Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses



Abstract
1. Introduction
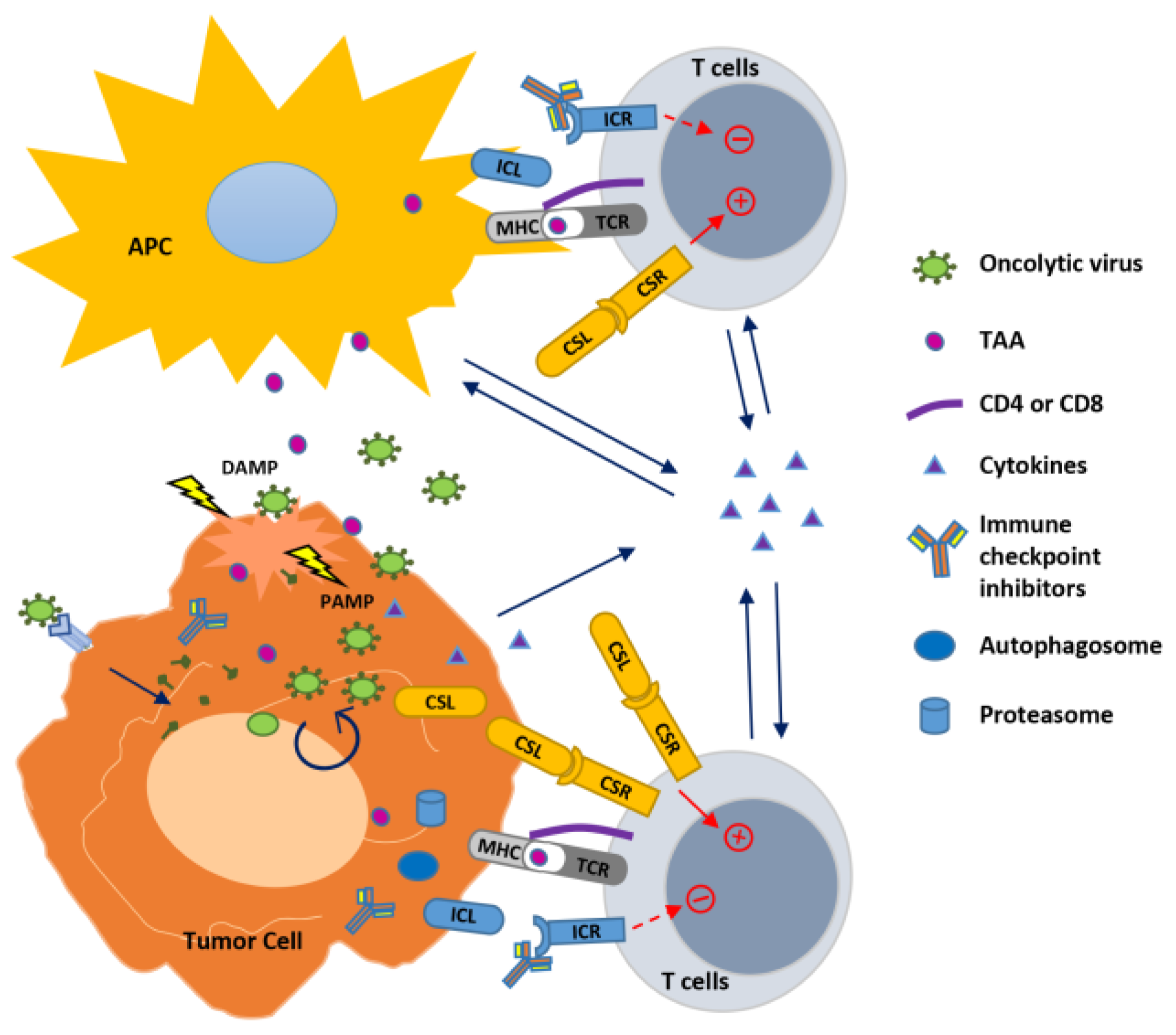
2. In Situ Autovaccination against Cancers Induced by Oncolytic Viruses
3. Strategies to Boost Oncolytic Virus-Induced Anti-Cancer Immunity
3.1. Oncolytic Viruses and Cytokines: Modulate the Innate and Adaptive Immune Response
3.2. Oncolytic Viruses and Immune Checkpoint Blockade: Release the “Brake” on T Cell Activation
3.3. Oncolytic Viruses and Immune Co-Stimulation: Hit the “Gas” for T Cell Activation
4. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Lengauer, T. Managing drug resistance in cancer: Lessons from HIV therapy. Nat. Rev. Cancer 2012, 12, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Dhupkar, P.; Gordon, N. Interleukin-2: Old and New Approaches to Enhance Immune-Therapeutic Efficacy. Adv. Exp. Med. Biol. 2017, 995, 33–51. [Google Scholar] [PubMed]
- Neri, D.; Sondel, P.M. Immunocytokines for cancer treatment: Past, present and future. Curr. Opin. Immunol. 2016, 40, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gomez-Manzano, C.; Rivera-Molina, Y.; Lang, F.F.; Conrad, C.A.; Fueyo, J. Oncolytic adenovirus research evolution: From cell-cycle checkpoints to immune checkpoints. Curr. Opin. Virol. 2015, 13, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Clise-Dwyer, K.; Ruisaard, K.E.; Fan, X.; Tian, W.; Gumin, J.; Lamfers, M.L.; Kleijn, A.; Lang, F.F.; Yung, W.K.; et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS ONE 2014, 9, e97407. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Binder, A.; Brody, J.D. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 2015, 9, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.H.; Campbell, J.S.; Pai, S.I.; Brody, J.D.; Kohrt, H.E. In-situ tumor vaccination: Bringing the fight to the tumor. Hum. Vaccines Immunother. 2015, 11, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.E.; Mossman, K.L. Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev. 2014, 25, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fueyo, J. Healing after death: Antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology 2014, 3, e947872. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Borrello, I.; Pardoll, D. GM-CSF-based cellular vaccines: A review of the clinical experience. Cytokine Growth Factor Rev. 2002, 13, 185–193. [Google Scholar] [CrossRef]
- Eklund, J.W.; Kuzel, T.M. A review of recent findings involving interleukin-2-based cancer therapy. Curr. Opin. Oncol. 2004, 16, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Portielje, J.E.; Gratama, J.W.; van Ojik, H.H.; Stoter, G.; Kruit, W.H. IL-12: A promising adjuvant for cancer vaccination. Cancer Immunol. Immunother. 2003, 52, 133–144. [Google Scholar] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. Paracrine Cytokine Adjuvants in Cancer-Immunotherapy. Annu. Rev. Immunol. 