cells through lysophosphatidic acid-mediated autocrine mechanism. Stem Cells 2016, 34, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.Y.; Kim, J.Y.; Pelletier, J.F.; Vanderhyden, B.C.; Bachvarov, D.R.; Tsang, B.K. AKT confers cisplatin chemoresistance in human gynecological carcinoma cells by modulating ppm1d stability. Mol. Carcinog. 2015, 54, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Chau, W.K.; Ip, C.K.; Mak, A.S.; Lai, H.C.; Wong, A.S. C-kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/BETA-catenin-ATP-binding cassette G2 signaling. Oncogene 2013, 32, 2767–2781. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Miyamoto, M.; Aoyama, T.; Soyama, H.; Goto, T.; Hirata, J.; Suzuki, A.; Nagaoka, I.; Tsuda, H.; Furuya, K.; et al. JAK2/STAT3 pathway as a therapeutic target in ovarian cancers. Oncol. Lett. 2018, 15, 5772–5780. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014, 14, 317. [Google Scholar] [CrossRef] [PubMed]
- Saini, U.; Suarez, A.A.; Naidu, S.; Wallbillich, J.J.; Bixel, K.; Wanner, R.A.; Bice, J.; Kladney, R.D.; Lester, J.; Karlan, B.Y.; et al. STAT3/PIAS3 levels serve as “early signature” genes in the development of high-grade serous carcinoma from the fallopian tube. Cancer Res. 2018, 78, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.G.; Mercado-Uribe, I.; Yang, G.; Bast, R.C., Jr.; Amin, H.M.; Lai, R.; Liu, J. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer 2006, 107, 2730–2740. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Ojeda, D.; Wu, R.; McLean, K.; Chen, Y.C.; Talpaz, M.; Yoon, E.; Cho, K.R.; Buckanovich, R.J. CD24+ ovarian cancer cells are enriched for cancer-initiating cells and dependent on JAK2 signaling for growth and metastasis. Mol. Cancer Ther. 2015, 14, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Rinkenbaugh, A.L.; Baldwin, A.S. The NF-κB pathway and cancer stem cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Torres, C.; Gaytan-Cervantes, J.; Vazquez-Santillan, K.; Mandujano-Tinoco, E.A.; Ceballos-Cancino, G.; Garcia-Venzor, A.; Zampedri, C.; Sanchez-Maldonado, P.; Mojica-Espinosa, R.; Jimenez-Hernandez, L.E.; et al. NF-κB participates in the stem cell phenotype of ovarian cancer cells. Arch. Med. Res. 2017, 48, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Leizer, A.L.; Alvero, A.B.; Fu, H.H.; Holmberg, J.C.; Cheng, Y.C.; Silasi, D.A.; Rutherford, T.; Mor, G. Regulation of inflammation by the NF-κB pathway in ovarian cancer stem cells. Am. J. Reprod. Immunol. 2011, 65, 438–447. [Google Scholar] [CrossRef] [PubMed]
- House, C.D.; Jordan, E.; Hernandez, L.; Ozaki, M.; James, J.M.; Kim, M.; Kruhlak, M.J.; Batchelor, E.; Elloumi, F.; Cam, M.C.; et al. NF-κB promotes ovarian tumorigenesis via classical pathways that support proliferative cancer cells and alternative pathways that support ALDH(+) cancer stem-like cells. Cancer Res. 2017, 77, 6927–6940. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, notch, and hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Thiaville, M.M.; Chen, L.; Stoeck, A.; Xuan, J.; Gao, M.; Shih, I.M.; Wang, T.L. Defining notch3 target genes in ovarian cancer. Cancer Res. 2012, 72, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Li, M.; Nakayama, K.; Mao, T.L.; Davidson, B.; Zhang, Z.; Kurman, R.J.; Eberhart, C.G.; Shih, I.M.; Wang, T.L. Notch3 gene amplification in ovarian cancer. Cancer Res. 2006, 66, 6312–6318. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Park, J.T.; Davidson, B.; Morin, P.J.; Shih, I.M.; Wang, T.L. Jagged-1 and notch3 juxtacrine loop regulates ovarian tumor growth and adhesion. Cancer Res. 2008, 68, 5716–5723. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Chen, X.; Trope, C.G.; Davidson, B.; Shih, I.M.; Wang, T.L. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am. J. Pathol. 2010, 177, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Li, Y.; Cao, X.; Du, J.; Huang, X. Nanog regulates epithelial-mesenchymal transition and chemoresistance in ovarian cancer. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.D.; Katre, A.A.; Goodman, B.; Han, H.D.; Nick, A.M.; Stone, R.L.; Coleman, R.L.; Alvarez, R.D.; Lopez-Berestein, G.; Sood, A.K.; et al. Targeting the notch ligand jagged1 in both tumor cells and stroma in ovarian cancer. Clin. Cancer Res. 2011, 17, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical Wnt signals in development and disease: Conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef] [PubMed]
