Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism



Abstract
1. Introduction
2. Overview on the Pharmacodynamics of Nucleobase and Nucleoside Analogues
3. Pharmacokinetic Resistance to Nucleobase/Nucleoside Analogues: Delivery
3.1. Bioavailability
3.2. Body Compartments as Sanctuaries
4. Pharmacokinetic Resistance to Nucleobase/Nucleoside Analogues: Stability
4.1. Deamination
4.2. Hydrogenation, Methylation, Deglycosylation
5. Tumour-Specific Resistance to Nucleobase/Nucleoside Analogues: Membrane Transport
6. Tumour-Specific Resistance to Nucleobase/Nucleoside Analogues: Intracellular Metabolism
6.1. Glycosylation of Nucleobases
6.2. Monophosphorylation
6.3. Diphosphorylation
6.4. Triphosphorylation
6.5. Intracellular Deamination
6.6. Dephosphorylation of Monophosphates
6.7. Dephosphorylation of Triphosphates
7. Tumour-Specific Resistance to Nucleobase/Nucleoside Analogues: Pharmacodynamics
8. Tumour-Specific Resistance to Nucleobase/Nucleoside Analogues: Tumour Biology
9. Clinical Resistance to Nucleobase/Nucleoside Analogues: Therapy-Limiting Toxicity
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Heitzeneder, S.; Mackall, C.L. Harnessing the Immunotherapy Revolution for the Treatment of Childhood Cancers. Cancer Cell 2017, 31, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Kisliuk, R.L. Synergistic interactions among antifolates. Pharmacol. Ther. 2000, 85, 183–190. [Google Scholar] [CrossRef]
- Shimizu, T.; Nakanishi, Y.; Nakagawa, Y.; Tsujino, I.; Takahashi, N.; Nemoto, N.; Hashimoto, S. Association between expression of thymidylate synthase, dihydrofolate reductase, and glycinamide ribonucleotide formyltransferase and efficacy of pemetrexed in advanced non-small cell lung cancer. Anticancer Res. 2012, 32, 4589–4596. [Google Scholar] [PubMed]
- Bertino, J.R. Cancer research: From folate antagonism to molecular targets. Best Pract. Res. Clin. Haematol. 2009, 22, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Hughes, A.N. Pemetrexed disodium, a novel antifolate with multiple targets. Lancet Oncol. 2001, 2, 298–306. [Google Scholar] [CrossRef]
- Rowland, P., 3rd; Pfeilsticker, J.; Hoffee, P.A. Adenosine deaminase gene amplification in deoxycoformycin-resistant mammalian cells. Arch. Biochem. Biophys. 1985, 239, 396–403. [Google Scholar] [CrossRef]
- Agarwal, R.P.; Blatt, J.; Miser, J.; Sallan, S.; Lipton, J.M.; Reaman, G.H.; Holcenberg, J.; Poplack, D.G. Clinical pharmacology of 9-beta-D-arabinofuranosyladenine in combination with 2′-deoxycoformycin. Cancer Res. 1982, 42, 3884–3886. [Google Scholar] [PubMed]
- Serdjebi, C.; Milano, G.; Ciccolini, J. Role of cytidine deaminase in toxicity and efficacy of nucleosidic analogs. Expert Opin. Drug Metab. Toxicol. 2015, 11, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Parmar, S.; Seeringer, A.; Denich, D.; Gartner, F.; Pitterle, K.; Syrovets, T.; Ohmle, B.; Stingl, J.C. Variability in transport and biotransformation of cytarabine is associated with its toxicity in peripheral blood mononuclear cells. Pharmacogenomics 2011, 12, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Saikawa, Y.; Ota, K.; Tanaka, M.; Nishimura, R.; Uehara, T.; Maeba, H.; Ito, T.; Sasaki, T.; Koizumi, S. A functional single-nucleotide polymorphism in the human cytidine deaminase gene contributing to ara-C sensitivity. Pharmacogenetics 2003, 13, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Falk, I.J.; Fyrberg, A.; Paul, E.; Nahi, H.; Hermanson, M.; Rosenquist, R.; Hoglund, M.; Palmqvist, L.; Stockelberg, D.; Wei, Y.; et al. Decreased survival in normal karyotype AML with single-nucleotide polymorphisms in genes encoding the AraC metabolizing enzymes cytidine deaminase and 5′-nucleotidase. Am. J. Hematol. 2013, 88, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Tibaldi, C.; Giovannetti, E.; Tiseo, M.; Leon, L.G.; D’Incecco, A.; Loosekoot, N.; Bartolotti, M.; Honeywell, R.; Cappuzzo, F.; Ardizzoni, A.; et al. Correlation of cytidine deaminase polymorphisms and activity with clinical outcome in gemcitabine-/platinum-treated advanced non-small-cell lung cancer patients. Ann. Oncol. 2012, 23, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Cahard, D.; McGuigan, C.; Balzarini, J. Aryloxy phosphoramidate triesters as pro-tides. Mini Rev. Med. Chem. 2004, 4, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Hori, H.; Ogawa, M.; Miyahara, M.; Kawasaki, H.; Taniguchi, N.; Komada, Y. Impact of cytidine deaminase activity on intrinsic resistance to cytarabine in carcinoma cells. Oncol. Rep. 2004, 12, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Kreis, W.; Budman, D.R.; Chan, K.; Allen, S.L.; Schulman, P.; Lichtman, S.; Weiselberg, L.; Schuster, M.; Freeman, J.; Akerman, S.; et al. Therapy of refractory/relapsed acute leukemia with cytosine arabinoside plus tetrahydrouridine (an inhibitor of cytidine deaminase)—A pilot study. Leukemia 1991, 5, 991–998. [Google Scholar] [PubMed]
- Lemaire, M.; Momparler, L.F.; Raynal, N.J.; Bernstein, M.L.; Momparler, R.L. Inhibition of cytidine deaminase by zebularine enhances the antineoplastic action of 5-aza-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2009, 63, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Cividini, F.; Filoni, D.N.; Pesi, R.; Allegrini, S.; Camici, M.; Tozzi, M.G. IMP-GMP specific cytosolic 5′-nucleotidase regulates nucleotide pool and prodrug metabolism. Biochim. Biophys. Acta 2015, 1850, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Marton, Z.; Rhimi, M.; Cros-Perrial, E.; Lionne, C.; Peyrottes, S.; Dumontet, C.; Aghajari, N.; Chaloin, L. Identification and characterization of inhibitors of cytoplasmic 5′-nucleotidase cN-II issued from virtual screening. Biochem. Pharmacol. 2013, 85, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Cros, E.; Thomas, X.; Jordheim, L.; Dumontet, C. The prognostic value of cN-II and cN-III enzymes in adult acute myeloid leukemia. Haematologica 2005, 90, 1699–1701. [Google Scholar] [PubMed]
- Negoro, E.; Yamauchi, T.; Urasaki, Y.; Nishi, R.; Hori, H.; Ueda, T. Characterization of cytarabine-resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. Int. J. Oncol. 2011, 38, 911–919. [Google Scholar] [PubMed]
- Galmarini, C.M.; Thomas, X.; Graham, K.; El Jafaari, A.; Cros, E.; Jordheim, L.; Mackey, J.R.; Dumontet, C. Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br. J. Haematol. 2003, 122, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Negoro, E.; Kishi, S.; Takagi, K.; Yoshida, A.; Urasaki, Y.; Iwasaki, H.; Ueda, T. Intracellular cytarabine triphosphate production correlates to deoxycytidine kinase/cytosolic 5′-nucleotidase II expression ratio in primary acute myeloid leukemia cells. Biochem. Pharmacol. 2009, 77, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Seve, P.; Mackey, J.R.; Isaac, S.; Tredan, O.; Souquet, P.J.; Perol, M.; Cass, C.; Dumontet, C. cN-II expression predicts survival in patients receiving gemcitabine for advanced non-small cell lung cancer. Lung Cancer 2005, 49, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Hunsucker, S.A.; Spychala, J.; Mitchell, B.S. Human cytosolic 5′-nucleotidase I: Characterization and role in nucleoside analog resistance. J. Biol. Chem. 2001, 276, 10498–10504. [Google Scholar] [CrossRef] [PubMed]
- Hnizda, A.; Fabry, M.; Moriyama, T.; Pachl, P.; Kugler, M.; Brinsa, V.; Ascher, D.B.; Carroll, W.L.; Novak, P.; Zaliova, M.; et al. Relapsed acute lymphoblastic leukemia-specific mutations in NT5C2 cluster into hotspots driving intersubunit stimulation. Leukemia 2018, 32, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.A.; Wang, J.; Hogan, L.E.; Yang, J.J.; Dandekar, S.; Patel, J.P.; Tang, Z.; Zumbo, P.; Li, S.; Zavadil, J.; et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Tzoneva, G.; Dieck, C.L.; Oshima, K.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Madubata, C.J.; Khiabanian, H.; Yu, J.; Waanders, E.; Iacobucci, I.; et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018, 553, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Tzoneva, G.; Perez-Garcia, A.; Carpenter, Z.; Khiabanian, H.; Tosello, V.; Allegretta, M.; Paietta, E.; Racevskis, J.; Rowe, J.M.; Tallman, M.S.; et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat. Med. 2013, 19, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Crews, K.R.; Pounds, S.; Cao, X.; Feldberg, T.; Ghodke, Y.; Gandhi, V.; Plunkett, W.; Dolan, M.E.; Hartford, C.; et al. Genetic variants in cytosolic 5′-nucleotidase II are associated with its expression and cytarabine sensitivity in HapMap cell lines and in patients with acute myeloid leukemia. J. Pharmacol. Exp. Ther. 2011, 339, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.S.; Koh, Y.; Ahn, K.S.; Lee, C.; Shin, H.D.; Yoon, S.S. NT5C3 polymorphisms and outcome of first induction chemotherapy in acute myeloid leukemia. Pharmacogenet. Genom. 2014, 24, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Edahiro, K.; Iimori, M.; Kobunai, T.; Morikawa-Ichinose, T.; Miura, D.; Kataoka, Y.; Niimi, S.; Wakasa, T.; Saeki, H.; Oki, E.; et al. Thymidine Kinase 1 Loss Confers Trifluridine Resistance without Affecting 5-Fluorouracil Metabolism and Cytotoxicity. Mol. Cancer Res. MCR 2018. [Google Scholar] [CrossRef] [PubMed]
- Mekras, J.A.; Boothman, D.A.; Perez, L.M.; Greer, S. Use of 5-fluorodeoxycytidine and tetrahydrouridine to exploit high levels of deoxycytidylate deaminase in tumors to achieve DNA- and target-directed therapies. Cancer Res. 1984, 44, 2551–2560. [Google Scholar] [PubMed]
- Heinemann, V.; Plunkett, W. Modulation of deoxynucleotide metabolism by the deoxycytidylate deaminase inhibitor 3,4,5,6-tetrahydrodeoxyuridine. Biochem. Pharmacol. 1989, 38, 4115–4121. [Google Scholar] [CrossRef]
- Klanova, M.; Lorkova, L.; Vit, O.; Maswabi, B.; Molinsky, J.; Pospisilova, J.; Vockova, P.; Mavis, C.; Lateckova, L.; Kulvait, V.; et al. Downregulation of deoxycytidine kinase in cytarabine-resistant mantle cell lymphoma cells confers cross-resistance to nucleoside analogs gemcitabine, fludarabine and cladribine, but not to other classes of anti-lymphoma agents. Mol. Cancer 2014, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Lorkova, L.; Scigelova, M.; Arrey, T.N.; Vit, O.; Pospisilova, J.; Doktorova, E.; Klanova, M.; Alam, M.; Vockova, P.; Maswabi, B.; et al. Detailed Functional and Proteomic Characterization of Fludarabine Resistance in Mantle Cell Lymphoma Cells. PLoS ONE 2015, 10, e0135314. [Google Scholar] [CrossRef] [PubMed]
- Geutjes, E.J.; Tian, S.; Roepman, P.; Bernards, R. Deoxycytidine kinase is overexpressed in poor outcome breast cancer and determines responsiveness to nucleoside analogs. Breast Cancer Res. Treat. 2012, 131, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Kurata, M.; Rathe, S.K.; Bailey, N.J.; Aumann, N.K.; Jones, J.M.; Veldhuijzen, G.W.; Moriarity, B.S.; Largaespada, D.A. Using genome-wide CRISPR library screening with library resistant DCK to find new sources of Ara-C drug resistance in AML. Sci. Rep. 2016, 6, 36199. [Google Scholar] [CrossRef] [PubMed]
- Malani, D.; Murumagi, A.; Yadav, B.; Kontro, M.; Eldfors, S.; Kumar, A.; Karjalainen, R.; Majumder, M.M.; Ojamies, P.; Pemovska, T.; et al. Enhanced sensitivity to glucocorticoids in cytarabine-resistant AML. Leukemia 2017, 31, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel-Eibrink, M.M.; Wiemer, E.A.; Kuijpers, M.; Pieters, R.; Sonneveld, P. Absence of mutations in the deoxycytidine kinase (dCK) gene in patients with relapsed and/or refractory acute myeloid leukemia (AML). Leukemia 2001, 15, 855–856. [Google Scholar] [CrossRef] [PubMed]
- Veuger, M.J.; Honders, M.W.; Landegent, J.E.; Willemze, R.; Barge, R.M. High incidence of alternatively spliced forms of deoxycytidine kinase in patients with resistant acute myeloid leukemia. Blood 2000, 96, 1517–1524. [Google Scholar] [PubMed]
- Medina-Sanson, A.; Ramirez-Pacheco, A.; Moreno-Guerrero, S.S.; Dorantes-Acosta, E.M.; Sanchez-Preza, M.; Reyes-Lopez, A. Role of Genetic Polymorphisms of Deoxycytidine Kinase and Cytidine Deaminase to Predict Risk of Death in Children with Acute Myeloid Leukemia. BioMed Res. Int. 2015, 2015, 309491. [Google Scholar] [CrossRef] [PubMed]