1995, 13, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimaki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T.; et al. A First in Human Phase 1 Study of CG0070, a GM-CSF Expressing Oncolytic Adenovirus, for the Treatment of Nonmuscle Invasive Bladder Cancer. J. Urol. 2012, 188, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Packiam, V.T.; Campanile, A.N.; Barocas, D.A.; Chamie, K.; Davis, R.L.; Kader, A.K.; Lamm, D.L.; Yeung, A.W.; Steinberg, G.D. A Phase II/III Trial of Cg0070, an Oncolytic Adenovirus, for Bcg-Refractory Non-Muscle-Invasive Bladder Cancer (Nmibc). J. Urol. 2016, 195, E142. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2798. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor–Encoding, Second-Generation Oncolytic Herpesvirus in Patients with Unresectable Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Rabkin, S.D.; Kojima, H.; Martuza, R.L. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum. Gene Ther. 1999, 10, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Park, B.H.; Hwang, T.; Liu, T.C.; Sze, D.Y.; Kim, J.S.; Kwon, H.C.; Oh, S.Y.; Han, S.Y.; Yoon, J.H.; Hong, S.H.; et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: A phase 1 trial. Lancet Oncol. 2008, 9, 533–542. [Google Scholar] [CrossRef]
- Lattime, E.C.; Mastrangelo, M.J. Intratumoral recombinant GM-CSF encoding vaccinia virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 2000, 7, S22. [Google Scholar]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Moon, A.; Burke, J.; Hwang, T.H.; Kirn, D.H. A Phase 2, Open-Label, Randomized Study of Pexa-Vec (JX-594) Administered by Intratumoral Injection in Patients with Unresectable Primary Hepatocellular Carcinoma. Methods Mol. Biol. 2015, 1317, 343–357. [Google Scholar] [PubMed]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Haenel, T.; Himbeck, R.; Scott, B.; Ramshaw, I.; Lake, R.A.; Harnett, G.; Phillips, P.; Morey, S.; Smith, D.; et al. Replication-restricted vaccinia as a cytokine gene therapy vector in cancer: Persistent transgene expression despite antibody generation. Cancer Gene Ther. 2000, 7, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with Irradiated Tumor-Cells Engineered to Secrete Murine Granulocyte-Macrophage Colony-Stimulating Factor Stimulates Potent, Specific, and Long-Lasting Antitumor Immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Sarkioja, M.; et al. Oncolytic Adenovirus Coding for Granulocyte Macrophage Colony-Stimulating Factor Induces Antitumoral Immunity in Cancer Patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, N.; Ge, Y.; Ennist, D.L.; Zhu, M.Z.; Mina, M.; Ganesh, S.; Reddy, P.S.; Yu, D.C. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor-armed oncolytic adenovirus for the treatment of bladder cancer. Clin. Cancer Res. 2006, 12, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEX(GM-CSF), a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- BLA125518 Talimogene Lahaparepvec FDA Briefing Document. 2015. Available online: https://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM469670.pdf (accessed on 17 May 2018).
