Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. 3D Skin Model
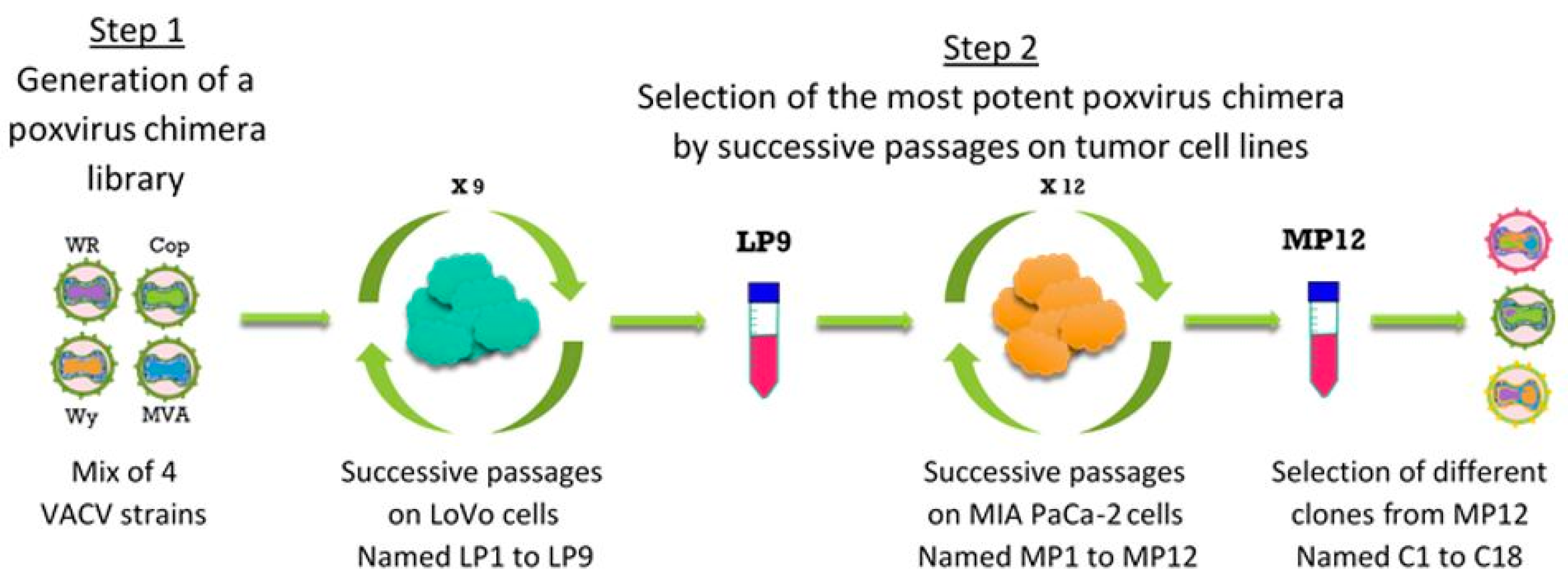
2.3. Directed Evolution for Selection of Chimeric Vaccinia Virus deVV5
2.4. Generation of deVV5-fcu1
2.5. In Vitro Cytotoxicity Assay
2.6. In Vitro Virus Yield Assay
2.7. In Vitro Cell Sensitivity to 5-FC
2.8. Cytosine Deaminase Enzymatic Assay
2.9. DNA Sequencing
2.10. Statistical Analyses
3. Results
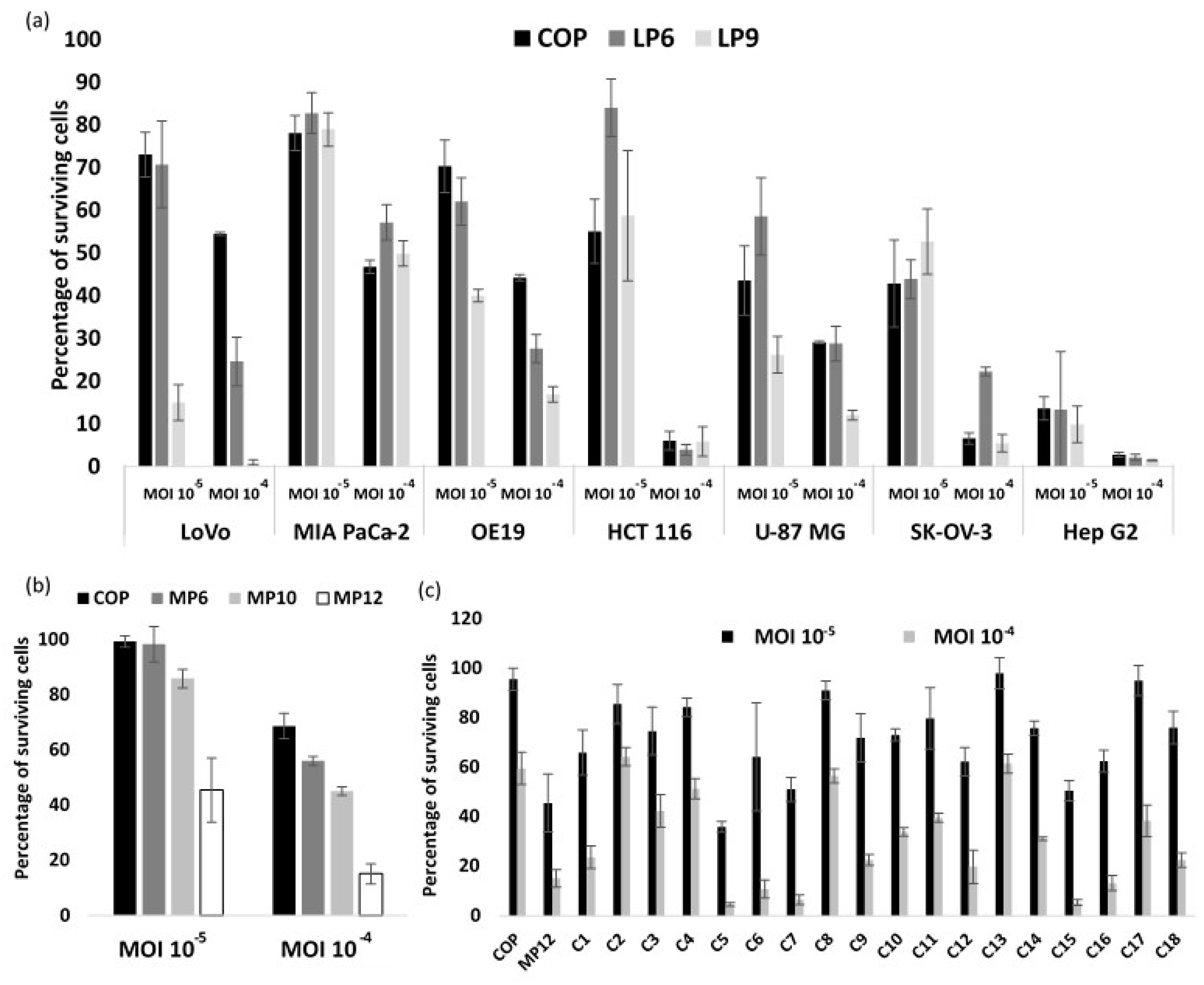
3.1. Oncolytic Activity of the Four VACV Parental Strains
3.2. VACV Shuffling: deVV5 Is a Chimeric Virus with Enhanced Oncolytic Potency In Vitro
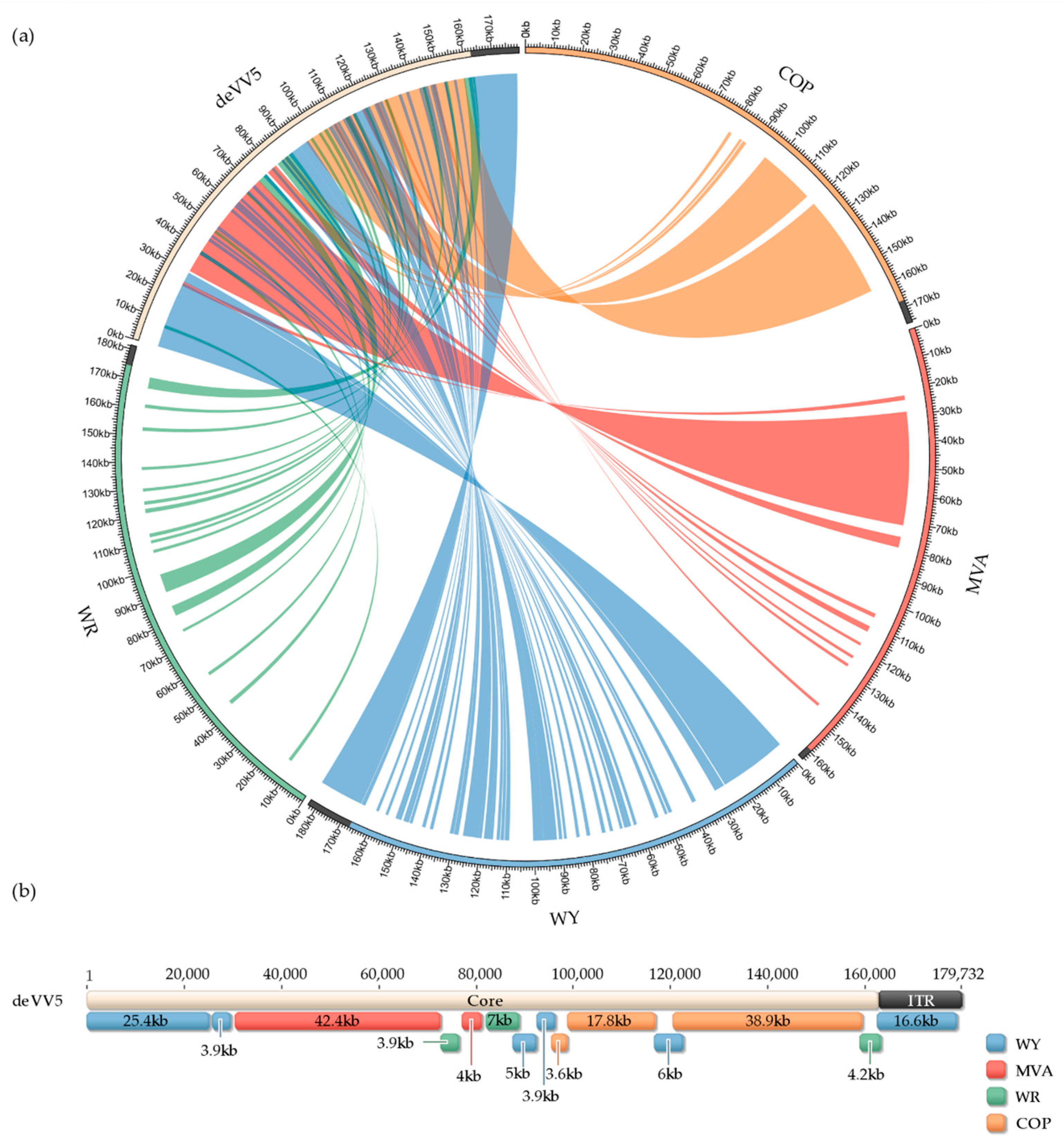
3.3. Genome Analysis
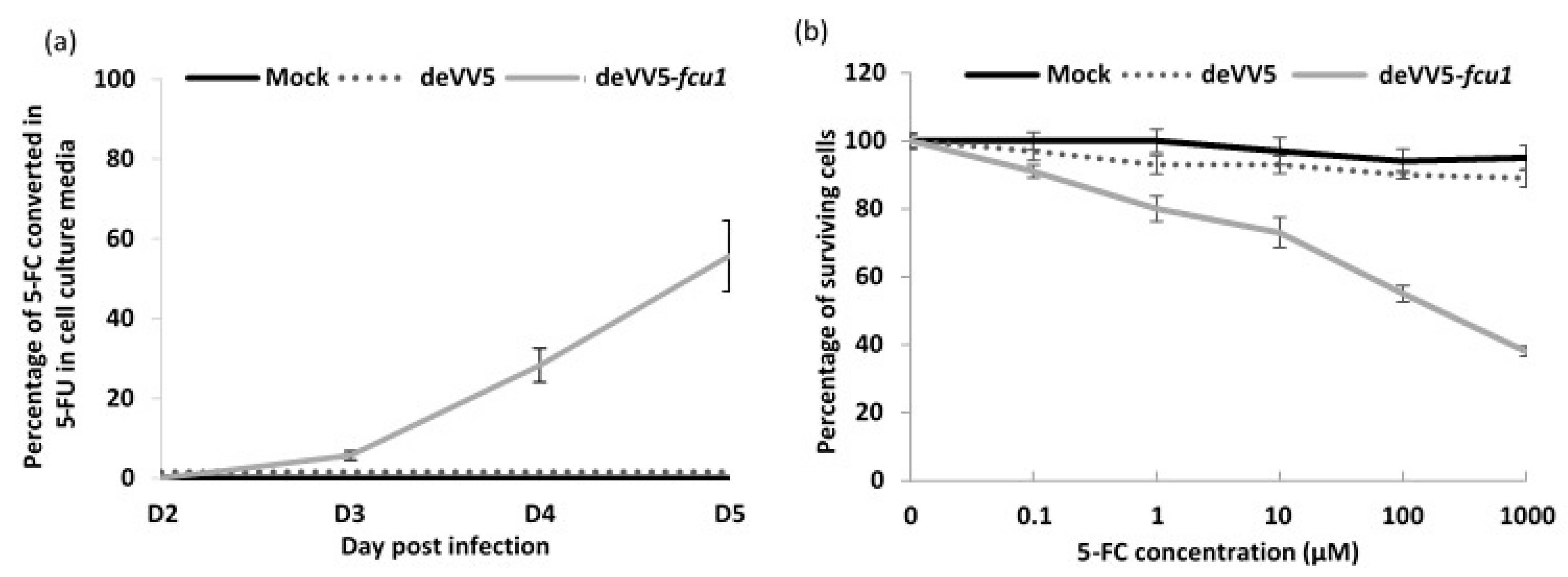
3.4. Arming of deVV5 Leads to Increased Potency
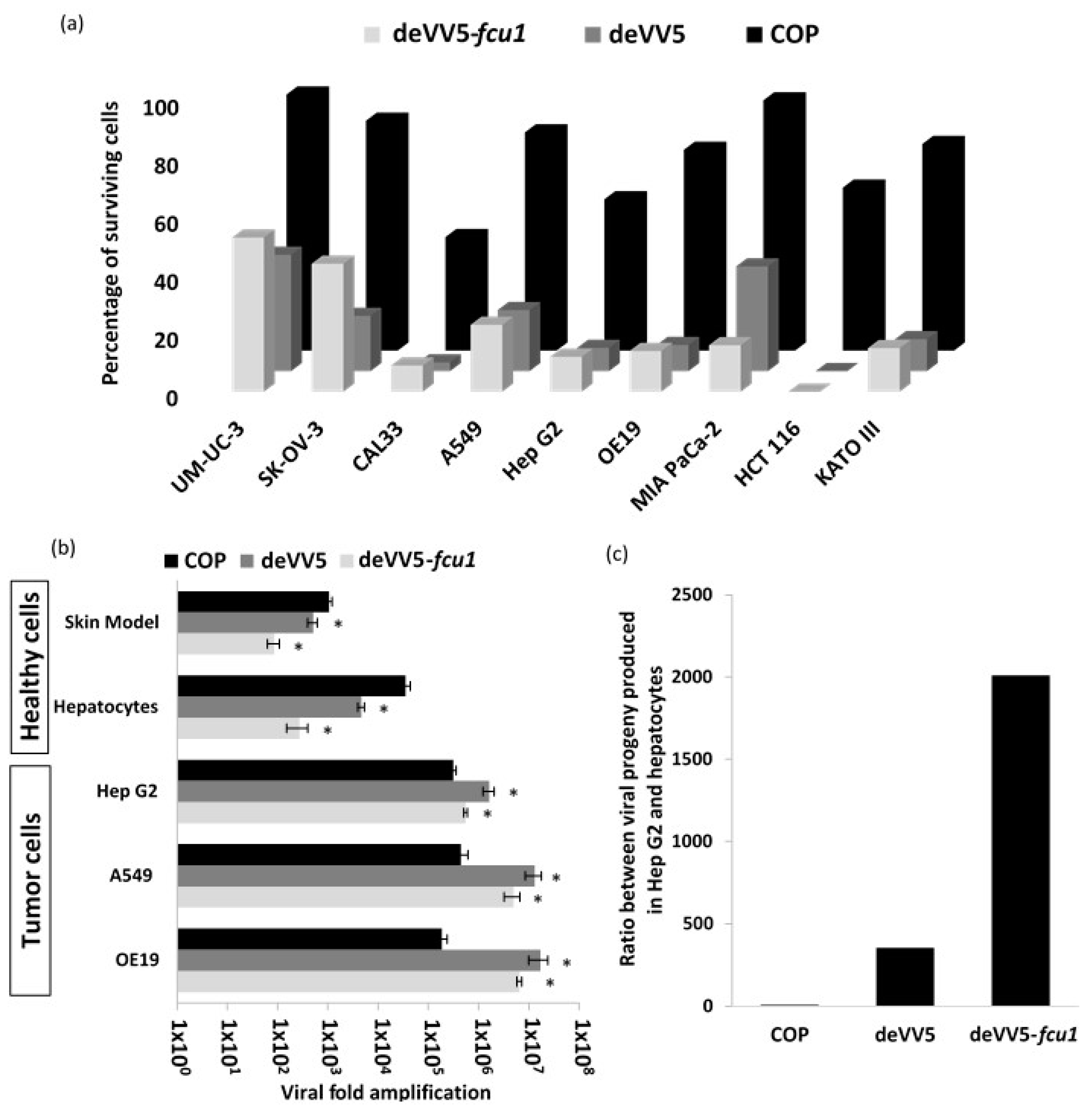
3.5. Increased Oncolytic and Replicative Efficiency of deVV5 and deVV5-fcu1
3.6. Replication of deVV5 and deVV5-fcu1 on Human Primary Cells
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Heinrich, B.; Klein, J.; Delic, M.; Goepfert, K.; Engel, V.; Geberzahn, L.; Lusky, M.; Erbs, P.; Preville, X.; Moehler, M. Immunogenicity of oncolytic vaccinia viruses JX-GFP and TG6002 in a human melanoma in vitro model: Studying immunogenic cell death, dendritic cell maturation and interaction with cytotoxic T lymphocytes. OncoTargets Ther. 2017, 10, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Yamazaki, T.; Remy, C.; Fahrner, C.; Gantzer, M.; Nourtier, V.; Préville, X.; Quemeneur, E.; Kepp, O.; Adam, J.; et al. Immune Checkpoint Blockade, Immunogenic Chemotherapy or IFN-α Blockade Boost the Local and Abscopal Effects of Oncolytic Virotherapy. Cancer Res. 2017, 77, 4146–4157. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, J.F.; de Vor, L.; Fouchier, R.A.; van den Hoogen, B.G. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D. Genetically Engineered Vaccinia Viruses as Agents for Cancer Treatment, Imaging, and Transgene Delivery. Front. Oncol. 2017, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Downs-Canner, S.; McCart, J.A.; Guo, Z.S.; Rao, U.N.; Ramalingam, L.; Thorne, S.H.; Jones, H.L.; Kalinski, P.; Wieckowski, E.; et al. First-in-man study of western reserve strain oncolytic vaccinia virus: Safety, systemic spread, and antitumor activity. Mol. Ther. 2015, 23, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, J.; Kintz, J.; Futin, N.; Findeli, A.; Cordier, P.; Schlesinger, Y.; Hoffmann, C.; Tosch, C.; Balloul, J.M.; Erbs, P. Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus. Gene Ther. 2008, 15, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J.; et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clin. Cancer Res. 2017, 23, 5696–5702. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Y.; Jeong, S.N.; Kang, D.H.; Heo, J. Evolutionary cancer-favoring engineered vaccinia virus for metastatic hepatocellular carcinoma. Oncotarget 2017, 8, 71489–71499. [Google Scholar] [CrossRef] [PubMed]
- Hruby, D.E. Vaccinia virus vectors: New strategies for producing recombinant vaccines. Clin. Microbiol. Rev. 1990, 3, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F. Genetic studies with mammalian poxviruses. II. Recombination between two strains of vaccinia virus in single HeLa cells. Virology 1959, 8, 499–507. [Google Scholar] [CrossRef]
- Woodroofe, G.M.; Fenner, F. Genetic studies with mammalian poxviruses. IV. Hybridization between several different poxviruses. Virology 1960, 12, 272–282. [Google Scholar] [CrossRef]
- Paszkowski, P.; Noyce, R.S.; Evans, D.H. Live-Cell Imaging of Vaccinia Virus Recombination. PLoS Pathog. 2016, 12, e1005824. [Google Scholar] [CrossRef] [PubMed]
- Koerber, J.T.; Maheshri, N.; Kaspar, B.K.; Schaffer, D.V. Construction of diverse adeno-associated viral libraries for directed evolution of enhanced gene delivery vehicles. Nat. Protoc. 2006, 1, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Bauzon, M.; Green, N.; Seymour, L.; Fisher, K.; Hermiston, T. OvAd1, a Novel, Potent, and Selective Chimeric Oncolytic Virus Developed for Ovarian Cancer by 3D-Directed Evolution. Mol. Ther. Oncolytics 2016, 4, 55–66. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.P.; Choi, A.H.; Kim, S.I.; Chaurasiya, S.; Lu, J.; Park, A.K.; Woo, Y.; Warner, S.G.; Fong, Y.; Chen, N.G. Novel oncolytic chimeric orthopoxvirus causes regression of pancreatic cancer xenografts and exhibits abscopal effect at a single low dose. J. Transl. Med. 2018, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sampedro, L.; Perdiguero, B.; Mejías-Pérez, E.; García-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Stickl, H.; Müller, H.K.; Danner, K.; Singer, H. The smallpox vaccination strain MVA: Marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism. Zentralbl. Bakteriol. B 1978, 167, 375–390. [Google Scholar] [PubMed]
- Erbs, P.; Findeli, A.; Kintz, J.; Cordier, P.; Hoffmann, C.; Geist, M.; Balloul, J.M. Modified vaccinia virus Ankara as a vector for suicide gene therapy. Cancer Gene Ther. 2008, 15, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Husseini, F.; Delord, J.P.; Fournel-Federico, C.; Guitton, J.; Erbs, P.; Homerin, M.; Halluard, C.; Jemming, C.; Orange, C.; Limacher, J.M.; et al. Vectorized gene therapy of liver tumors: Proof-of-concept of TG4023 (MVA-FCU1) in combination with flucytosine. Ann. Oncol. 