- Gabor, K.M.; Schermann, G.; Lautner-Csorba, O.; Rarosi, F.; Erdelyi, D.J.; Endreffy, E.; Berek, K.; Bartyik, K.; Bereczki, C.; Szalai, C.; et al. Impact of single nucleotide polymorphisms of cytarabine metabolic genes on drug toxicity in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2015, 62, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Spasokoukotskaja, T.; Sasvari-Szekely, M.; Keszler, G.; Albertioni, F.; Eriksson, S.; Staub, M. Treatment of normal and malignant cells with nucleoside analogues and etoposide enhances deoxycytidine kinase activity. Eur. J. Cancer 1999, 35, 1862–1867. [Google Scholar] [CrossRef]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. FDA approvals usher in the post-interferon era in HCV. Nat. Biotechnol. 2014, 32, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Tobias, S.C.; Borch, R.F. Synthesis and biological evaluation of a cytarabine phosphoramidate prodrug. Mol. Pharm. 2004, 1, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Cros, E.; Gouy, M.H.; Galmarini, C.M.; Peyrottes, S.; Mackey, J.; Perigaud, C.; Dumontet, C. Characterization of a gemcitabine-resistant murine leukemic cell line: Reversion of in vitro resistance by a mononucleotide prodrug. Clin. Cancer Res. 2004, 10, 5614–5621. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Uzui, K.; Nishi, R.; Shigemi, H.; Ueda, T. Cytarabine-resistant leukemia cells are moderately sensitive to clofarabine in vitro. Anticancer Res. 2014, 34, 1657–1662. [Google Scholar] [PubMed]
- Yamauchi, T.; Uzui, K.; Nishi, R.; Shigemi, H.; Ueda, T. Reduced drug incorporation into DNA and antiapoptosis as the crucial mechanisms of resistance in a novel nelarabine-resistant cell line. BMC Cancer 2014, 14, 547. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Johansson, M.; Permert, J.; Karlsson, A. Enhanced cytotoxicity of nucleoside analogs by overexpression of mitochondrial deoxyguanosine kinase in cancer cell lines. J. Biol. Chem. 1998, 273, 14707–14711. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, D.; Henricks, L.M.; Sonke, G.S.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiader, C.R.; Jennings, B.A.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef]
- Del Re, M.; Restante, G.; Di Paolo, A.; Crucitta, S.; Rofi, E.; Danesi, R. Pharmacogenetics and Metabolism from Science to Implementation in Clinical Practice: The Example of Dihydropyrimidine Dehydrogenase. Curr. Pharm. Des. 2017, 23, 2028–2034. [Google Scholar] [PubMed]
- Rivera, E.; Chang, J.C.; Semiglazov, V.; Burdaeva, O.; Kirby, M.G.; Spector, T. Eniluracil plus 5-fluorouracil and leucovorin: Treatment for metastatic breast cancer patients in whom capecitabine treatment rapidly failed. Clin. Breast Cancer 2014, 14, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Yen-Revollo, J.L.; Goldberg, R.M.; McLeod, H.L. Can inhibiting dihydropyrimidine dehydrogenase limit hand-foot syndrome caused by fluoropyrimidines? Clin. Cancer Res. 2008, 14, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, H.J. Dihydropyrimidine dehydrogenase inhibition as a strategy for the oral administration of 5-fluorouracil: Utility in the treatment of advanced colorectal cancer. Anti-Cancer Drugs 2003, 14, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mizutani, Y.; Shiraishi, T.; Nakamura, T.; Mikami, K.; Takaha, N.; Okihara, K.; Kawauchi, A.; Sakai, T.; Miki, T. The significance of the expression of dihydropyrimidine dehydrogenase in prostate cancer. BJU Int. 2007, 99, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Shen, N.; Pang, J.; Molina, J.R.; Yang, P.; Liu, S. The DNA Methyltransferase DNMT1 and Tyrosine-Protein Kinase KIT Cooperatively Promote Resistance to 5-Aza-2′-deoxycytidine (Decitabine) and Midostaurin (PKC412) in Lung Cancer Cells. J. Biol. Chem. 2015, 290, 18480–18494. [Google Scholar] [CrossRef] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Ladner, R.D.; Lynch, F.J.; Groshen, S.; Xiong, Y.P.; Sherrod, A.; Caradonna, S.J.; Stoehlmacher, J.; Lenz, H.J. dUTP nucleotidohydrolase isoform expression in normal and neoplastic tissues: Association with survival and response to 5-fluorouracil in colorectal cancer. Cancer Res. 2000, 60, 3493–3503. [Google Scholar] [PubMed]
- An, Q.; Robins, P.; Lindahl, T.; Barnes, D.E. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007, 67, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Hagenkort, A.; Paulin, C.B.J.; Desroses, M.; Sarno, A.; Wiita, E.; Mortusewicz, O.; Koolmeister, T.; Loseva, O.; Jemth, A.S.; Almlof, I.; et al. dUTPase inhibition augments replication defects of 5-Fluorouracil. Oncotarget 2017, 8, 23713–23726. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, S.; Miyakoshi, H.; Yokogawa, T.; Chong, K.T.; Taguchi, J.; Muto, T.; Endoh, K.; Yano, W.; Wakasa, T.; Ueno, H.; et al. Discovery of a novel class of potent human deoxyuridine triphosphatase inhibitors remarkably enhancing the antitumor activity of thymidylate synthase inhibitors. J. Med. Chem. 2012, 55, 2970–2980. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, H.; Miyahara, S.; Yokogawa, T.; Endoh, K.; Muto, T.; Yano, W.; Wakasa, T.; Ueno, H.; Chong, K.T.; Taguchi, J.; et al. 1,2,3-Triazole-containing uracil derivatives with excellent pharmacokinetics as a novel class of potent human deoxyuridine triphosphatase inhibitors. J. Med. Chem. 2012, 55, 6427–6437. [Google Scholar] [CrossRef] [PubMed]
- Yano, W.; Yokogawa, T.; Wakasa, T.; Yamamura, K.; Fujioka, A.; Yoshisue, K.; Matsushima, E.; Miyahara, S.; Miyakoshi, H.; Taguchi, J.; et al. TAS-114, a First-in-Class Dual dUTPase/DPD Inhibitor, Demonstrates Potential to Improve Therapeutic Efficacy of Fluoropyrimidine-based Chemotherapy. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Karim, H.; Hashemi, J.; Larsson, C.; Moshfegh, A.; Fotoohi, A.K.; Albertioni, F. The pattern of gene expression and gene dose profiles of 6-Mercaptopurine- and 6-Thioguanine-resistant human leukemia cells. Biochem. Biophys. Res. Commun. 2011, 411, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Fotoohi, A.K.; Lindqvist, M.; Peterson, C.; Albertioni, F. Involvement of the concentrative nucleoside transporter 3 and equilibrative nucleoside transporter 2 in the resistance of T-lymphoblastic cell lines to thiopurines. Biochem. Biophys. Res. Commun. 2006, 343, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Sandvold, M.L.; Myhren, F.; Jacobsen, T.F.; Giles, F.; Rizzieri, D.A. Anti proliferative activity of ELACY (CP-4055) in combination with cloretazine (VNP40101M), idarubicin, gemcitabine, irinotecan and topotecan in human leukemia and lymphoma cells. Leuk. Lymphoma 2008, 49, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Rizzieri, D.; Vey, N.; Thomas, X.; Huguet-Rigal, F.; Schlenk, R.F.; Krauter, J.; Kindler, T.; Gjertsen, B.T.; Blau, I.W.; Jacobsen, T.F.; et al. A phase II study of elacytarabine in combination with idarubicin and of human equilibrative nucleoside transporter 1 expression in patients with acute myeloid leukemia and persistent blasts after the first induction course. Leuk. Lymphoma 2014, 55, 2114–2119. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Rosenblat, T.; Arellano, M.; Gobbi, M.; Altman, J.K.; Montesinos, P.; O’Connell, C.; Solomon, S.R.; Pigneux, A.; Vey, N.; et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol. 2014, 32, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.L.; Mackey, J.R.; Baldwin, S.A.; Young, J.D.; Cass, C.E. The role of membrane transporters in cellular resistance to anticancer nucleoside drugs. Cancer Treat. Res. 2002, 112, 27–47. [Google Scholar] [PubMed]
- Hubeek, I.; Peters, G.J.; Broekhuizen, R.; Zwaan, C.M.; Kaaijk, P.; van Wering, E.S.; Gibson, B.E.; Creutzig, U.; Janka-Schaub, G.E.; den Boer, M.L.; et al. In vitro sensitivity and cross-resistance to deoxynucleoside analogs in childhood acute leukemia. Haematologica 2006, 91, 17–23. [Google Scholar] [PubMed]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Visser, F.; King, K.M.; Baldwin, S.A.; Young, J.D.; Cass, C.E. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 2007, 26, 85–110. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.A.; Garcia, P.; Bizama, C.; Leal, J.L.; Riquelme, I.; Weber, H.; Macanas, P.; Aguayo, G.; Vinuela, E.; Roa, J.C.; et al. Low expression of equilibrative nucleoside transporter 1 is associated with poor prognosis in chemotherapy-naive pT2 gallbladder adenocarcinoma patients. Histopathology 2016, 68, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Greenhalf, W.; Ghaneh, P.; Neoptolemos, J.P.; Palmer, D.H.; Cox, T.F.; Lamb, R.F.; Garner, E.; Campbell, F.; Mackey, J.R.; Costello, E.; et al. Pancreatic cancer hENT1 expression and survival from gemcitabine in patients from the ESPAC-3 trial. J. Natl. Cancer Inst. 2014, 106, djt347. [Google Scholar] [CrossRef] [PubMed]
- Mohelnikova-Duchonova, B.; Brynychova, V.; Hlavac, V.; Kocik, M.; Oliverius, M.; Hlavsa, J.; Honsova, E.; Mazanec, J.; Kala, Z.; Melichar, B.; et al. The association between the expression of solute carrier transporters and the prognosis of pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 72, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, M.; Schirripa, M.; Cao, S.; Zhang, W.; Yang, D.; Dadduzio, V.; Salvatore, L.; Borelli, B.; Pietrantonio, F.; Ning, Y.; et al. Potential role of polymorphisms in the transporter genes ENT1 and MATE1/OCT2 in predicting TAS-102 efficacy and toxicity in patients with refractory metastatic colorectal cancer. Eur. J. Cancer 2017, 86, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Lemstrova, R.; Soucek, P.; Melichar, B.; Mohelnikova-Duchonova, B. Role of solute carrier transporters in pancreatic cancer: A review. Pharmacogenomics 2014, 15, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Asher, N.; Heled, M.; Cohen, S.B.; Risch, A.; Rund, D. Polymorphisms in transporter and phase II metabolism genes as potential modifiers of the predisposition to and treatment outcome of de novo acute myeloid leukemia in Israeli ethnic groups. Leuk. Res. 2008, 32, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Thomas, X.; Calvo, F.; Rousselot, P.; Rabilloud, M.; El Jaffari, A.; Cros, E.; Dumontet, C. In vivo mechanisms of resistance to cytarabine in acute myeloid leukaemia. Br. J. Haematol. 2002, 117, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Cho, K.M.; Kim, H.J.; Kim, Y.K.; Kim, N.Y.; Kim, H.J.; Lee, T.H.; Hwang, S.Y.; Kim, T.S. Concentrative nucleoside transporter 3 as a prognostic indicator for favorable outcome of t(8;21)-positive acute myeloid leukemia patients after cytarabine-based chemotherapy. Oncol. Rep. 2015, 34, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, M.; Corrales-Alfaro, C.; Gutierrez-Hernandez, O.; Diaz-Chavez, J.; Labardini-Mendez, J.; Vidal-Millan, S.; Herrera, L.A. Expression Levels of Human Equilibrative Nucleoside Transporter 1 and Deoxycytidine Kinase Enzyme as Prognostic Factors in Patients with Acute Myeloid Leukemia Treated with Cytarabine. Chemotherapy 2016, 61, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Zhu, J.; Chen, F.; Xiao, F.; Huang, H.; Han, X.; Zhong, L.; Zhong, H.; Xu, L.; Ni, B.; et al. SLC29A1 single nucleotide polymorphisms as independent prognostic predictors for survival of patients with acute myeloid leukemia: An in vitro study. J. Exp. Clin. Cancer Res. 2014, 33, 90. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.M.; Gati, W.P.; Paterson, A.R. Enhancement of retention and cytotoxicity of 2-chlorodeoxyadenosine in cultured human leukemic lymphoblasts by nitrobenzylthioinosine, an inhibitor of equilibrative nucleoside transport. Leukemia 2000, 14, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Huismans, D.R.; Loonen, A.H.; Peters, G.J.; Hahlen, K.; van der Does-van den Berg, A.; van Wering, E.R.; Veerman, A.J. Hypoxanthine-guanine phosphoribosyl-transferase in childhood leukemia: Relation with immunophenotype, in vitro drug resistance and clinical prognosis. Int. J. Cancer 1992, 51, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Zimm, S.; Reaman, G.; Murphy, R.F.; Poplack, D.G. Biochemical parameters of mercaptopurine activity in patients with acute lymphoblastic leukemia. Cancer Res. 1986, 46, 1495–1498. [Google Scholar] [PubMed]