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Reichard, K.W.; Lorence, R.M.; Cascino, C.J.; Peeples, M.E.; Walter, R.J.; Fernando, M.B.; Reyes, H.M.; Greager, J.A. Newcastle-Disease Virus Selectively Kills Human Tumor-Cells. J. Surg. Res. 1992, 52, 448–453. [Google Scholar] [CrossRef]
- Lech, P.J.; Russell, S.J. Use of attenuated paramyxoviruses for cancer therapy. Expert Rev. Vaccines 2010, 9, 1275–1302. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, W.J.; Xu, Q.; Harper, J.; Carroll, D.; Galinski, M.S.; Suzich, J.; Jin, H. Genetic Modification of Oncolytic Newcastle Disease Virus for Cancer Therapy. J. Virol. 2016, 90, 5343–5352. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.B.; Chamberlain, R.S.; Bronte, V.; Carroll, M.W.; Irvine, K.R.; Moss, B.; Rosenberg, S.A.; Restifo, N.P. IL-12 is an effective adjuvant to recombinant vaccinia virus-based tumor vaccines—Enhancement by simultaneous B7-1 expression. J. Immunol. 1996, 156, 3357–3365. [Google Scholar] [PubMed]
- Strengell, M.; Lehtonen, A.; Matikainen, S.; Julkunen, I. IL-21 enhances SOCS gene expression and inhibits LPS-induced cytokine production in human monocyte-derived dendritic cells. J. Leukoc. Biol. 2006, 79, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Carr, A.L.; Donald, E.J.; Skitzki, J.J.; Okuyama, R.; Stoolman, L.M.; Chang, A.E. Synergistic effects of IL-12 and IL-18 in skewing tumor-reactive T-cell responses towards a type 1 pattern. Cancer Res. 2005, 65, 1063–1070. [Google Scholar] [PubMed]
- Kaufman, H.L.; Flanagan, K.; Lee, C.S.D.; Perretta, D.J.; Horig, H. Insertion of interleukin-2 (IL-2) and interleukin-12 (IL-12) genes into vaccinia virus results in effective anti-tumor responses without toxicity. Vaccine 2002, 20, 1862–1869. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Nagai, N.; Ohkusu, K.; Ueda, H.; Okamura, H.; Nakanishi, K. LPS-stimulated SJL macrophages produce IL-12 and IL-18 that inhibit IgE production in vitro by induction of IFN-gamma production from CD3(int)IL-2R beta(+) T cells. J. Immunol. 1998, 161, 1483–1492. [Google Scholar] [PubMed]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-gamma production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar] [PubMed]
- Addison, C.L.; Bramson, J.L.; Hitt, M.M.; Muller, W.J.; Gauldie, J.; Graham, F.L. Intratumoral coinjection of adenoviral vectors expressing IL-2 and IL-12 results in enhanced frequency of regression of injected and untreated distal tumors. Gene Ther. 1998, 5, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Lee, J.S.; Zhang, S.N.; Park, J.; Sonn, C.H.; Lee, K.M.; Yun, C.O. Oncolytic adenovirus co-expressing IL-12 and IL-18 improves tumor-specific immunity via differentiation of T cells expressing IL-12R beta(2) or IL-18R alpha. Gene Ther. 2011, 18, 898–942. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, J.H.; Choi, K.J.; Choi, I.K.; Kim, H.; Cho, S.; Cho, B.C.; Yun, C.O. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 2006, 12, 5859–5868. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Zhang, S.N.; Choi, K.J.; Choi, I.K.; Kim, J.H.; Lee, M.G.; Kim, H.; Yun, C.O. Therapeutic and Tumor-specific Immunity Induced by Combination of Dendritic Cells and Oncolytic Adenovirus Expressing IL-12 and 4-1BBL. Mol. Ther. 2010, 18, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Li, Y.; Oh, E.; Kim, J.; Yun, C.O. Oncolytic Adenovirus Expressing IL-23 and p35 Elicits IFN-gamma- and TNF-alpha-Co-Producing T Cell-Mediated Antitumor Immunity. PLoS ONE 2013, 8, e67512. [Google Scholar]