2017, 28, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Quoix, E.; Lena, H.; Losonczy, G.; Forget, F.; Chouaid, C.; Papai, Z.; Gervais, R.; Ottensmeier, C.; Szczesna, A.; Kazarnowicz, A.; et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): Results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016, 17, 212–223. [Google Scholar] [CrossRef]
- Erbs, P.; Regulier, E.; Kintz, J.; Leroy, P.; Poitevin, Y.; Exinger, F.; Jund, R.; Mehtali, M. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000, 60, 3813–3822. [Google Scholar] [PubMed]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Ricordel, M.; Foloppe, J.; Pichon, C.; Sfrontato, N.; Antoine, D.; Tosch, C.; Cochin, S.; Cordier, P.; Quemeneur, E.; Camus-Bouclainville, C.; et al. Cowpox Virus: A New and Armed Oncolytic Poxvirus. Mol. Ther. Oncolytics 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.R.; Horvath, A.; Annels, N.E.; Pencavel, T.; Metcalf, S.; Seth, R.; Peschard, P.; Price, T.; Coffin, R.S.; Mostafid, H.; et al. Combination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancer. Br. J. Cancer 2012, 106, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Liikanen, I.; Guse, K.; Foloppe, J.; Sloniecka, M.; Diaconu, I.; Rantanen, V.; Eriksson, M.; Hakkarainen, T.; Lusky, M.; et al. Targeted chemotherapy for head and neck cancer with a chimeric oncolytic adenovirus coding for bifunctional suicide