- Brackett, J.; Schafer, E.S.; Leung, D.H.; Bernhardt, M.B. Use of allopurinol in children with acute lymphoblastic leukemia to reduce skewed thiopurine metabolism. Pediatr. Blood Cancer 2014, 61, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Seinen, M.L.; van Asseldonk, D.P.; de Boer, N.K.; Losekoot, N.; Smid, K.; Mulder, C.J.; Bouma, G.; Peters, G.J.; van Bodegraven, A.A. The effect of allopurinol and low-dose thiopurine combination therapy on the activity of three pivotal thiopurine metabolizing enzymes: Results from a prospective pharmacological study. J. Crohn’s Colitis 2013, 7, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.L.; Gearry, R.B.; Barclay, M.L.; Kennedy, M.A. IMPDH1 promoter mutations in a patient exhibiting azathioprine resistance. Pharmacogenom. J. 2007, 7, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Adam de Beaumais, T.; Fakhoury, M.; Medard, Y.; Azougagh, S.; Zhang, D.; Yakouben, K.; Jacqz-Aigrain, E. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. Br. J. Clin. Pharmacol. 2011, 71, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Gerbek, T.; Ebbesen, M.; Nersting, J.; Frandsen, T.L.; Appell, M.L.; Schmiegelow, K. Role of TPMT and ITPA variants in mercaptopurine disposition. Cancer Chemother. Pharmacol. 2018, 81, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Franca, R.; Rebora, P.; Bertorello, N.; Fagioli, F.; Conter, V.; Biondi, A.; Colombini, A.; Micalizzi, C.; Zecca, M.; Parasole, R.; et al. Pharmacogenetics and induction/consolidation therapy toxicities in acute lymphoblastic leukemia patients treated with AIEOP-BFM ALL 2000 protocol. Pharmacogenom. J. 2017, 17, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Matimba, A.; Li, F.; Livshits, A.; Cartwright, C.S.; Scully, S.; Fridley, B.L.; Jenkins, G.; Batzler, A.; Wang, L.; Weinshilboum, R.; et al. Thiopurine pharmacogenomics: Association of SNPs with clinical response and functional validation of candidate genes. Pharmacogenomics 2014, 15, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.; Jemth, A.S.; Hagenkort, A.; Page, B.D.; Gustafsson, R.; Griese, J.J.; Gad, H.; Valerie, N.C.; Desroses, M.; Bostrom, J.; et al. Crystal structure, biochemical and cellular activities demonstrate separate functions of MTH1 and MTH2. Nat. Commun. 2015, 6, 7871. [Google Scholar] [CrossRef] [PubMed]
- Braunagel, D.; Schaich, M.; Kramer, M.; Dransfeld, C.L.; Ehninger, G.; Mahlknecht, U. The T_T genotype within the NME1 promoter single nucleotide polymorphism-835 C/T is associated with an increased risk of cytarabine induced neurotoxicity in patients with acute myeloid leukemia. Leuk. Lymphoma 2012, 53, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.W.; Chen, P.; Zhang, D.Y.; Yan, H.; Liu, H.; Cen, L.N.; Liu, Y.L.; Cao, S.; Zhou, G.; Zeng, H.; et al. Association of genetic polymorphisms in genes involved in Ara-C and dNTP metabolism pathway with chemosensitivity and prognosis of adult acute myeloid leukemia (AML). J. Transl. Med. 2018, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Meier, C. Nucleoside diphosphate and triphosphate prodrugs—An unsolvable task? Antivir. Chem. Chemother. 2017, 25, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 2012, 26, 1006. [Google Scholar] [CrossRef] [PubMed]
- Goan, Y.G.; Zhou, B.; Hu, E.; Mi, S.; Yen, Y. Overexpression of ribonucleotide reductase as a mechanism of resistance to 2,2-difluorodeoxycytidine in the human KB cancer cell line. Cancer Res. 1999, 59, 4204–4207. [Google Scholar] [PubMed]
- Davidson, J.D.; Ma, L.; Flagella, M.; Geeganage, S.; Gelbert, L.M.; Slapak, C.A. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004, 64, 3761–3766. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, S.; Nakamori, S.; Tsujie, M.; Takahashi, Y.; Okami, J.; Yoshioka, S.; Yamasaki, M.; Marubashi, S.; Takemasa, I.; Miyamoto, A.; et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer 2007, 120, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Guittet, O.; Lepoivre, M.; Galmarini, C.M.; Dumontet, C. Increased expression of the large subunit of ribonucleotide reductase is involved in resistance to gemcitabine in human mammary adenocarcinoma cells. Mol. Cancer Ther. 2005, 4, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.M.; Eijk, P.P.; Ruiz van Haperen, V.W.; Smid, K.; Veerman, G.; Hubeek, I.; van den Ijssel, P.; Ylstra, B.; Peters, G.J. In vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit M1 as the major determinant. Cancer Res. 2005, 65, 9510–9516. [Google Scholar] [CrossRef] [PubMed]
- Itoi, T.; Sofuni, A.; Fukushima, N.; Itokawa, F.; Tsuchiya, T.; Kurihara, T.; Moriyasu, F.; Tsuchida, A.; Kasuya, K. Ribonucleotide reductase subunit M2 mRNA expression in pretreatment biopsies obtained from unresectable pancreatic carcinomas. J. Gastroenterol. 2007, 42, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ku, J.H.; Kwak, C.; Kim, H.H.; Lee, E.; Keam, B.; Kim, T.M.; Heo, D.S.; Lee, S.H.; Moon, K.C. Predictive and Prognostic Value of Ribonucleotide Reductase Regulatory Subunit M1 and Excision Repair Cross-Complementation Group 1 in Advanced Urothelial Carcinoma (UC) Treated with First-Line Gemcitabine Plus Platinum Combination Chemotherapy. PLoS ONE 2015, 10, e0133371. [Google Scholar] [CrossRef] [PubMed]
- Bepler, G.; Kusmartseva, I.; Sharma, S.; Gautam, A.; Cantor, A.; Sharma, A.; Simon, G. RRM1 modulated in vitro and in vivo efficacy of gemcitabine and platinum in non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 4731–4737. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Danenberg, K.D.; Alberola, V.; Bepler, G.; Sanchez, J.J.; Camps, C.; Provencio, M.; Isla, D.; Taron, M.; Diz, P.; et al. Ribonucleotide reductase messenger RNA expression and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin. Cancer Res. 2004, 10, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Kohya, N.; Kai, K.; Ohtaka, K.; Hashiguchi, K.; Hiraki, M.; Kitajima, Y.; Tokunaga, O.; Noshiro, H.; Miyazaki, K. Ribonucleotide reductase subunit M1 assessed by quantitative double-fluorescence immunohistochemistry predicts the efficacy of gemcitabine in biliary tract carcinoma. Int. J. Oncol. 2010, 37, 845–852. [Google Scholar] [PubMed]
- Ferrandina, G.; Mey, V.; Nannizzi, S.; Ricciardi, S.; Petrillo, M.; Ferlini, C.; Danesi, R.; Scambia, G.; Del Tacca, M. Expression of nucleoside transporters, deoxycitidine kinase, ribonucleotide reductase regulatory subunits, and gemcitabine catabolic enzymes in primary ovarian cancer. Cancer Chemother. Pharmacol. 2010, 65, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Boukovinas, I.; Papadaki, C.; Mendez, P.; Taron, M.; Mavroudis, D.; Koutsopoulos, A.; Sanchez-Ronco, M.; Sanchez, J.J.; Trypaki, M.; Staphopoulos, E.; et al. Tumor BRCA1, RRM1 and RRM2 mRNA expression levels and clinical response to first-line gemcitabine plus docetaxel in non-small-cell lung cancer patients. PLoS ONE 2008, 3, e3695. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.O.; Jeong, J.Y.; Kim, M.R.; Cho, H.J.; Ju, J.Y.; Kwon, Y.S.; Oh, I.J.; Kim, K.S.; Kim, Y.I.; Lim, S.C.; et al. Efficacy of gemcitabine in patients with non-small cell lung cancer according to promoter polymorphisms of the ribonucleotide reductase M1 gene. Clin. Cancer Res. 2008, 14, 3083–3088. [Google Scholar] [CrossRef] [PubMed]
- Gotanda, K.; Hirota, T.; Matsumoto, N.; Ieiri, I. MicroRNA-433 negatively regulates the expression of thymidylate synthase (TYMS) responsible for 5-fluorouracil sensitivity in HeLa cells. BMC Cancer 2013, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Rudd, S.G.; Ljungblad, L.; Sanjiv, K.; Myrberg, I.H.; Paulin, C.B.; Heshmati, Y.; Hagenkort, A.; Kutzner, J.; Page, B.D.; et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat. Med. 2017, 23, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Rudd, S.G.; Sanjiv, K.; Kutzner, J.; Myrberg, I.H.; Paulin, C.B.J.; Olsen, T.K.; Helleday, T.; Henter, J.I.; Schaller, T. With me or against me: Tumor suppressor and drug resistance activities of SAMHD1. Exp. Hematol. 2017, 52, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, S.A.; Matheson, E.C.; Hall, A.G.; Hogarth, L.A. The clinical impact of thiopurine methyltransferase polymorphisms on thiopurine treatment. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Blaker, P.A.; Arenas-Hernandez, M.; Smith, M.A.; Shobowale-Bakre, E.A.; Fairbanks, L.; Irving, P.M.; Sanderson, J.D.; Marinaki, A.M. Mechanism of allopurinol induced TPMT inhibition. Biochem. Pharmacol. 2013, 86, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Pan, L.; Yang, X.; Liu, Z.; Huang, L.; Wen, C.; Lin, A.; Liu, H. Thymidine phosphorylase expression and prognosis in colorectal cancer treated with 5-fluorouracil-based chemotherapy: A meta-analysis. Mol. Clin. Oncol. 2017, 7, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Rosen, L.S.; Yoshida, K.; Rasco, D.; Shapiro, G.I.; Sun, W. A phase 1 study of the pharmacokinetics of nucleoside analog trifluridine and thymidine phosphorylase inhibitor tipiracil (components of TAS-102) vs trifluridine alone. Investig. New Drugs 2017, 35, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Lemery, S.J.; Khasar, S.; Wearne, E.; Helms, W.S.; Yuan, W.; He, K.; Cao, X.; Yu, J.; Zhao, H.; et al. FDA Approval Summary: TAS-102. Clin. Cancer Res. 2017, 23, 2924–2927. [Google Scholar] [CrossRef] [PubMed]
- Nadella, P.; Shapiro, C.; Otterson, G.A.; Hauger, M.; Erdal, S.; Kraut, E.; Clinton, S.; Shah, M.; Stanek, M.; Monk, P.; et al. Pharmacobiologically based scheduling of capecitabine and docetaxel results in antitumor activity in resistant human malignancies. J. Clin. Oncol. 2002, 20, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Nakagawa, F.; Kobunai, T.; Takechi, T. Trifluridine/tipiracil overcomes the resistance of human gastric 5-fluorouracil-refractory cells with high thymidylate synthase expression. Oncotarget 2018, 9, 13438–13450. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Kazuno, H.; Emura, T.; Tsujimoto, H.; Suzuki, N.; Fukushima, M. Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int. J. Oncol. 2000, 17, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.Y.; Lee, J.S.; Min, H.Y.; Lee, H.Y. Acquired resistance to 5-fluorouracil via HSP90/Src-mediated increase in thymidylate synthase expression in colon cancer. Oncotarget 2015, 6, 32622–32633. [Google Scholar] [CrossRef] [PubMed]
- Fazzone, W.; Wilson, P.M.; Labonte, M.J.; Lenz, H.J.; Ladner, R.D. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int. J. Cancer 2009, 125, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gao, F.; Zhang, X.P. miR-203 enhances chemosensitivity to 5-fluorouracil by targeting thymidylate synthase in colorectal cancer. Oncol. Rep. 2015, 33, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Calascibetta, A.; Martorana, A.; Cabibi, D.; Aragona, F.; Sanguedolce, R. Relationship between thymidylate synthase and p53 and response to FEC versus taxane adjuvant chemotherapy for breast carcinoma. J. Chemother. 2011, 23, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.A.; Kim, M.; Husain, K.; Kang, T. Regulation of purine nucleotide synthesis in human B lymphoblasts with both hypoxanthine-guanine phosphoribosyltransferase deficiency and phosphoribosylpyrophosphate synthetase superactivity. J. Biol. Chem. 1992, 267, 4317–4321. [Google Scholar] [PubMed]