- Freytag, S.O.; Barton, K.N.; Zhang, Y. Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Ther. 2013, 20, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Stafman, L.L.; Garner, E.F.; Mruthyunjayappa, S.; Stewart, J.E.; Friedman, G.K.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; Beierle, E.A. Effect of Repeat Dosing of Engineered Oncolytic Herpes Simplex Virus on Preclinical Models of Rhabdomyosarcoma. Transl. Oncol. 2016, 9, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.D.; Meza-Perez, S.; Bevis, K.S.; Randall, T.D.; Gillespie, G.Y.; Langford, C.; Alvarez, R.D. IL-12 Expressing oncolytic herpes simplex virus promotes anti-tumor activity and immunologic control of metastatic ovarian cancer in mice. J. Ovarian Res. 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Gillory, L.A.; Megison, M.L.; Stewart, J.E.; Mroczek-Musulman, E.; Nabers, H.C.; Waters, A.M.; Kelly, V.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PLoS ONE 2013, 8, e77753. [Google Scholar] [CrossRef] [PubMed]
- Jackaman, C.; Nelson, D.J. Cytokine-armed vaccinia virus infects the mesothelioma tumor microenvironment to overcome immune tolerance and mediate tumor resolution. Cancer Gene Ther. 2010, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Wanna, G.B.; Choi, B.; Aguila, D.; Ebert, O.; Genden, E.M.; Woo, S.L. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope 2007, 117, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Janke, M.; Fournier, P.; Schirrmacher, V. Recombinant Newcastle disease virus expressing human interleukin-2 serves as a potential candidate for tumor therapy. Virus Res. 2008, 136, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Janke, M.; Peeters, B.; Zhao, H.; De Leeuw, O.; Moorman, R.; Arnold, A.; Ziouta, Y.; Fournier, P.; Schirrmacher, V. Activation of human T cells by a tumor vaccine infected with recombinant Newcastle disease virus producing IL-2. Int. J. Oncol. 2008, 33, 823–832. [Google Scholar] [PubMed]
- Vigil, A.; Park, M.S.; Martinez, O.; Chua, M.A.; Xiao, S.; Cros, J.F.; Martinez-Sobrido, L.; Woo, S.L.C.; Garcia-Sastre, A. Use of reverse genetics to enhance the oncolytic properties of newcastle disease virus. Cancer Res. 2007, 67, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Z.; He, J.J.; An, Y.; Wang, X.; Liu, Y.Y.; Yan, S.J.; Ye, X.L.; Qi, J.Y.; Zhu, S.L.; Yu, Q.Z.; et al. Recombinant Newcastle disease virus (NDV/Anh-IL-2) expressing human IL-2 as a potential candidate for suppresses growth of hepatoma therapy. J. Pharmacol. Sci. 2016, 132, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.P.; Tian, G.Y.; Liu, Y.Y.; He, J.J.; Gao, X.Y.; Yu, Y.H.; Liu, X.; Zhang, X.; Sun, T.; Liu, S.Q.; et al. Recombinant Newcastle Disease Virus Encoding IL-12 and/or IL-2 as Potential Candidate for Hepatoma Carcinoma Therapy. Technol. Cancer Res. Treat. 2016, 15, Np83–Np94. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, T.; Kato, T.; Igarashi, O.; Inoue, T.; Nariuchi, H., 2nd. Signal Activity of Il-12 on the Proliferation and Il-2r Expression of T-Helper Cell-1 Clone. J. Immunol. 1994, 152, 4919–4928. [Google Scholar] [PubMed]
- Frucht, D.M.; Fukao, T.; Bogdan, C.; Schindler, H.; O’Shea, J.J.; Koyasu, S. IFN-gamma-production by antigen-presenting cells: Mechanisms emerge. Trends Immunol. 2001, 22, 556–560. [Google Scholar] [CrossRef]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Nimal, S.; McCormick, A.L.; Thomas, M.S.; Heath, A.W. An interferon gamma-gp120 fusion delivered as a DNA vaccine induces enhanced priming. Vaccine 2005, 23, 3984–3990. [Google Scholar] [CrossRef] [PubMed]