protein FCU1. Clin. Cancer Res. 2010, 16, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Quirin, C.; Rohmer, S.; Fernández-Ulibarri, I.; Behr, M.; Hesse, A.; Engelhardt, S.; Erbs, P.; Enk, A.H.; Nettelbeck, D.M. Selectivity and efficiency of late transgene expression by transcriptionally targeted oncolytic adenoviruses are dependent on the transgene insertion strategy. Hum. Gene Ther. 2011, 22, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, J.K.; Bossow, S.; Grossardt, C.; Sawall, S.; Kupsch, J.; Erbs, P.; Hassel, J.C.; von Kalle, C.; Enk, A.H.; Nettelbeck, D.M.; et al. Chemovirotherapy of malignant melanoma with a targeted and armed oncolytic measles virus. J. Investig. Dermatol. 2013, 133, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Hammer, K.; Kazcorowski, A.; Liu, L.; Behr, M.; Schemmer, P.; Herr, I.; Nettelbeck, D.M. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int. J. Cancer 2015, 137, 978–990. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | MVA | WY | WR | COP |
|---|---|---|---|---|
| A549 | 94.5 ± 3.6 | 90.2 ± 3.2 | 58.6 ± 3.7 | 47.8 ± 4.8 |
| LoVo | 98.6 ± 2.7 | 90.4 ± 5.9 | 70.8 ± 6.2 | 73.1 ± 5.2 |
| MIA PaCa-2 | 103.2 ± 4.2 | 92.8 ± 2.9 | 89.4 ± 2.4 | 78.1 ± 1.5 |
| U-87 MG | 102.8 ± 4.8 | 64.6 ± 1.6 | 41.0 ± 0.8 | 43.6 ± 0.3 |
| Hep G2 | 97.3 ± 3.9 | 66.8 ± 4.9 | 70.9 ± 9.4 | 13.6 ± 2.7 |