- Humeniuk, R.; Menon, L.G.; Mishra, P.J.; Gorlick, R.; Sowers, R.; Rode, W.; Pizzorno, G.; Cheng, Y.C.; Kemeny, N.; Bertino, J.R.; et al. Decreased levels of UMP kinase as a mechanism of fluoropyrimidine resistance. Mol. Cancer Ther. 2009, 8, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.I.; Kim, K.K.; Choi, H.; Kim, S.; Jang, K.T.; Yi, J.H.; Park, Y.S.; Park, J.O.; Lee, S.Y. Effect of genetic polymorphisms on therapeutic response and clinical outcomes in pancreatic cancer patients treated with gemcitabine. Pharmacogenomics 2012, 13, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.S.; Shin, E.S.; Nam, H.S.; Yi, H.G.; Cho, J.H.; Kim, C.S.; Kim, H.J.; Lee, J.E. Differential effect of polymorphisms of CMPK1 and RRM1 on survival in advanced non-small cell lung cancer patients treated with gemcitabine or taxane/cisplatinum. J. Thorac. Oncol. 2011, 6, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Tsutani, Y.; Yoshida, K.; Sanada, Y.; Wada, Y.; Konishi, K.; Fukushima, M.; Okada, M. Decreased orotate phosphoribosyltransferase activity produces 5-fluorouracil resistance in a human gastric cancer cell line. Oncol. Rep. 2008, 20, 1545–1551. [Google Scholar] [PubMed]
- Im, Y.S.; Shin, H.K.; Kim, H.R.; Jeong, S.H.; Kim, S.R.; Kim, Y.M.; Lee, D.H.; Jeon, S.H.; Lee, H.W.; Choi, J.K. Enhanced cytotoxicity of 5-FU by bFGF through up-regulation of uridine phosphorylase 1. Mol. Cells 2009, 28, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Renck, D.; Machado, P.; Souto, A.A.; Rosado, L.A.; Erig, T.; Campos, M.M.; Farias, C.B.; Roesler, R.; Timmers, L.F.; de Souza, O.N.; et al. Design of novel potent inhibitors of human uridine phosphorylase-1: Synthesis, inhibition studies, thermodynamics, and in vitro influence on 5-fluorouracil cytotoxicity. J. Med. Chem. 2013, 56, 8892–8902. [Google Scholar] [CrossRef] [PubMed]
- Cividini, F.; Pesi, R.; Chaloin, L.; Allegrini, S.; Camici, M.; Cros-Perrial, E.; Dumontet, C.; Jordheim, L.P.; Tozzi, M.G. The purine analog fludarabine acts as a cytosolic 5′-nucleotidase II inhibitor. Biochem. Pharmacol. 2015, 94, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.; Perrino, F.W. The effects of cytosine arabinoside on RNA-primed DNA synthesis by DNA polymerase alpha-primase. J. Biol. Chem. 1995, 270, 26664–26669. [Google Scholar] [CrossRef] [PubMed]
- Mikita, T.; Beardsley, G.P. Functional consequences of the arabinosylcytosine structural lesion in DNA. Biochemistry 1988, 27, 4698–4705. [Google Scholar] [CrossRef] [PubMed]
- Perrino, F.W.; Mekosh, H.L. Incorporation of cytosine arabinoside monophosphate into DNA at internucleotide linkages by human DNA polymerase alpha. J. Biol. Chem. 1992, 267, 23043–23051. [Google Scholar] [PubMed]
- Tsuda, M.; Terada, K.; Ooka, M.; Kobayashi, K.; Sasanuma, H.; Fujisawa, R.; Tsurimoto, T.; Yamamoto, J.; Iwai, S.; Kadoda, K.; et al. The dominant role of proofreading exonuclease activity of replicative polymerase epsilon in cellular tolerance to cytarabine (Ara-C). Oncotarget 2017, 8, 33457–33474. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Loegering, D.; Powell, H.L.; Flatten, K.; Arlander, S.J.; Dai, N.T.; Heldebrant, M.P.; Vroman, B.T.; Smith, B.D.; Karp, J.E.; et al. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood 2005, 106, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Dijkwel, P.A.; Wanka, F. Enhanced release of nascent single strands from DNA synthesized in the presence of arabinosylcytosine. Biochim. Biophys. Acta 1978, 520, 461–471. [Google Scholar] [CrossRef]
- Graham, F.L.; Whitmore, G.F. Studies in mouse L-cells on the incorporation of 1-beta-D-arabinofuranosylcytosine into DNA and on inhibition of DNA polymerase by 1-beta-D-arabinofuranosylcytosine 5′-triphosphate. Cancer Res. 1970, 30, 2636–2644. [Google Scholar] [PubMed]
- Ross, D.D.; Chen, S.R.; Cuddy, D.P. Effects of 1-beta-D-arabinofuranosylcytosine on DNA replication intermediates monitored by pH-step alkaline elution. Cancer Res. 1990, 50, 2658–2666. [Google Scholar] [PubMed]
- Ross, D.D.; Cuddy, D.P.; Cohen, N.; Hensley, D.R. Mechanistic implications of alterations in HL-60 cell nascent DNA after exposure to 1-beta-D-arabinofuranosylcytosine. Cancer Chemother. Pharmacol. 1992, 31, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chubb, S.; Plunkett, W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J. Biol. Chem. 1990, 265, 16617–16625. [Google Scholar] [PubMed]
- Xie, C.; Plunkett, W. Metabolism and actions of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-adenine in human lymphoblastoid cells. Cancer Res. 1995, 55, 2847–2852. [Google Scholar] [PubMed]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar] [PubMed]
- Plunkett, W.; Huang, P.; Gandhi, V. Preclinical characteristics of gemcitabine. Anti-Cancer Drugs 1995, 6 (Suppl. S6), 7–13. [Google Scholar] [CrossRef] [PubMed]
- Azuma, A.; Hanaoka, K.; Kurihara, A.; Kobayashi, T.; Miyauchi, S.; Kamo, N.; Tanaka, M.; Sasaki, T.; Matsuda, A. Nucleosides and nucleotides. 141. Chemical stability of a new antitumor nucleoside, 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine in alkaline medium: Formation of 2′-C-cyano-2′-deoxy-1-beta-D-ribo-pentofuranosylcytosine and its antitumor activity. J. Med. Chem. 1995, 38, 3391–3397. [Google Scholar] [PubMed]
- Azuma, A.; Huang, P.; Matsuda, A.; Plunkett, W. 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine: A novel anticancer nucleoside analog that causes both DNA strand breaks and G(2) arrest. Mol. Pharmacol. 2001, 59, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Cuffari, C.; Li, D.Y.; Mahoney, J.; Barnes, Y.; Bayless, T.M. Peripheral blood mononuclear cell DNA 6-thioguanine metabolite levels correlate with decreased interferon-gamma production in patients with Crohn’s disease on AZA therapy. Dig. Dis. Sci. 2004, 49, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Nelson, J.A.; Cheng, Y.C.; Anderson, R.S.; Beattie, K.L. 2′-Deoxy-6-thioguanosine 5′-triphosphate as a substrate for purified human DNA polymerases and calf thymus terminal deoxynucleotidyltransferase in vitro. Mol. Pharmacol. 1991, 40, 508–514. [Google Scholar] [PubMed]
- Nelson, J.A.; Carpenter, J.W.; Rose, L.M.; Adamson, D.J. Mechanisms of action of 6-thioguanine, 6-mercaptopurine, and 8-azaguanine. Cancer Res. 1975, 35, 2872–2878. [Google Scholar] [PubMed]
- Warren, D.J.; Andersen, A.; Slordal, L. Quantitation of 6-thioguanine residues in peripheral blood leukocyte DNA obtained from patients receiving 6-mercaptopurine-based maintenance therapy. Cancer Res. 1995, 55, 1670–1674. [Google Scholar] [PubMed]
- Karran, P.; Attard, N. Thiopurines in current medical practice: Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Yamada, M.; Masaki, S.; Saneyoshi, M. Utilization of 2′-deoxy-6-thioguanosine 5′-triphosphate in DNA synthesis in vitro by DNA polymerase alpha from calf thymus. Cancer Res. 1979, 39, 3955–3958. [Google Scholar] [PubMed]
- Aquilina, G.; Giammarioli, A.M.; Zijno, A.; Di Muccio, A.; Dogliotti, E.; Bignami, M. Tolerance to O6-methylguanine and 6-thioguanine cytotoxic effects: A cross-resistant phenotype in N-methylnitrosourea-resistant Chinese hamster ovary cells. Cancer Res. 1990, 50, 4248–4253. [Google Scholar] [PubMed]
- Swann, P.F.; Waters, T.R.; Moulton, D.C.; Xu, Y.Z.; Zheng, Q.; Edwards, M.; Mace, R. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 1996, 273, 1109–1111. [Google Scholar] [CrossRef] [PubMed]
- Karran, P.; Bignami, M. DNA damage tolerance, mismatch repair and genome instability. BioEssays 1994, 16, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Stojic, L.; Mojas, N.; Cejka, P.; Di Pietro, M.; Ferrari, S.; Marra, G.; Jiricny, J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004, 18, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Berry, S.E.; Desai, A.B.; Kinsella, T.J. DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin. Cancer Res. 2003, 9, 2327–2334. [Google Scholar] [PubMed]
- Vernole, P.; Pepponi, R.; D’Atri, S. Role of mismatch repair in the induction of chromosomal aberrations and sister chromatid exchanges in cells treated with different chemotherapeutic agents. Cancer Chemother. Pharmacol. 2003, 52, 185–192. [Google Scholar] [PubMed]
- Matheson, E.C.; Hall, A.G. Assessment of mismatch repair function in leukaemic cell lines and blasts from children with acute lymphoblastic leukaemia. Carcinogenesis 2003, 24, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Tsukada, J.; Kominato, Y.; Tanaka, Y. Reduced expression of human mismatch repair genes in adult T-cell leukemia. Am. J. Hematol. 2005, 78, 100–107. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Dai, X.; Yuan, B.; Wang, Y. Effects of 6-thioguanine and S6-methylthioguanine on transcription in vitro and in human cells. J. Biol. Chem. 2012, 287, 40915–40923. [Google Scholar] [CrossRef] [PubMed]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R.; et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Investig. 2003, 111, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.; Momparler, R.L. Incorporation of 5-Aza-2′-deoxycytidine-5′-triphosphate into DNA. Interactions with mammalian DNA polymerase alpha and DNA methylase. Mol. Pharmacol. 1983, 24, 109–114. [Google Scholar] [PubMed]
- Chen, L.; MacMillan, A.M.; Chang, W.; Ezaz-Nikpay, K.; Lane, W.S.; Verdine, G.L. Direct identification of the active-site nucleophile in a DNA (cytosine-5)-methyltransferase. Biochemistry 1991, 30, 11018–11025. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.V.; Norment, A.; Garrett, C.E. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. USA 1984, 81, 6993–6997. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Frankhouser, D.; Murphy, M.; Tam, H.H.; Rodriguez, B.; Curfman, J.; Trimarchi, M.; Geyer, S.; Wu, Y.Z.; Whitman, S.P.; et al. Genome-wide methylation profiling in decitabine-treated patients with acute myeloid leukemia. Blood 2012, 120, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Orta, M.L.; Calderon-Montano, J.M.; Dominguez, I.; Pastor, N.; Burgos-Moron, E.; Lopez-Lazaro, M.; Cortes, F.; Mateos, S.; Helleday, T. 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 2013, 41, 5827–5836. [Google Scholar] [CrossRef] [PubMed]
- Palii, S.S.; Van Emburgh, B.O.; Sankpal, U.T.; Brown, K.D.; Robertson, K.D. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 2008, 28, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol. Pharmacol. 1990, 38, 567–572. [Google Scholar] [PubMed]
- Wang, J.; Lohman, G.J.; Stubbe, J. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. USA 2007, 104, 14324–14329. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Brignole, E.J.; Long, M.J.; Chittuluru, J.; Drennan, C.L.; Asturias, F.J.; Stubbe, J. Clofarabine targets the large subunit (alpha) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem. Biol. 2012, 19, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Stubbe, J. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. USA 2011, 108, 9815–9820. [Google Scholar] [CrossRef] [PubMed]
- Wisitpitthaya, S.; Zhao, Y.; Long, M.J.; Li, M.; Fletcher, E.A.; Blessing, W.A.; Weiss, R.S.; Aye, Y. Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition. ACS Chem. Biol. 2016, 11, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Tay, B.S.; Lilley, R.M.; Murray, A.W.; Atkinson, M.R. Inhibition of phosphoribosyl pyrophosphate amidotransferase from Ehrlich ascites-tumour cells by thiopurine nucleotides. Biochem. Pharmacol. 1969, 18, 936–938. [Google Scholar] [CrossRef]
- Tidd, D.M.; Paterson, A.R. Distinction between inhibition of purine nucleotide synthesis and the delayed cytotoxic reaction of 6-mercaptopurine. Cancer Res. 1974, 34, 733–737. [Google Scholar] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.J.; Ladner, R.D. Standing the test of time: Targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Costi, M.P.; Ferrari, S.; Venturelli, A.; Calo, S.; Tondi, D.; Barlocco, D. Thymidylate synthase structure, function and implication in drug discovery. Curr. Med. Chem. 2005, 12, 2241–2258. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.V.; McHenry, C.S.; Sommer, H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 1974, 13, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Sommer, H.; Santi, D.V. Purification and amino acid analysis of an active site peptide from thymidylate synthetase containing covalently bound 5-fluoro-2′-deoxyuridylate and methylenetetrahydrofolate. Biochem. Biophys. Res. Commun. 1974, 57, 689–695. [Google Scholar] [CrossRef]
- Morgan, R.G. Leucovorin enhancement of the effects of the fluoropyrimidines on thymidylate synthase. Cancer 1989, 63, 1008–1012. [Google Scholar] [CrossRef]