- Heath, A.W.; Devey, M.E.; Brown, I.N.; Richards, C.E.; Playfair, J.H.L. Interferon-Gamma as an Adjuvant in Immunocompromised Mice. Immunology 1989, 67, 520–524. [Google Scholar] [PubMed]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Falls, T.; Twumasi-Boateng, K.; St-Germain, L.E.; Marguerie, M.; Garcia, V.; Selman, M.; Jennings, V.A.; Pettigrew, J.; et al. Oncolytic vesicular stomatitis virus expressing interferon-gamma has enhanced therapeutic activity. Mol. Ther. Oncolytics 2016, 3, 16001. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpel, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H. Costimulation of Lymphocytes-T—The Role of Cd28, Ctla-4, and B7/Bb1 in Interleukin-2 Production and Immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- Schneider, H.; Mandelbrot, D.A.; Greenwald, R.J.; Ng, F.; Lechler, R.; Sharpe, A.H.; Rudd, C.E. Cutting edge: CTLA-4 (CD152) differentially regulates mitogen-activated protein kinases (extracellular signal-regulated kinase and c-Jun N-terminal kinase) in CD4(+) T cells from receptor/ligand-deficient mice. J. Immunol. 2002, 169, 3475–3479. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hodi, F.S.; Weber, J.S.; Allison, J.P.; Urba, W.J.; Robert, C.; O’Day, S.J.; Hoos, A.; Humphrey, R.; Berman, D.M.; et al. Development of ipilimumab: A novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. N. Y. Acad. Sci. 2013, 1291, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Neyns, B.; Linette, G. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Thompson, J.A.; Berman, D.; Siegal, J.; Minor, D.; Amin, A.; Ron, I.; Ridolfi, R.; Assi, H.; Hamid, O.; Weber, J. Effect of prior treatment status on the efficacy and safety of ipilimumab monotherapy in treatment-naive and previously treated patients with advanced melanoma. J. Clin. Oncol. 2008, 26, 4134–4140. [Google Scholar] [CrossRef]
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Barmada, M.A.; Bergman, I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009, 16, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.J.; Sampath, P.; Hou, W.Z.; Thorne, S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Cockle, J.V.; Rajani, K.; Zaidi, S.; Kottke, T.; Thompson, J.; Diaz, R.M.; Shim, K.; Peterson, T.; Parney, I.F.; Short, S.; et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro-Oncology 2016, 18, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.; Chemova, T.; Iwai, Y.; Malenkovich, N.; Long, A.; Bourque, K.; Boussiotis, V.; Nishimura, H.; Honjo, T.; et al. PD-L2, a novel B7 homologue, is a second ligand for PD-1 and inhibits T cell activation. Faseb J. 2001, 15, A345. [Google Scholar]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun. Rev. 2013, 12, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.D.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.F.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chung, Y.; Bishop, C.; Daugherty, B.; Chute, H.; Hoist, P.; Kurahara, C.; Lott, F.; Sun, N.; Welcher, A.A.; et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl. Acad. Sci. USA 2006, 103, 11695–11700. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol. Ther. 2016, 24, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Vile, R.G.; Melcher, A.; Chester, J.; Pandha, H.S. Clinical trials with oncolytic reovirus: Moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev. 2010, 21, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.; McManus, D.; Lefebvre, C.; Hu, K.; Falls, T.; Atkins, H.; Bell, J.C.; McCart, J.A.; Mahoney, D.; Stojdl, D.F. Identification of Genetically Modified Maraba Virus as an Oncolytic Rhabdovirus. Mol. Ther. 2010, 18, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.G.; Zhang, L.; Bridle, B.W.; Stephenson, K.B.; Resseguier, J.; Hanson, S.; Chen, L.; Kazdhan, N.; Bramson, J.L.; Stojdl, D.F.; et al. Maraba virus as a potent oncolytic vaccine vector. Mol. Ther. 2014, 22, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.S.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Loskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, E.R.; Redmond, W.L. A Review of Current Biologics Targeting OX40, 4–1BB, CD27, and GITR. Am. J. Hematol. Oncol. 2017, 13, 4–15. [Google Scholar]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Howland, L.J.; Flynn, J.K.; West, A.C.; Devaud, C.; Duong, C.P.; Stewart, T.J.; Westwood, J.A.; Guo, Z.S.; Bartlett, D.L.; et al. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res. 2012, 72, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim-Schulze, S.; Kim, D.W.; Kaufman, H.L. Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4-1BB ligand. Cancer Res. 2009, 69, 8516–8525. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Milenova, I.; Wenthe, J.; Stahle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin. Cancer Res. 2017, 23, 5846–5857. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Balasubramanian, S.; Liu, W.; Chu, X.; Wang, H.; Taparowsky, E.J.; Fu, Y.X.; Choi, Y.; Walsh, M.C.; Li, X.C. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat. Immunol. 2012, 13, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, J.; Tanaka, J.; Kim, J.A.; Rothchild, K.; Weinberg, A.; Shu, S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000, 60, 5514–5521. [Google Scholar] [PubMed]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liakou, C.I.; Kamat, A.; Pettaway, C.; Ward, J.F.; Tang, D.N.; Sun, J.; Jungbluth, A.A.; Troncoso, P.; Logothetis, C.; et al. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 2729–2734. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Quezada, S.A.; Allison, J.P. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr. Opin. Immunol. 2010, 22, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kroenke, M.A.; Eto, D.; Locci, M.; Cho, M.; Davidson, T.; Haddad, E.K.; Crotty, S. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J. Immunol. 2012, 188, 3734–3744. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W. Oncolytic Virotherapy: A Contest between Apples and Oranges. Mol. Ther. 2017, 25, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Karube, H.; Aruga, A. A comparative Safety profile assessment of oncolytic virus therapy based on clinical Trials. Ther. Innov. Regul. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.; Braidwood, L.; Brown, S.M. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: Redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther. 2008, 15, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Krimmel, J.; Zhang, Z.; Hu, Z.; Seth, P. Systemic delivery of a novel liver-detargeted oncolytic adenovirus causes reduced liver toxicity but maintains the antitumor response in a breast cancer bone metastasis model. Hum. Gene. Ther. 2011, 22, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, Z.; Yang, Y.; Hu, Z.; Wang, C.H.; Morgan, M.; Wu, Y.; Hutten, R.; Xiao, X.; Stock, S.; et al. Ad5/48 hexon oncolytic virus expressing sTGFbetaRIIFc produces reduced hepatic and systemic toxicities and inhibits prostate cancer bone metastases. Mol. Ther. 2014, 22, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.S.; Lemoine, N.R.; Wang, Y. Systemic delivery of oncolytic viruses: Hopes and hurdles. Adv. Virol. 2012, 2012, 805629. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hall, R.R.; Lesniak, M.S.; Ahmed, A.U. Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses 2015, 7, 6200–6217. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cytokine | Virus | Modification in Viral Genome | Tested Disease | Route of Administration | Clinical Status |
|---|---|---|---|---|---|
| GM-CSF | Human adenovirus 5 (ONCOS-102) | 24-bp deletion in E1A; modified fiber with a serotype 3 knob | solid tumors refractory to available treatments | Intratumoral and intravenous | Phase I [36] |
| Human adenovirus 5 (CG0070) | E2F-1 promoter /E1A gene, human GM-CSF insertion | Non-muscle invasive bladder cancer after BCG failure | Bladder instillation | Phase II/III [37,38] | |
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Unresected stage IIIB/C to IV melanoma with various metastasis | Intratumoral | Approved in the USA and Europe [39,40,41,42] | |