- Gavert, N.; Ben-Ze’ev, A. Beta-catenin signaling in biological control and cancer. J. Cell. Biochem. 2007, 102, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a signaling in cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.J.; Watanabe, A.; Howell, S.B. LGR5 and LGR6 in stem cell biology and ovarian cancer. Oncotarget 2018, 9, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Flesken-Nikitin, A.; Hwang, C.I.; Cheng, C.Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Tan, S.; Singh, G.; Rizk, P.; Swathi, Y.; Tan, T.Z.; Huang, R.Y.; Leushacke, M.; Barker, N. LGR5 marks stem/progenitor cells in ovary and tubal epithelia. Nat. Cell Biol. 2014, 16, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. Wnt signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Barbolina, M.V.; Burkhalter, R.J.; Stack, M.S. Diverse mechanisms for activation of Wnt signalling in the ovarian tumour microenvironment. Biochem. J. 2011, 437, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Meng, E.; Reed, E.; Shevde, L.A.; Rocconi, R.P. Hedgehog signaling pathway regulates the growth of ovarian cancer spheroid forming cells. Int. J. Oncol. 2011, 39, 797–804. [Google Scholar] [PubMed]
- Bhattacharya, R.; Kwon, J.; Ali, B.; Wang, E.; Patra, S.; Shridhar, V.; Mukherjee, P. Role of hedgehog signaling in ovarian cancer. Clin. Cancer Res. 2008, 14, 7659–7666. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Gavin, E.; Das, S.; Amable, L.; Shevde, L.A.; Reed, E. Inhibition of gli1 results in altered c-JUN activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum-DNA adduct repair. Oncogene 2012, 31, 4718–4724. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yan, L.; Lu, C.; Zhang, C.; Zhu, F.; Yang, J.; Jing, H.; Zhang, Y.; Qiao, J.; Guo, H. Activation of hedgehog signaling and its association with cisplatin resistance in ovarian epithelial tumors. Oncol. Lett. 2018, 15, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Matsui, W. Targeting hedgehog—A cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Type of Protein | Suspected Role in Stem Cells | References |
|---|---|---|---|
| CD24 | Cell surface transmembrane glycoprotein | Stem gene expression, tumor initiation, chemoresistance, stem cell maintenance | [46,53,61,62] |
| CD44 | Cell surface transmembrane glycoprotein (hyaluronic acid receptor) | Chemoresistance, tumor initiation, stem gene expression, spheroid formation | [13,46,53,61,62,63,64,65,66,67] |
| cKit/CD117 | Tyrosine kinase receptor | Chemoresistance, stem cell maintenance, tumor initiation | [11,53,59,61,68,69] |
| PROM1/CD133 | Cell surface transmembrane glycoprotein | Tumor initiation, chemoresistance, spheroid formation, high cell proliferation | [13,46,53,61,62,70,71,72,73,74,75,76] |
| ALDH1 | Cytosolic aldehyde dehydrogenase enzyme | Tumor initiation, chemoresistance, spheroid formation | [46,53,61,75,77,78] |
| ROR1 | Tyrosine kinase receptor | Spheroid formation, tumor initiation, proliferation | [79,80] |
| SOX2 | Transcription factor | Stem cell maintenance, self-renewal | [8,81,82,83,84] |
| NANOG | Transcription factor | Stem cell maintenance, self-renewal, chemoresistance | [8,53,61,66,81,82,83] |
| POU5F1/OCT4 | Transcription factor | Tumor initiation, chemoresistance | [8,53,61,81,82,83] |
| MYC | Transcription factor | Tumor initiation, chemoresistance | [85,86] |
| EpCAM | Cell surface membrane glycoprotein | Tumor initiation, spheroid formation, proliferation | [13,46,53,61,62] |
| MDR1/ABCB1 | ATP binding cassette transporter | Chemoresistance | [46,49,53,61,66,87,88,89,90,91] |
| ABCG2 | ATP binding cassette transporter | Chemoresistance | [46,49,53,61,87,88,90,91] |
| Pathway | Gene | Potential Therapeutics in Trials |
|---|---|---|
| PI3K/PTEN/AKT | AKT1 | BKM120, Everdimus, Perifosine |
| PTEN | ||
| PPMID | ||
| Jak/STAT | STAT3 | |
| JAK2 | ||
| NFκB | RelA | |
| RelB | ||
| IKK | ||
| IκBα | ||
| TNFα | ||
| Notch | Notch3 | γ-secretase inhibitors, γ-secretase modifiers, Notch soluble decoys, negative regulatory region monoclonal antibodies |
| Jagged1 | ||
| Wnt | β-catenin | NSC668036, FJ9, Frizzled receptor antibodies, Thiazoldinedone, Suldinac |
| Wnt5A | ||
| Disheveled | ||
| Frizzled | ||
| Hedgehog | Patched | HPI-1, HPI-2, HPI-3, HPI-4, GDC-0449 |
| Gli1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, L.; Cowden Dahl, K.D. Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer? Cancers 2018, 10, 241. https://doi.org/10.3390/cancers10080241
Roy L, Cowden Dahl KD. Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer? Cancers. 2018; 10(8):241. https://doi.org/10.3390/cancers10080241
Chicago/Turabian StyleRoy, Lynn, and Karen D. Cowden Dahl. 2018. "Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer?" Cancers 10, no. 8: 241. https://doi.org/10.3390/cancers10080241
APA StyleRoy, L., & Cowden Dahl, K. D. (2018). Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer? Cancers, 10(8), 241. https://doi.org/10.3390/cancers10080241