- Temmink, O.H.; Comijn, E.M.; Fukushima, M.; Peters, G.J. Intracellular thymidylate synthase inhibition by trifluorothymidine in FM3A cells. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.S. On the nature of thymineless death. Ann. N. Y. Acad. Sci. 1971, 186, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Goulian, M.; Bleile, B.M.; Dickey, L.M.; Grafstrom, R.H.; Ingraham, H.A.; Neynaber, S.A.; Peterson, M.S.; Tseng, B.Y. Mechanism of thymineless death. Adv. Exp. Med. Biol. 1986, 195, 89–95. [Google Scholar] [PubMed]
- Houghton, J.A.; Tillman, D.M.; Harwood, F.G. Ratio of 2′-deoxyadenosine-5′-triphosphate/thymidine-5′-triphosphate influences the commitment of human colon carcinoma cells to thymineless death. Clin. Cancer Res. 1995, 1, 723–730. [Google Scholar] [PubMed]
- Caradonna, S.J.; Cheng, Y.C. The role of deoxyuridine triphosphate nucleotidohydrolase, uracil-DNA glycosylase, and DNA polymerase alpha in the metabolism of FUdR in human tumor cells. Mol. Pharmacol. 1980, 18, 513–520. [Google Scholar] [PubMed]
- Suzuki, N.; Emura, T.; Fukushima, M. Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int. J. Oncol. 2011, 39, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yoshida, S.; Saneyoshi, M.; Yamaguchi, T. Utilization of 5-fluoro-2′-deoxyuridine triphosphate and 5-fluoro-2′-deoxycytidine triphosphate in DNA synthesis by DNA polymerases alpha and beta from calf thymus. Cancer Res. 1981, 41, 4132–4135. [Google Scholar] [PubMed]
- Yoshida, S.; Masaki, S. Utilization in vitro of deoxyuridine triphosphate in DNA synthesis by DNA polymerases alpha and beta from calf thymus. Biochim. Biophys. Acta 1979, 561, 396–402. [Google Scholar] [CrossRef]
- Hagen, L.; Pena-Diaz, J.; Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Genomic uracil and human disease. Exp. Cell. Res. 2006, 312, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, M.D.; Wilson, D.M., 3rd. Participation of DNA repair in the response to 5-fluorouracil. Cell. Mol. Life Sci. 2009, 66, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Baerenfaller, K.; Jiricny, J. 5-Fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology 2007, 133, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Iwaizumi, M.; Yamada, H.; Sugiyama, T.; Hamaya, Y.; Furuta, T.; Kanaoka, S.; Sugimura, H.; Miyajima, H.; Osawa, S.; et al. MBD4 frameshift mutation caused by DNA mismatch repair deficiency enhances cytotoxicity by trifluridine, an active antitumor agent of TAS-102, in colorectal cancer cells. Oncotarget 2018, 9, 11477–11488. [Google Scholar] [CrossRef] [PubMed]
- Sjolund, A.B.; Senejani, A.G.; Sweasy, J.B. MBD4 and TDG: Multifaceted DNA glycosylases with ever expanding biological roles. Mutat. Res. 2013, 743–744, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Drablos, F.; Slupphaug, G. Uracil in DNA—Occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [PubMed]
- Doong, S.L.; Dolnick, B.J. 5-Fluorouracil substitution alters pre-mRNA splicing in vitro. J. Biol. Chem. 1988, 263, 4467–4473. [Google Scholar] [PubMed]
- Glazer, R.I.; Lloyd, L.S. Association of cell lethality with incorporation of 5-fluorouracil and 5-fluorouridine into nuclear RNA in human colon carcinoma cells in culture. Mol. Pharmacol. 1982, 21, 468–473. [Google Scholar] [PubMed]
- Geoffroy, F.J.; Allegra, C.J.; Sinha, B.; Grem, J.L. Enhanced cytotoxicity with interleukin-1 alpha and 5-fluorouracil in HCT116 colon cancer cells. Oncol. Res. 1994, 6, 581–591. [Google Scholar] [PubMed]
- Pettersen, H.S.; Visnes, T.; Vagbo, C.B.; Svaasand, E.K.; Doseth, B.; Slupphaug, G.; Kavli, B.; Krokan, H.E. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011, 39, 8430–8444. [Google Scholar] [CrossRef] [PubMed]
- Ison, G.; Beaver, J.A.; McGuinn, W.D., Jr.; Palmby, T.R.; Dinin, J.; Charlab, R.; Marathe, A.; Jin, R.; Liu, Q.; Chen, X.H.; et al. FDA Approval: Uridine Triacetate for the Treatment of Patients Following Fluorouracil or Capecitabine Overdose or Exhibiting Early-Onset Severe Toxicities Following Administration of These Drugs. Clin. Cancer Res. 2016, 22, 4545–4549. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Johnson, L.A.; Jacobson, P.A.; Kachler, S.; Kirstein, M.N.; Lamba, J.; Klotz, K.N. Cytotoxic purine nucleoside analogues bind to A1, A2A, and A3 adenosine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Komi, Y.; Ohno, O.; Suzuki, Y.; Shimamura, M.; Shimokado, K.; Umezawa, K.; Kojima, S. Inhibition of tumor angiogenesis by targeting endothelial surface ATP synthase with sangivamycin. Jpn. J. Clin. Oncol. 2007, 37, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Priebe, T.; Kandil, O.; Nakic, M.; Pan, B.F.; Nelson, J.A. Selective modulation of antibody response and natural killer cell activity by purine nucleoside analogues. Cancer Res. 1988, 48, 4799–4803. [Google Scholar] [PubMed]
- Priebe, T.; Platsoucas, C.D.; Nelson, J.A. Adenosine receptors and modulation of natural killer cell activity by purine nucleosides. Cancer Res. 1990, 50, 4328–4331. [Google Scholar] [PubMed]
- Maaser, K.; Hopfner, M.; Kap, H.; Sutter, A.P.; Barthel, B.; von Lampe, B.; Zeitz, M.; Scherubl, H. Extracellular nucleotides inhibit growth of human oesophageal cancer cells via P2Y(2)-receptors. Br. J. Cancer 2002, 86, 636–644. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Balzarini, J.; Madej, D.; Hansske, F.; Robins, M.J. Nucleic acid related compounds. 51. Synthesis and biological properties of sugar-modified analogues of the nucleoside antibiotics tubercidin, toyocamycin, sangivamycin, and formycin. J. Med. Chem. 1987, 30, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.B.; Glazer, R.I. Cytotoxicity and the inhibition of ribosomal RNA processing in human colon carcinoma cells. Mol. Pharmacol. 1985, 27, 308–313. [Google Scholar] [PubMed]
- Finch, R.A.; Revankar, G.R.; Chan, P.K. Structural and functional relationships of toyocamycin on NPM-translocation. Anti-Cancer Drug Des. 1997, 12, 205–215. [Google Scholar]
- Mooberry, S.L.; Stratman, K.; Moore, R.E. Tubercidin stabilizes microtubules against vinblastine-induced depolymerization, a taxol-like effect. Cancer Lett. 1995, 96, 261–266. [Google Scholar] [CrossRef]
- Awrich, A.; Fletcher, W.S.; Klotz, J.H.; Minton, J.P.; Hill, G.J., 2nd; Aust, J.B.; Grage, T.B.; Multhauf, P.M. 5-FU versus combination therapy with tubercidin, streptozotocin, and 5-FU in the treatment of pancreatic carcinomas: COG protocol 7230. J. Surg. Oncol. 1979, 12, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Cavins, J.A.; Hall, T.C.; Olson, K.B.; Khung, C.L.; Horton, J.; Colsky, J.; Shadduck, R.K. Initial toxicity study of sangivamycin (NSC-65346). Cancer Chemother. Rep. 1967, 51, 197–200. [Google Scholar] [PubMed]
- Wilson, W.L. Phase I study with toyocamycin (NSC-63701). Cancer Chemother. Rep. 1968, 52, 301–303. [Google Scholar] [PubMed]
- Loomis, C.R.; Bell, R.M. Sangivamycin, a nucleoside analogue, is a potent inhibitor of protein kinase C. J. Biol. Chem. 1988, 263, 1682–1692. [Google Scholar] [PubMed]
- Burgess, G.H.; Bloch, A.; Stoll, H.; Milgrom, H.; Helm, F.; Klein, E. Effect of topical tubercidin on basal cell carcinomas and actinic keratoses. Cancer 1974, 34, 250–253. [Google Scholar] [CrossRef]
- Kraybill, W.G., Jr.; Anderson, D.D.; Lindell, T.D.; Fletcher, W.S. Islet cell carcinoma of the pancreas: Effective therapy with 5-fluorouracil, streptozotocin, and tubercidin. Am. Surg. 1976, 42, 467–470. [Google Scholar] [PubMed]
- Gupta, R.S.; Mehta, K.D. Genetic and biochemical studies on mutants of CHO cells resistant to 7-deazapurine nucleosides: Differences in the mechanisms of action of toyocamycin and tubercidin. Biochem. Biophys. Res. Commun. 1984, 120, 88–95. [Google Scholar] [CrossRef]
- Johnston, J.B. Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leuk. Lymphoma 2011, 52 (Suppl. S2), 43–45. [Google Scholar] [CrossRef] [PubMed]
- Bosanquet, A.G.; Richards, S.M.; Wade, R.; Else, M.; Matutes, E.; Dyer, M.J.; Rassam, S.M.; Durant, J.; Scadding, S.M.; Raper, S.L.; et al. Drug cross-resistance and therapy-induced resistance in chronic lymphocytic leukaemia by an enhanced method of individualised tumour response testing. Br. J. Haematol. 2009, 146, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Homminga, I.; Zwaan, C.M.; Manz, C.Y.; Parker, C.; Bantia, S.; Smits, W.K.; Higginbotham, F.; Pieters, R.; Meijerink, J.P. In vitro efficacy of forodesine and nelarabine (ara-G) in pediatric leukemia. Blood 2011, 118, 2184–2190. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Uzui, K.; Nishi, R.; Tasaki, T.; Ueda, T. A nelarabine-resistant T-lymphoblastic leukemia CCRF-CEM variant cell line is cross-resistant to the purine nucleoside phosphorylase inhibitor forodesine. Anticancer Res. 2014, 34, 4885–4892. [Google Scholar] [PubMed]
- Balis, F.M.; Holcenberg, J.S.; Bleyer, W.A. Clinical pharmacokinetics of commonly used anticancer drugs. Clin. Pharmacokinet. 1983, 8, 202–232. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L. Therapeutic drug monitoring of antimetabolic cytotoxic drugs. Br. J. Clin. Pharmacol. 1999, 47, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Budman, D.R. Capecitabine. Investig. New Drugs 2000, 18, 355–363. [Google Scholar] [CrossRef]
- Braess, J.; Freund, M.; Hanauske, A.; Heil, G.; Kaufmann, C.; Kern, W.; Schussler, M.; Hiddemann, W.; Schleyer, E. Oral cytarabine ocfosfate in acute myeloid leukemia and non-Hodgkin’s lymphoma—Phase I/II studies and pharmacokinetics. Leukemia 1998, 12, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Koolen, S.L.; Witteveen, P.O.; Jansen, R.S.; Langenberg, M.H.; Kronemeijer, R.H.; Nol, A.; Garcia-Ribas, I.; Callies, S.; Benhadji, K.A.; Slapak, C.A.; et al. Phase I study of Oral gemcitabine prodrug (LY2334737) alone and in combination with erlotinib in patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kantarjian, H.; Plunkett, W. Sapacitabine for cancer. Expert Opin. Investig. Drugs 2012, 21, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Plunkett, W.; Rodriguez, C.O., Jr.; Nowak, B.J.; Du, M.; Ayres, M.; Kisor, D.F.; Mitchell, B.S.; Kurtzberg, J.; Keating, M.J. Compound GW506U78 in refractory hematologic malignancies: Relationship between cellular pharmacokinetics and clinical response. J. Clin. Oncol. 1998, 16, 3607–3615. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.G.; Leyland-Jones, B.; Cheson, B.D. Fludarabine phosphate: A synthetic purine antimetabolite with significant activity against lymphoid malignancies. J. Clin. Oncol. 1991, 9, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Nagura, E.; Yamada, K.; Hamajima, N. Leukemia relapse in long-term survivors of acute leukemia. Cancer 1985, 56, 88–94. [Google Scholar] [CrossRef]
- Riccardi, R.; Vigersky, R.A.; Barnes, S.; Bleyer, W.A.; Poplack, D.G. Methotrexate levels in the interstitial space and seminiferous tubule of rat testis. Cancer Res. 1982, 42, 1617–1619. [Google Scholar] [PubMed]
- Kalaria, R.N.; Harik, S.I. Nucleoside transporter of cerebral microvessels and choroid plexus. J. Neurochem. 1986, 47, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Varatharajan, L.; Thomas, S.A. The transport of anti-HIV drugs across blood-CNS interfaces: Summary of current knowledge and recommendations for further research. Antivir. Res. 2009, 82, A99–A109. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Tsuji, A. Transporter-mediated permeation of drugs across the blood-brain barrier. J. Pharm. Sci. 2000, 89, 1371–1388. [Google Scholar] [CrossRef]
- Saidijam, M.; Karimi Dermani, F.; Sohrabi, S.; Patching, S.G. Efflux proteins at the blood-brain barrier: Review and bioinformatics analysis. Xenobiotica 2018, 48, 506–532. [Google Scholar] [CrossRef] [PubMed]