| HCT 116 | 99.2 ± 5.4 | 91.5 ± 3.4 | 94.6 ± 3.4 | 55.1 ± 7.5 |
| OE19 | 105.2 ± 6.2 | 78.9 ± 2.2 | 77.6 ± 3.3 | 70.3 ± 0.7 |
| SK-OV-3 | 95.4 ± 7.3 | 59.8 ± 1.8 | 99.7 ± 1.5 | 42.9 ± 1.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricordel, M.; Foloppe, J.; Antoine, D.; Findeli, A.; Kempf, J.; Cordier, P.; Gerbaud, A.; Grellier, B.; Lusky, M.; Quemeneur, E.; et al. Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency. Cancers 2018, 10, 231. https://doi.org/10.3390/cancers10070231
Ricordel M, Foloppe J, Antoine D, Findeli A, Kempf J, Cordier P, Gerbaud A, Grellier B, Lusky M, Quemeneur E, et al. Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency. Cancers. 2018; 10(7):231. https://doi.org/10.3390/cancers10070231
Chicago/Turabian StyleRicordel, Marine, Johann Foloppe, Delphine Antoine, Annie Findeli, Juliette Kempf, Pascale Cordier, Aude Gerbaud, Benoit Grellier, Monika Lusky, Eric Quemeneur, and et al. 2018. "Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency" Cancers 10, no. 7: 231. https://doi.org/10.3390/cancers10070231
APA StyleRicordel, M., Foloppe, J., Antoine, D., Findeli, A., Kempf, J., Cordier, P., Gerbaud, A., Grellier, B., Lusky, M., Quemeneur, E., & Erbs, P. (2018). Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency. Cancers, 10(7), 231. https://doi.org/10.3390/cancers10070231