- Lindemalm, S.; Liliemark, J.; Larsson, B.S.; Albertioni, F. Distribution of 2-chloro-2′-deoxyadenosine, 2-chloro-2′-arabino-fluoro-2′-deoxyadenosine, fludarabine and cytarabine in mice: A whole-body autoradiography study. Med. Oncol. 1999, 16, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Yoshikawa, T.; Kang, Y.S.; Miller, L.P. Blood-brain barrier transport and brain metabolism of adenosine and adenosine analogs. J. Pharmacol. Exp. Ther. 1994, 268, 14–18. [Google Scholar] [PubMed]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Hustu, H.O.; Aur, R.J.; Verzosa, M.S.; Simone, J.V.; Pinkel, D. Prevention of central nervous system leukemia by irradiation. Cancer 1973, 32, 585–597. [Google Scholar] [CrossRef]
- Wahidijat, I.; Markum, A.H.; Abdulsalam, M.; Muslichan, S.; Odang, O. Prophylactic treatment of meningeal leukemia in children by intrathecal methotrexate. Acta Haematol. 1972, 47, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Pratt, C.B. Intrathecal arabinosyl cytosine in meningeal leukemia. Cancer 1970, 25, 531–534. [Google Scholar] [CrossRef]
- Abromowitch, M.; Ochs, J.; Pui, C.H.; Kalwinsky, D.; Rivera, G.K.; Fairclough, D.; Look, A.T.; Hustu, H.O.; Murphy, S.B.; Evans, W.E.; et al. High-dose methotrexate improves clinical outcome in children with acute lymphoblastic leukemia: St. Jude Total Therapy Study X. Med. Pediatr. Oncol. 1988, 16, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Morra, E.; Lazzarino, M.; Inverardi, D.; Brusamolino, E.; Orlandi, E.; Canevari, A.; Pagnucco, G.; Bernasconi, C. Systemic high-dose ara-C for the treatment of meningeal leukemia in adult acute lymphoblastic leukemia and non-Hodgkin’s lymphoma. J. Clin. Oncol. 1986, 4, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Felix, A.; Leblanc, T.; Petit, A.; Nelkem, B.; Bertrand, Y.; Gandemer, V.; Sirvent, A.; Paillard, C.; Schmitt, C.; Rohrlich, P.S.; et al. Acute Myeloid Leukemia With Central Nervous System Involvement in Children: Experience From the French Protocol Analysis ELAM02. J. Pediatr. Hematol. Oncol. 2018, 40, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.H.; Frei, E., 3rd. Clinical pharmacology of 1-beta-d-arabinofuranosyl cytosine. Clin. Pharmacol. Ther. 1971, 12, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Robak, P.; Robak, T. Older and new purine nucleoside analogs for patients with acute leukemias. Cancer Treat. Rev. 2013, 39, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Bouffard, D.Y.; Laliberte, J.; Momparler, R.L. Kinetic studies on 2′,2′-difluorodeoxycytidine (Gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem. Pharmacol. 1993, 45, 1857–1861. [Google Scholar] [CrossRef]
- Mahfouz, R.Z.; Jankowska, A.; Ebrahem, Q.; Gu, X.; Visconte, V.; Tabarroki, A.; Terse, P.; Covey, J.; Chan, K.; Ling, Y.; et al. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin. Cancer Res. 2013, 19, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Serdjebi, C.; Seitz, J.F.; Ciccolini, J.; Duluc, M.; Norguet, E.; Fina, F.; Lacarelle, B.; Ouafik, L.; Dahan, L. Rapid deaminator status is associated with poor clinical outcome in pancreatic cancer patients treated with a gemcitabine-based regimen. Pharmacogenomics 2013, 14, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Varatharajan, S.; Abbas, S.; Zhang, W.; Shaji, R.V.; Ahmed, R.; Abraham, A.; George, B.; Srivastava, A.; Chandy, M.; et al. Cytidine deaminase genetic variants influence RNA expression and cytarabine cytotoxicity in acute myeloid leukemia. Pharmacogenomics 2012, 13, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Serdjebi, C.; Ciccolini, J.; Fina, F.; Delarue, A.; Verschuur, A.; Lacarelle, B.; Ouafik, L.; Andre, N. Recipient/donor contradictory genotypes with impact on drug pharmacogenetics after liver transplant: A deadly gift? Pharmacogenet. Genom. 2014, 24, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Fanciullino, R.; Farnault, L.; Donnette, M.; Imbs, D.C.; Roche, C.; Venton, G.; Berda-Haddad, Y.; Ivanov, V.; Ciccolini, J.; Ouafik, L.; et al. CDA as a predictive marker for life-threatening toxicities in patients with AML treated with cytarabine. Blood Adv. 2018, 2, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, W.; Liliemark, J.O.; Adams, T.M.; Nowak, B.; Estey, E.; Kantarjian, H.; Keating, M.J. Saturation of 1-beta-D-arabinofuranosylcytosine 5′-triphosphate accumulation in leukemia cells during high-dose 1-beta-D-arabinofuranosylcytosine therapy. Cancer Res. 1987, 47, 3005–3011. [Google Scholar] [PubMed]
- Veltkamp, S.A.; Beijnen, J.H.; Schellens, J.H. Prolonged versus standard gemcitabine infusion: Translation of molecular pharmacology to new treatment strategy. Oncologist 2008, 13, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.M.; Quebbeman, E.; Anderson, T. 5-Fluorouracil by protracted venous infusion. A review of current progress. Oncology 1989, 46, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zimm, S.; Collins, J.M.; Miser, J.; Chatterji, D.; Poplack, D.G. Cytosine arabinoside cerebrospinal fluid kinetics. Clin. Pharmacol. Ther. 1984, 35, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Bomgaars, L.; Geyer, J.R.; Franklin, J.; Dahl, G.; Park, J.; Winick, N.J.; Klenke, R.; Berg, S.L.; Blaney, S.M. Phase I trial of intrathecal liposomal cytarabine in children with neoplastic meningitis. J. Clin. Oncol. 2004, 22, 3916–3921. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chatelut, E.; Kim, J.C.; Howell, S.B.; Cates, C.; Kormanik, P.A.; Chamberlain, M.C. Extended CSF cytarabine exposure following intrathecal administration of DTC 101. J. Clin. Oncol. 1993, 11, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Ploylearmsaeng, S.A.; Fuhr, U.; Jetter, A. How may anticancer chemotherapy with fluorouracil be individualised? Clin. Pharmacokinet. 2006, 45, 567–592. [Google Scholar] [CrossRef] [PubMed]
- Capitain, O.; Boisdron-Celle, M.; Poirier, A.L.; Abadie-Lacourtoisie, S.; Morel, A.; Gamelin, E. The influence of fluorouracil outcome parameters on tolerance and efficacy in patients with advanced colorectal cancer. Pharmacogenom. J. 2008, 8, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Deenen, M.J.; Tol, J.; Burylo, A.M.; Doodeman, V.D.; de Boer, A.; Vincent, A.; Guchelaar, H.J.; Smits, P.H.; Beijnen, J.H.; Punt, C.J.; et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin. Cancer Res. 2011, 17, 3455–3468. [Google Scholar] [CrossRef] [PubMed]
- Buchel, B.; Rhyn, P.; Schurch, S.; Buhr, C.; Amstutz, U.; Largiader, C.R. LC-MS/MS method for simultaneous analysis of uracil, 5,6-dihydrouracil, 5-fluorouracil and 5-fluoro-5,6-dihydrouracil in human plasma for therapeutic drug monitoring and toxicity prediction in cancer patients. Biomed. Chromatogr. BMC 2013, 27, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Krynetski, E.Y.; Krynetskaia, N.F.; Yanishevski, Y.; Evans, W.E. Methylation of mercaptopurine, thioguanine, and their nucleotide metabolites by heterologously expressed human thiopurine S-methyltransferase. Mol. Pharmacol. 1995, 47, 1141–1147. [Google Scholar] [PubMed]
- Rieger, H.; Schmidt, P.; Schaeffeler, E.; Abe, M.; Schiffhauer, M.; Schwab, M.; von Ahsen, N.; Zurek, G.; Kirchherr, H.; Shipkova, M.; et al. Validation of a high-performance liquid chromatography method for thiopurine S-methyltransferase activity in whole blood using 6-mercaptopurine as substrate. Clin. Chem. Lab. Med. 2018, 56, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Karim, H.; Appell, M.L.; Fotoohi, A. Comparison of three methods for measuring thiopurine methyltransferase activity in red blood cells and human leukemia cells. J. Chromatogr. 2013, 939, 80–85. [Google Scholar] [CrossRef] [PubMed]
- McLeod, H.L.; Krynetski, E.Y.; Relling, M.V.; Evans, W.E. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia 2000, 14, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Schmiegelow, K.; Forestier, E.; Kristinsson, J.; Soderhall, S.; Vettenranta, K.; Weinshilboum, R.; Wesenberg, F. Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: Results from the NOPHO ALL-92 study. Leukemia 2009, 23, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.N.; Grell, K.; Nersting, J.; Abrahamsson, J.; Lund, B.; Kanerva, J.; Jonsson, O.G.; Vaitkeviciene, G.; Pruunsild, K.; Hjalgrim, L.L.; et al. DNA-thioguanine nucleotide concentration and relapse-free survival during maintenance therapy of childhood acute lymphoblastic leukaemia (NOPHO ALL2008): A prospective substudy of a phase 3 trial. Lancet Oncol. 2017, 18, 515–524. [Google Scholar] [CrossRef]
- Young, J.D. The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: A 30-year collaborative odyssey. Biochem. Soc. Trans. 2016, 44, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Asp. Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, M.W.; Ng, A.M.; Yao, S.Y.; Graham, K.; Loewen, S.K.; Smith, K.M.; Ritzel, R.G.; Mowles, D.A.; Carpenter, P.; Chen, X.Z.; et al. Molecular identification and characterization of novel human and mouse concentrative Na+-nucleoside cotransporter proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). J. Biol. Chem. 2001, 276, 2914–2927. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. The plasma membrane monoamine transporter (PMAT): Structure, function, and role in organic cation disposition. Clin. Pharmacol. Ther. 2016, 100, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Marechal, R.; Mackey, J.R.; Lai, R.; Demetter, P.; Peeters, M.; Polus, M.; Cass, C.E.; Young, J.; Salmon, I.; Deviere, J.; et al. Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clin. Cancer Res. 2009, 15, 2913–2919. [Google Scholar] [CrossRef] [PubMed]
- Drenberg, C.D.; Gibson, A.A.; Pounds, S.B.; Shi, L.; Rhinehart, D.P.; Li, L.; Hu, S.; Du, G.; Nies, A.T.; Schwab, M.; et al. OCTN1 Is a High-Affinity Carrier of Nucleoside Analogues. Cancer Res. 2017, 77, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Tan, T.M. Role of glutathione in the multidrug resistance protein 4 (MRP4/ABCC4)-mediated efflux of cAMP and resistance to purine analogues. Biochem. J. 2002, 361, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kock, K.; Ritter, C.A.; Chen, Z.S.; Grube, M.; Jedlitschky, G.; Illmer, T.; Ayres, M.; Beck, J.F.; Siegmund, W.; et al. Expression of ABCC-type nucleotide exporters in blasts of adult acute myeloid leukemia: Relation to long-term survival. Clin. Cancer Res. 2009, 15, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Schuetz, J.D. ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem. Pharmacol. 2012, 83, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, R.L. Biochemical and clinical pharmacology of 5-fluorouracil. Oncology 1998, 12, 13–18. [Google Scholar] [PubMed]
- Kanzaki, A.; Takebayashi, Y.; Bando, H.; Eliason, J.F.; Watanabe Si, S.; Miyashita, H.; Fukumoto, M.; Toi, M.; Uchida, T. Expression of uridine and thymidine phosphorylase genes in human breast carcinoma. Int. J. Cancer 2002, 97, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Temmink, O.H.; de Bruin, M.; Laan, A.C.; Turksma, A.W.; Cricca, S.; Masterson, A.J.; Noordhuis, P.; Peters, G.J. The role of thymidine phosphorylase and uridine phosphorylase in (fluoro)pyrimidine metabolism in peripheral blood mononuclear cells. Int.J. Biochem. Cell Biol. 2006, 38, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Bronckaers, A.; Gago, F.; Balzarini, J.; Liekens, S. The dual role of thymidine phosphorylase in cancer development and chemotherapy. Med. Res. Rev. 2009, 29, 903–953. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, Y.; Tanaka, T.; Nakano, T.; Matsui, H.; Nasu, T.; Koike, S.; Kakehata, S.; Ito, T.; Goto, K. Orotate phosphoribosyltransferase localizes to the Golgi complex and its expression levels affect the sensitivity to anti-cancer drug 5-fluorouracil. Biomed. Res. 2015, 36, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Yasumatsu, R.; Nakashima, T.; Komune, S. Overexpression of the orotate phosphoribosyl-transferase gene enhances the effect of 5-Fluorouracil in head and neck squamous cell carcinoma in vitro. J. Oncol. 2012, 2012, 649605. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, T.; Umeki, M.; Miyake, H.; Iida, T.; Okumura, M.; Ohno, K.; Sakamoto, M.; Miyoshi, N.; Takahashi, M.; Tsumura, H.; et al. Impact of 5-fluorouracil metabolizing enzymes on chemotherapy in patients with resectable colorectal cancer. Oncol. Rep. 2014, 32, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Komori, S.; Osada, S.; Tomita, H.; Nishio, K.; Kumazawa, I.; Tachibana, S.; Tsuchiya, J.; Yoshida, K. Predictive value of orotate phosphoribosyltransferase in colorectal cancer patients receiving 5-FU-based chemotherapy. Mol. Clin. Oncol. 2013, 1, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, T.; Nishina, T.; Hyodo, I.; Moriwaki, T.; Endo, S.; Nasu, J.; Hori, S.; Matsuura, B.; Hiasa, Y.; Onji, M. High orotate phosphoribosyltransferase gene expression predicts complete response to chemoradiotherapy in patients with squamous cell carcinoma of the esophagus. Oncology 2009, 76, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, W.; Takahashi, T.; Suto, K.; Sasaki, Y.; Hirayama, R. Orotate phosphoribosyltransferase gene polymorphism predicts toxicity in patients treated with bolus 5-fluorouracil regimen. Clin. Cancer Res. 2006, 12, 3928–3934. [Google Scholar] [CrossRef] [PubMed]
- Elgemeie, G.H. Thioguanine, mercaptopurine: Their analogs and nucleosides as antimetabolites. Curr. Pharm. Des. 2003, 9, 2627–2642. [Google Scholar] [CrossRef] [PubMed]
- Monnat, R.J., Jr. Molecular analysis of spontaneous hypoxanthine phosphoribosyltransferase mutations in thioguanine-resistant HL-60 human leukemia cells. Cancer Res. 1989, 49, 81–87. [Google Scholar] [PubMed]
- Verhoef, V.; Sarup, J.; Fridland, A. Identification of the mechanism of activation of 9-beta-D-arabinofuranosyladenine in human lymphoid cells using mutants deficient in nucleoside kinases. Cancer Res. 1981, 41, 4478–4483. [Google Scholar] [PubMed]
- Hurley, M.C.; Lin, B.; Fox, I.H. Regulation of deoxyadenosine and nucleoside analog phosphorylation by human placental adenosine kinase. J. Biol. Chem. 1985, 260, 15675–15681. [Google Scholar] [PubMed]
- Kees, U.R.; Ford, J.; Dawson, V.M.; Piall, E.; Aherne, G.W. Development of resistance to 1-beta-D-arabinofuranosylcytosine after high-dose treatment in childhood lymphoblastic leukemia: Analysis of resistance mechanism in established cell lines. Cancer Res. 1989, 49, 3015–3019. [Google Scholar] [PubMed]
- Colly, L.P.; Peters, W.G.; Richel, D.; Arentsen-Honders, M.W.; Starrenburg, C.W.; Willemze, R. Deoxycytidine kinase and deoxycytidine deaminase values correspond closely to clinical response to cytosine arabinoside remission induction therapy in patients with acute myelogenous leukemia. Semin. Oncol. 1987, 14, 257–261. [Google Scholar] [PubMed]
- Genini, D.; Adachi, S.; Chao, Q.; Rose, D.W.; Carrera, C.J.; Cottam, H.B.; Carson, D.A.; Leoni, L.M. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood 2000, 96, 3537–3543. [Google Scholar] [PubMed]
- Sakamoto, K.; Yokogawa, T.; Ueno, H.; Oguchi, K.; Kazuno, H.; Ishida, K.; Tanaka, N.; Osada, A.; Yamada, Y.; Okabe, H.; et al. Crucial roles of thymidine kinase 1 and deoxyUTPase in incorporating the antineoplastic nucleosides trifluridine and 2′-deoxy-5-fluorouridine into DNA. Int. J. Oncol. 2015, 46, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Munch-Petersen, B.; Herrstrom Sjoberg, A.; Hellman, U.; Bergman, T.; Jornvall, H.; Eriksson, S. Human thymidine kinase 2: Molecular cloning and characterisation of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett. 1999, 443, 170–174. [Google Scholar] [CrossRef]
- Priego, E.M.; Karlsson, A.; Gago, F.; Camarasa, M.J.; Balzarini, J.; Perez-Perez, M.J. Recent advances in thymidine kinase 2 (TK2) inhibitors and new perspectives for potential applications. Curr. Pharm. Des. 2012, 18, 2981–2994. [Google Scholar] [CrossRef] [PubMed]
- Meier, C. cycloSal-pronucleotides design of chemical trojan horses. Mini Rev. Med. Chem. 2002, 2, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.Y.; Dutschman, G.E.; Lam, W.; Jiang, Z.; Cheng, Y.C. Characterization of human UMP/CMP kinase and its phosphorylation of D- and L-form deoxycytidine analogue monophosphates. Cancer Res. 2002, 62, 1624–1631. [Google Scholar] [PubMed]
- Liou, J.Y.; Lai, H.R.; Hsu, C.H.; Chang, W.L.; Hsieh, M.J.; Huang, Y.C.; Cheng, Y.C. Modulation of human UMP/CMP kinase affects activation and cellular sensitivity of deoxycytidine analogs. Biochem. Pharmacol. 2010, 79, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, H.; Kurasawa, M.; Yanagisawa, M.; Sato, Y.; Harada, N.; Mori, K. Predictive markers of capecitabine sensitivity identified from the expression profile of pyrimidine nucleoside-metabolizing enzymes. Oncol. Rep. 2013, 29, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Johansson, M.; Karlsson, A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem. 2008, 283, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Temmink, O.H.; Emura, T.; de Bruin, M.; Fukushima, M.; Peters, G.J. Therapeutic potential of the dual-targeted TAS-102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci. 2007, 98, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Okabe-Kado, J.; Kasukabe, T.; Honma, Y. Differentiation inhibitory factor Nm23 as a prognostic factor for acute myeloid leukemia. Leuk. Lymphoma 1998, 32, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Okabe-Kado, J.; Wakimoto, N.; Kobayashi, H.; Sakashita, A.; Maseki, N.; Nakamaki, T.; Hino, K.; Tomoyasu, S.; Tsuruoka, N.; et al. Evaluation by multivariate analysis of the differentiation inhibitory factor nm23 as a prognostic factor in acute myelogenous leukemia and application to other hematologic malignancies. Blood 1998, 91, 1845–1851. [Google Scholar] [PubMed]
- Bach, E.; Krahl, R.; Lange, T.; Schuler, F.; Al-Ali, H.; Buchner, T.; Haferlach, T.; Dolken, G.; Niederwieser, D.; Cross, M. Delayed processing of bone marrow samples reveals a prognostic pattern of NME mRNA expression in cytogenetically normal acute myeloid leukemia. Leuk. Lymphoma 2012, 53, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Steuart, C.D.; Burke, P.J. Cytidine deaminase and the development of resistance to arabinosyl cytosine. Nat. New Biol. 1971, 233, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Hubeek, I.; Stam, R.W.; Peters, G.J.; Broekhuizen, R.; Meijerink, J.P.; van Wering, E.R.; Gibson, B.E.; Creutzig, U.; Zwaan, C.M.; Cloos, J.; et al. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br. J. Cancer 2005, 93, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Niitsu, N.; Ishii, Y.; Matsuda, A.; Honma, Y. Induction of differentiation of acute promyelocytic leukemia cells by a cytidine deaminase-resistant analogue of 1-beta-D-arabinofuranosylcytosine, 1-(2-deoxy-2-methylene-beta-D-erythro-pentofuranosyl)cytidine. Cancer Res. 2001, 61, 178–185. [Google Scholar] [PubMed]
- Miwa, M.; Eda, H.; Ura, M.; Ouchi, K.F.; Keith, D.D.; Foley, L.H.; Ishitsuka, H. High susceptibility of human cancer xenografts with higher levels of cytidine deaminase to a 2′-deoxycytidine antimetabolite, 2′-deoxy-2′-methylidenecytidine. Clin. Cancer Res. 1998, 4, 493–497. [Google Scholar] [PubMed]
- Chiba, P.; Tihan, T.; Szekeres, T.; Salamon, J.; Kraupp, M.; Eher, R.; Koller, U.; Knapp, W. Concordant changes of pyrimidine metabolism in blasts of two cases of acute myeloid leukemia after repeated treatment with ara-C in vivo. Leukemia 1990, 4, 761–765. [Google Scholar] [PubMed]
- Gilbert, J.A.; Salavaggione, O.E.; Ji, Y.; Pelleymounter, L.L.; Eckloff, B.W.; Wieben, E.D.; Ames, M.M.; Weinshilboum, R.M. Gemcitabine pharmacogenomics: Cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin. Cancer Res. 2006, 12, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Elander, N.O.; Aughton, K.; Ghaneh, P.; Neoptolemos, J.P.; Palmer, D.H.; Cox, T.F.; Campbell, F.; Costello, E.; Halloran, C.M.; Mackey, J.R.; et al. Intratumoural expression of deoxycytidylate deaminase or ribonuceotide reductase subunit M1 expression are not related to survival in patients with resected pancreatic cancer given adjuvant chemotherapy. Br. J. Cancer 2018, 118, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.S.; Rosing, H.; Schellens, J.H.; Beijnen, J.H. Deoxyuridine analog nucleotides in deoxycytidine analog treatment: Secondary active metabolites? Fund. Clin. Pharmacol. 2011, 25, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fridley, B.; Kalari, K.; Jenkins, G.; Batzler, A.; Safgren, S.; Hildebrandt, M.; Ames, M.; Schaid, D.; Wang, L. Gemcitabine and cytosine arabinoside cytotoxicity: Association with lymphoblastoid cell expression. Cancer Res. 2008, 68, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Valerie, N.C.K.; Helleday, T. Pathways controlling dNTP pools to maintain genome stability. DNA Repair 2016, 44, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Requena, C.E.; Perez-Moreno, G.; Horvath, A.; Vertessy, B.G.; Ruiz-Perez, L.M.; Gonzalez-Pacanowska, D.; Vidal, A.E. The nucleotidohydrolases DCTPP1 and dUTPase are involved in the cellular response to decitabine. Biochem. J. 2016, 473, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Almqvist, H.; Axelsson, H.; Jafari, R.; Dan, C.; Mateus, A.; Haraldsson, M.; Larsson, A.; Martinez Molina, D.; Artursson, P.; Lundback, T.; et al. CETSA screening identifies known and novel thymidylate synthase inhibitors and slow intracellular activation of 5-fluorouracil. Nat. Commun. 2016, 7, 11040. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.M.; Fazzone, W.; LaBonte, M.J.; Lenz, H.J.; Ladner, R.D. Regulation of human dUTPase gene expression and p53-mediated transcriptional repression in response to oxaliplatin-induced DNA damage. Nucleic Acids Res. 2009, 37, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Hoglund, A.; Jacques, S.A.; Johansson, L.; Calderon-Montano, J.M.; Claesson, M.; Loseva, O.; Valerie, N.C.K.; Lundback, T.; Piedrafita, J.; et al. Discovery of the First Potent and Selective Inhibitors of Human dCTP Pyrophosphatase 1. J. Med. Chem. 2016, 59, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Corson, T.W.; Cavga, H.; Aberle, N.; Crews, C.M. Triptolide directly inhibits dCTP pyrophosphatase. Chembiochem 2011, 12, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, Y.; Naito, T.; Onodera, M.; Kuroha, M.; Kimura, T.; Shiga, H.; Endo, K.; Negoro, K.; Kinouchi, Y.; Shimosegawa, T. NUDT15 R139C causes thiopurine-induced early severe hair loss and leukopenia in Japanese patients with IBD. Pharmacogenom. J. 2016, 16, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Landier, W.; Yang, W.; Liu, C.; Hageman, L.; Cheng, C.; Pei, D.; Chen, Y.; Crews, K.R.; Kornegay, N.; et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J. Clin. Oncol. 2015, 33, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.K.; Hong, M.; Baek, J.; Choi, H.; Zhao, W.; Jung, Y.; Haritunians, T.; Ye, B.D.; Kim, K.J.; Park, S.H.; et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat. Genet. 2014, 46, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Nishii, R.; Perez-Andreu, V.; Yang, W.; Klussmann, F.A.; Zhao, X.; Lin, T.N.; Hoshitsuki, K.; Nersting, J.; Kihira, K.; et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat. Genet. 2016, 48, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Valerie, N.C.; Hagenkort, A.; Page, B.D.; Masuyer, G.; Rehling, D.; Carter, M.; Bevc, L.; Herr, P.; Homan, E.; Sheppard, N.G.; et al. NUDT15 Hydrolyzes 6-Thio-DeoxyGTP to Mediate the Anticancer Efficacy of 6-Thioguanine. Cancer Res. 2016, 76, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Nishii, R.; Moriyama, T.; Janke, L.J.; Yang, W.; Suiter, C.C.; Lin, T.N.; Li, L.; Kihira, K.; Toyoda, H.; Hofmann, U.; et al. Preclinical evaluation of NUDT15-guided thiopurine therapy and its effects on toxicity and antileukemic efficacy. Blood 2018, 131, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; McLennan, A.G.; Ying, K.; Wang, Z.; Gu, S.; Jin, H.; Wu, C.; Liu, W.; Yuan, Y.; Tang, R.; et al. Cloning, expression, and characterization of a human inosine triphosphate pyrophosphatase encoded by the itpa gene. J. Biol. Chem. 2001, 276, 18695–18701. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.R.; Waisertreiger, I.S.; Lopez-Bertoni, H.; Luo, X.; Pavlov, Y.I. Pivotal role of inosine triphosphate pyrophosphatase in maintaining genome stability and the prevention of apoptosis in human cells. PLoS ONE 2012, 7, e32313. [Google Scholar] [CrossRef] [PubMed]
- Stocco, G.; Crews, K.R.; Evans, W.E. Genetic polymorphism of inosine-triphosphate-pyrophosphatase influences mercaptopurine metabolism and toxicity during treatment of acute lymphoblastic leukemia individualized for thiopurine-S-methyl-transferase status. Expert Opin. Drug Saf. 2010, 9, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Marinaki, A.M.; Duley, J.A.; Arenas, M.; Ansari, A.; Sumi, S.; Lewis, C.M.; Shobowale-Bakre, M.; Fairbanks, L.D.; Sanderson, J. Mutation in the ITPA gene predicts intolerance to azathioprine. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Van Dieren, J.M.; Hansen, B.E.; Kuipers, E.J.; Nieuwenhuis, E.E.; Van der Woude, C.J. Meta-analysis: Inosine triphosphate pyrophosphatase polymorphisms and thiopurine toxicity in the treatment of inflammatory bowel disease. Aliment. Pharmacol. Ther. 2007, 26, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.; Herold, N. The Early Bird Catches the Worm—Can Evolution Teach us Lessons in Fighting HIV? Curr. HIV Res. 2016, 14, 183–210. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.H.; Kunzelmann, S.; Webb, M.R.; Taylor, I.A. A continuous enzyme-coupled assay for triphosphohydrolase activity of HIV-1 restriction factor SAMHD1. Antimicrob. Agents Chemother. 2015, 59, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Rudd, S.G.; Sanjiv, K.; Kutzner, J.; Bladh, J.; Paulin, C.B.J.; Helleday, T.; Henter, J.I.; Schaller, T. SAMHD1 protects cancer cells from various nucleoside-based antimetabolites. Cell Cycle 2017, 16, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Schaller, T.; Herold, N. SAMHD1 is a barrier to antimetabolite-based cancer therapies. Mol. Cell. Oncol. 2017, 4, e1287554. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Oellerich, T.; Baldauf, H.M.; Schwarz, S.M.; Thomas, D.; Flick, R.; Bohnenberger, H.; Kaderali, L.; Stegmann, L.; Cremer, A.; et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat. Med. 2017, 23, 250. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Rudd, S.; Myrberg, I.H.; Sanjiv, K.; Ljungblad, L.; Kutzner, J.; Grander, D.; Kogner, P.; Rassidakis, G.; Helleday, T.; et al. Development of a Small Molecule Inhibitor for SAMHD1 to Improve Outcome for Patients with Acute Myelogenous Leukaemia and Relapsed T-Lymphoblastic Malignancies Treated with Nucleoside Analogues. Pediatr. Blood Cancer 2017, 64, S135. [Google Scholar]
- Seamon, K.J.; Hansen, E.C.; Kadina, A.P.; Kashemirov, B.A.; McKenna, C.E.; Bumpus, N.N.; Stivers, J.T. Small molecule inhibition of SAMHD1 dNTPase by tetramer destabilization. J. Am. Chem. Soc. 2014, 136, 9822–9825. [Google Scholar] [CrossRef] [PubMed]
- Seamon, K.J.; Stivers, J.T. A High-Throughput Enzyme-Coupled Assay for SAMHD1 dNTPase. J. Biomol. Screen 2015, 20, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Vena, F.; Li Causi, E.; Rodriguez-Justo, M.; Goodstal, S.; Hagemann, T.; Hartley, J.A.; Hochhauser, D. The MEK1/2 Inhibitor Pimasertib Enhances Gemcitabine Efficacy in Pancreatic Cancer Models by Altering Ribonucleotide Reductase Subunit-1 (RRM1). Clin. Cancer Res. 2015, 21, 5563–5577. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, J.H.; Jung, Y.; Kim, J.H.; Jong, H.S.; Kim, T.Y.; Bang, Y.J. Histone deacetylase inhibitor enhances 5-fluorouracil cytotoxicity by down-regulating thymidylate synthase in human cancer cells. Mol. Cancer Ther. 2006, 5, 3085–3095. [Google Scholar] [CrossRef] [PubMed]
- Meyers, M.; Hwang, A.; Wagner, M.W.; Bruening, A.J.; Veigl, M.L.; Sedwick, W.D.; Boothman, D.A. A role for DNA mismatch repair in sensing and responding to fluoropyrimidine damage. Oncogene 2003, 22, 7376–7388. [Google Scholar] [CrossRef] [PubMed]
- Triemer, T.; Messikommer, A.; Glasauer, S.M.K.; Alzeer, J.; Paulisch, M.H.; Luedtke, N.W. Superresolution imaging of individual replication forks reveals unexpected prodrug resistance mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, E1366–E1373. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Clarke, M.L.; Falette, N.; Puisieux, A.; Mackey, J.R.; Dumontet, C. Expression of a non-functional p53 affects the sensitivity of cancer cells to gemcitabine. Int. J. Cancer 2002, 97, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, A.R.; Sherrington, P.D.; Cawley, J.C. The effect of p53 dysfunction on purine analogue cytotoxicity in chronic lymphocytic leukaemia. Br. J. Haematol. 1999, 106, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Zoppoli, G.; Regairaz, M.; Leo, E.; Reinhold, W.C.; Varma, S.; Ballestrero, A.; Doroshow, J.H.; Pommier, Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. USA 2012, 109, 15030–15035. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol 2015, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Zawacka-Pankau, J.; Selivanova, G. Pharmacological reactivation of p53 as a strategy to treat cancer. J. Intern. Med. 2015, 277, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.W.; Thomas, A.; Murai, J.; Trepel, J.B.; Bates, S.E.; Rajapakse, V.N.; Pommier, Y. Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors. Clin. Cancer Res. 2018, 24, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, Y.; Kato, Y.; Ura, M.; Horii, I.; Ishitsuka, H.; Kusuhara, H.; Sugiyama, Y. A physiologically based pharmacokinetic analysis of capecitabine, a triple prodrug of 5-FU, in humans: The mechanism for tumor-selective accumulation of 5-FU. Pharm. Res. 2001, 18, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Ciccolini, J.; Serdjebi, C.; Le Thi Thu, H.; Lacarelle, B.; Milano, G.; Fanciullino, R. Nucleoside analogs: Ready to enter the era of precision medicine? Expert Opin. Drug Metab. Toxicol. 2016, 12, 865–877. [Google Scholar] [CrossRef] [PubMed]




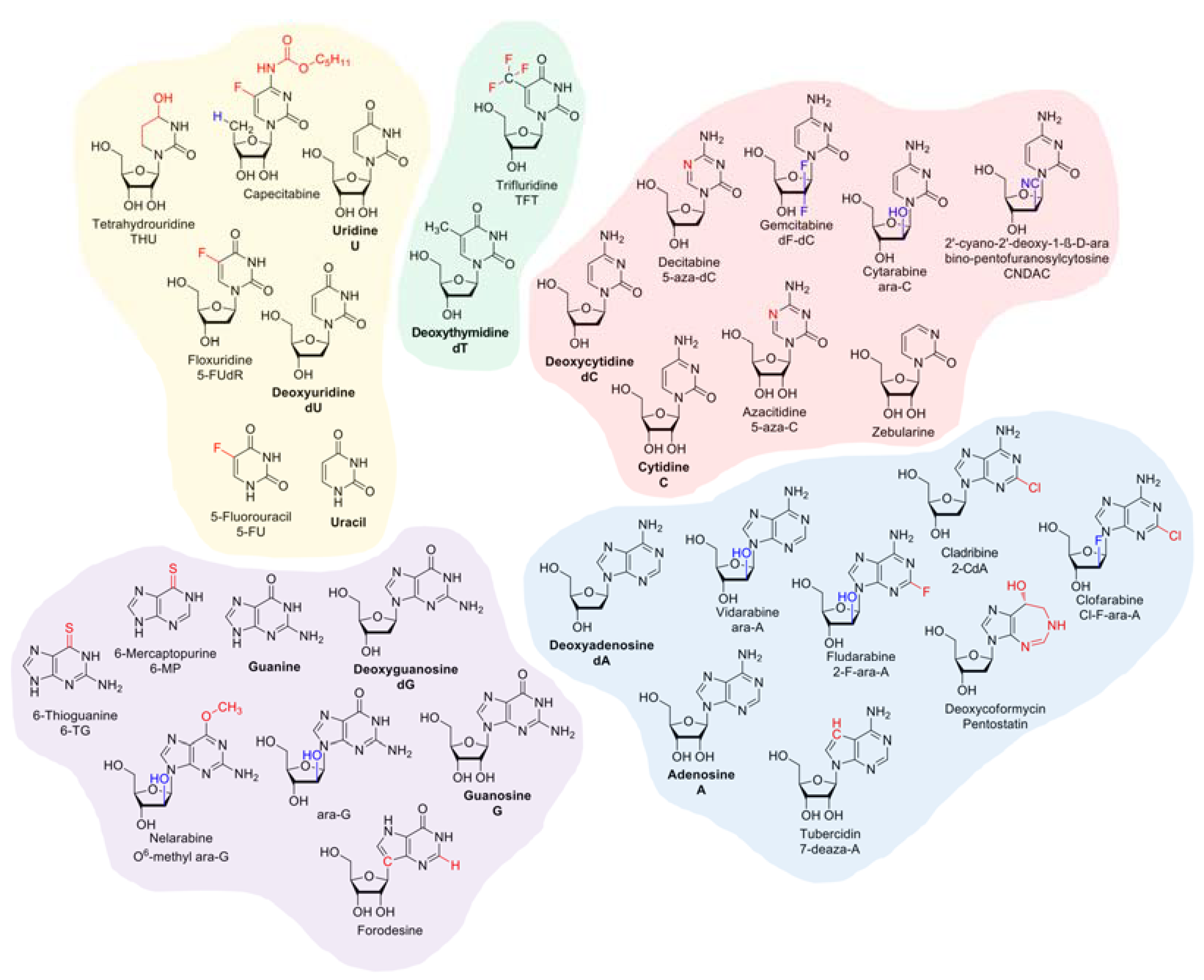{kind=link}
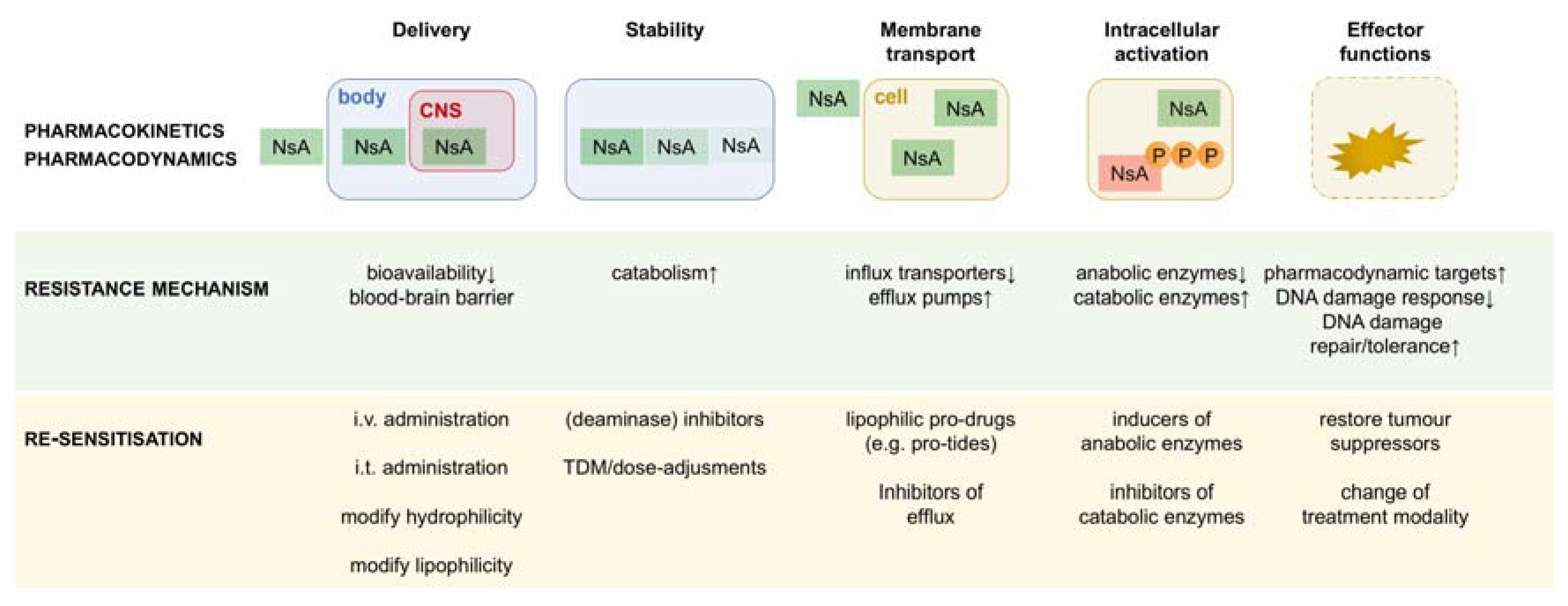{kind=link}
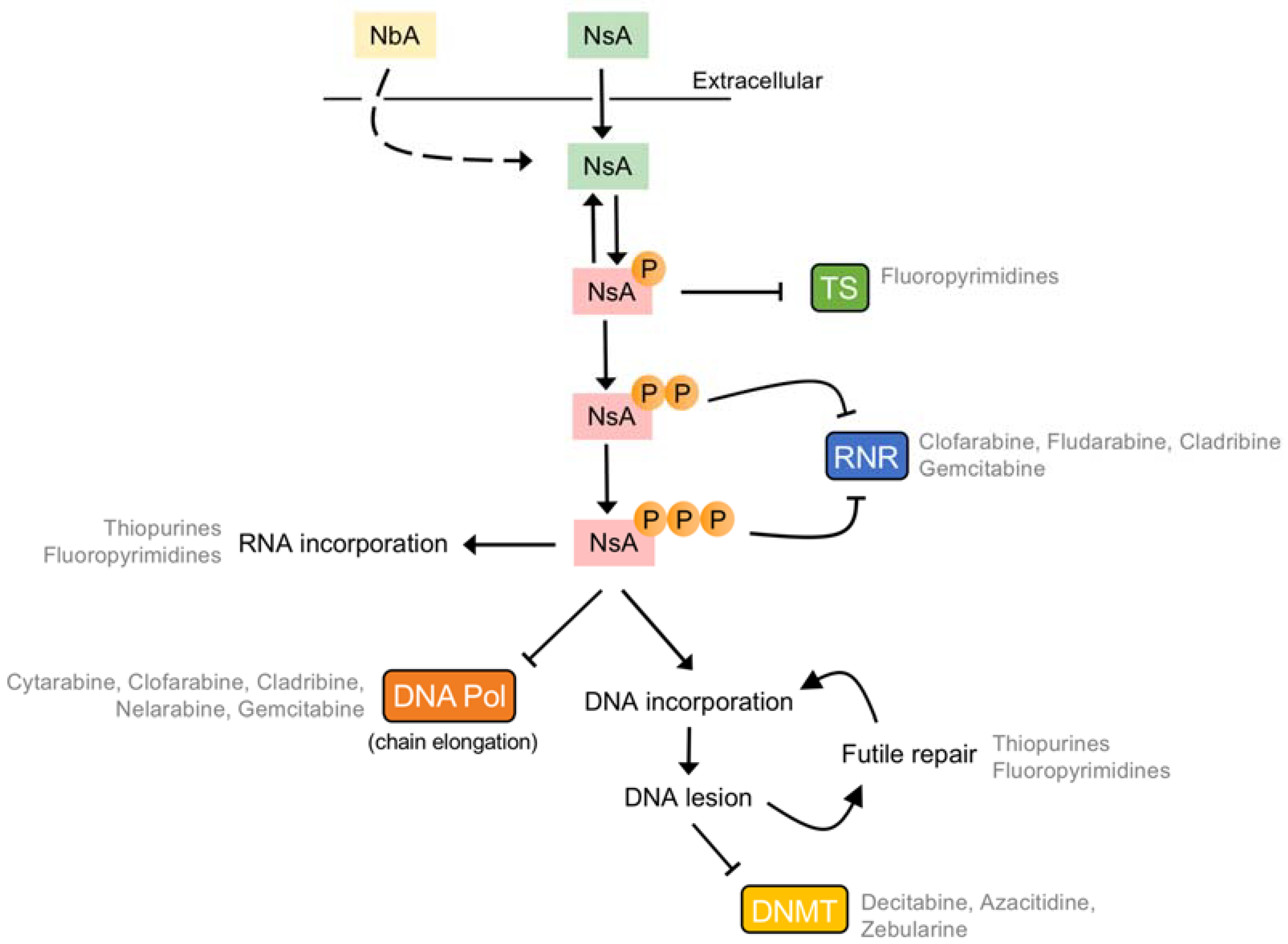{kind=link}
| Protein | Properties | Resistance Mechanism | Re-Sensitisation | ||
|---|---|---|---|---|---|
| Name | Abbrev. | Gene | |||
| Adenosine deaminase | ADA | ADA | Deamination of adenosine analogues | ADA amplification [9] | ADA inhibitors (pentostatin) [10], ADA-resistant nucleosides |
| Cytidine deaminase | CDA | CDA | Deamination of (deoxy)cytidine analogues | Polymorphisms [11,12,13,14,15], Overexpression [16,17]. | CDA inhibitors [18,19] |
| Cytosolic 5′-nucleotidase I, II and III | cN-I/II/III | NT5C1–3 | Dephosphorylation of nucleoside analogue monophosphates | Over-expression [20,21,22,23,24,25,26,27], gain-of-function mutations [20,28,29,30,31], polymorphisms [14,32,33] | |
| Cytosolic thymidine kinase | TK1 | TK1 | Monophosphorylation of thymidine analogues | Loss of TK1 expression confers resistance to trifluridine [34] | |
| dCMP deaminase | DCTD | DCTD | Deaminates monophosphates of cytidine analogues | DCTD inhibitor [35,36] | |
| Deoxycytidine kinase | dCK | DCK | Monophosphorylation of pyrimidine and purine nucleoside analogues | Downregulation [37,38,39,40,41,42], Alternative splicing resulting in reduced dCK activity [43], Polymorphisms [44,45] | Etoposide to increase dCK activity [46], Pro-tide chemistry [16,47,48,49,50,51] |
| Deoxyguanosine kinase | dGK | DGUOK | Monophosphorylation of purine nucleoside analogues | Activity/expression [52,53,54] | |
| Dihydropyrimidine dehydrogenase | DPD | DYPD | Reduction of uracil and thymine analogues | Polymorphisms [55,56] | DPD inhibitors 5-Chloro-2,4-dihydroxypyridine, Elinuracil [57,58,59,60] |
| DNA methylatransferases | DNMTs | DNMT1, DNMT3A, DNMT3B, DNMT3L | Methylation of 5′ cytosine in GpC dinucleotides | Increased activity/expression of DNMT1 [61] | Change to Zebularine [62] |
| dUTP diphosphatase | dUTPase | DUT | Dephosphorylation of dUTP analogues | Overexpression [63,64] | dUTPase inhibitors [65,66,67,68] |
| Guanosine monophosphate synthetase | GMPS | GMPS | Conversion of TXMP to TGMP | Downregulation [69] | |
| Human concentrative nucleoside transporters 1–3 | hCNT1–3 | SLC28A1–3 | Unidirectional membrane transport | Downregulation [70] | Lipophilic modifications [71,72,73], pro-tide chemistry [16,47,48,49,50,51] |
| Human equilibrative nucleoside transporters 1–4 | hENT1–4 | SLC29A1–4 | Bi-directional membrane transport | Low expression [74,75,76,77,78,79,80,81], Polymorphisms [70,82,83,84,85,86,87] | Efflux inhibitors [88], Lipophilic modifications [71,72,73], Pro-tide chemistry [16,47,48,49,50,51] |
| Hypoxanthine guanosine phosphoribosyltransferase | HGPRT | HPRT1 | Converts thiopurines 6-MP and 6-TG into 6-thioinosine monophosphate (TIMP) and 6-thioguanosine monophosphate (TGMP) | Decreased activity/expression [89,90] | Allopurinol to increase HGPRT activity [91,92] |
| Inosine monophosphate dehydrogenase | IMPDH | IMPDH1 | Conversion of TIMP to thioxanthosine monophosphate (TXMP) | Loss or reduction of activity [93] | |
| Inosine triphosphatase | ITPA | ITPA | Regulates sanitation of the endogenous non-canonical (deoxy)nucleotide triphosphates (deoxy)inosine and (deoxy)xanthosine triphosphate | Polymorphisms [94,95,96,97] | |
| Nucleotide diphosphatase NUDT15 | NUDT15 | NUDT15 | Dephosphorylation of thiopurine triphosphates | High expression [98] | |
| Nucleotide diphosphate kinase 1 and 2 | NDPK1/2 | NME1/2 | Phosphorylation of nucleoside analogue diphosphates | Polymorphisms [99,100] | Lipophilising diphosphate analogues [101] |
| Phosphoribosyl pyrophosphate amidotransferase | PPAT | PPAT | Purine biosynthesis | Increased activity of PPAT [102] | |
| Purine nucleoside phosphorylase | PNP | PNP | De-glycosylation of guanosine/inosine analogues | ||
| Ribonucleotide reductase | RNR | RRM1, RRM2, RRM2B | Reduction of nucleoside diphosphates (NDPs) to deoxy-NDPs (dNDPs) | Overexpression [103,104,105,106,107,108,109,110,111,112,113,114], Polymorphisms [115] | MEK-ERK inhibitors increase dF-dC sensitivity by reducing RNR expression [116] |
| SAM and HD domain protein 1 | SAMHD1 | SAMHD1 | Dephosphorylation of dNTP analogues | High expression [117,118] | Use of viral protein X to inhibit SAMHD1 [117], Small-molecule inhibitors [103,104,105] |
| Thiopurine S-methyltransferase | TPMT | TPMT | Methylation of thiopurines | Polymorphisms [119] | Xanthine oxidase inhibitors (allopurinol) [120] |
| Thymidine phosphorylase | TP | TYMP | Glycosylation of 5-FU, De-glycosylation of thymidine analogues | Low expression in tumour tissue (for 5-FU treatment) [121], High plasma activity (for TFT treatment) [122,123] | Taxanes to increase expression (for 5-FU treatment) [124], Inhibitor tipiracil (for TFT treatment) [122,123,125,126] |
| Thymidylate synthase | TS | TYMS | Reductive methylation of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) | Overexpression [116,127,128,129,130] | Histone deacetylase inhibitors (HDACi) reduce TYMS expression and synergise with fluoropyrimidines [130,131] |
| UMP/CMP kinase | UCK | CMPK1 | Phosphorylates cytidine and uridine analogue monophosphates | Downregulation [132], Polymorphisms [133,134] | Lipophilising diphosphate analogues [101] |
| Uridine monophosphate synthetase | UMPS | UMPS | Conversion of 5-FU to 5-FUMP | Downregulation [135] | |
| Uridine phosphorylase 1 | UP1 | UPP1 | Glycosylation of 5-FU, De-glycosylation of 5-FdUrd | Expression [136] | UP1 inhibitors (cell-line dependent effects) [137] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsesmetzis, N.; Paulin, C.B.J.; Rudd, S.G.; Herold, N. Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism. Cancers 2018, 10, 240. https://doi.org/10.3390/cancers10070240
Tsesmetzis N, Paulin CBJ, Rudd SG, Herold N. Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism. Cancers. 2018; 10(7):240. https://doi.org/10.3390/cancers10070240
Chicago/Turabian StyleTsesmetzis, Nikolaos, Cynthia B. J. Paulin, Sean G. Rudd, and Nikolas Herold. 2018. "Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism" Cancers 10, no. 7: 240. https://doi.org/10.3390/cancers10070240
APA StyleTsesmetzis, N., Paulin, C. B. J., Rudd, S. G., & Herold, N. (2018). Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism. Cancers, 10(7), 240. https://doi.org/10.3390/cancers10070240