| Vaccinia virus (JX-594) | Thymidine kinase, human GM-CSF, lacZ insertion | Various cancers in adult and pediatric patients | Intravenous | Phase III trial [43,44,45,46,47] | |
| IL-12 | Human adenovirus 5 (Ad5-yCD/mutTKSR39rep-hIL12) | IL-12, yeast cytosine deaminase (CD), TKSR39 (thymidine kinase mutant) insertions | Non-small cell lung carcinoma (NSCLC) Prostate cancer | Intratumoral | Phase I (NSCLC) Phase II (prostate cancer) |
| HSV-1 (M032) | Deletion of ICP34.5, IL-12 insertion | Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, Gliosarcoma | Intracerebral | Phase I [48] | |
| IL-2 | Vaccinia virus (VV-IL-2) | Deletion of thymidine kinase, insertion of IL-2 | malignant mesothelioma | Intratumoral | Small pilot study with six patients [49] |
| Antibodies | Virus | Modification in Viral Genome | Tested Disease | Route of Administration | Clinical Status |
|---|---|---|---|---|---|
| Anti-CTLA-4 | VSV | Breast cancer | Intraperitoneally | Pre-clinical [102] | |
| NDV | Colon carcinoma and melanoma | Intratumoral dose of OV followed by intraperitoneal ICIs | Pre-clinical [16] | ||
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Malignant melanoma | Intratumoral dose of OV followed by intravenous ICIs | Phase II [118] | |
| Human adenovirus 5 (Ad5/3-Delta24aCTLA4) | 24-bp deletion in E1A; modified fiber with a serotype 3 knob; anti-CTLA-4 mAb insertion | Advanced solid tumors | Subcutaneous dose of OV followed by intraperitoneal ICIs | Pre-clinical [119] | |
| Anti-CTLA-4 + anti-CD25 | Vaccinia virus | Renal adenocarcinoma | Intravenous dose of OV followed by intraperitoneal ICIs | Pre-clinical [103] | |
| Anti-CTLA-4 + anti-PD-1 | VSV (VSV-HIF-2a, VSV-Sox-10, VSV-c-Myc) | c-Myc, HIF-2α, and Sox-10 insertion | Glioma | Intravenous dose of OV followed by intracranial ICIs | Pre-clinical [104] |
| Maraba virus MG1 | Triple-negative breast cancer | Intratumoral or intravenous dose of OV followed by intraperitoneal ICIs | Pre-clinical [117] | ||
| Anti-CTLA-4 or anti-PD-L1 | Measles virus (MV-aCTLA-4, MV-aPD-L1) | Anti-CTLA-4 p4F10-γ1 or anti-PD-L1 mAb insertion | Melanoma | Intratumoral injection of OV | Pre-clinical [120] |
| Anti-PD-1 | Reovirus | Melanoma | Intratumoral dose of OV followed by systemic ICIs | Pre-clinical [113] | |
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Unresectable Stage IIIB to IVM1c Melanoma | Intratumoral dose of OV followed by intravenous ICIs | Phase Ib/3 | |
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Head and neck squamous cell carcinoma | Intratumoral dose of OV followed by intravenous ICIs | Phase Ib/3 | |
| Human adenovirus 5 (DNX-2401) | 24-bp deletion in E1A, RGD-4C motif insertion in fiber | Recurrent glioblastoma or gliosarcoma | Intratumoral dose of OV followed by intravenous ICIs | Phase II | |
| Human adenovirus 5 (ONCOS-102) | Insertion of human GM-CSF | Advanced or Unresectable Melanoma | Intratumoral dose of OV followed by intravenous ICIs | Phase I | |
| Maraba virus (MG1-MAGEA3) | Insertion of human melanoma antigen A3 (MAGE-A3) | Non-Small Cell Lung Cancer | Intratumoral dose of OV followed by intravenous ICIs | Phase I/II | |
| Human adenovirus 5 (ADV/HSV-tk) | Insertion of herpes simplex virus thymidine kinase (HSV-tk) | Metastatic triple negative breast cancer and metastatic non-small cell lung cancer | Intratumoral dose of OV followed by intravenous ICIs | Phase II |
| Virus | Modification in Viral Genome | Tested Disease | Route of Administration | Clinical Status |
|---|---|---|---|---|
| Vaccinia virus | Sarcoma and breast cancer | OV: intratumoral; anti-4-1BB: intraperitoneal | Pre-clinical [124] | |
| Vaccinia virus (rV-4-1BBL) | Insertion of 4-1BBL | Melanoma | Intratumoral | Pre-clinical [125] |
| Human adenovirus 5 (Ad-ΔB7/IL-12/4-1BBL) | Insertion of IL-12 and 4-1BB | Melanoma | Intratumoral | Pre-clinical [72] |
| Human adenovirus 5 (LOAd703) | Co-insertion of CD40L and 4-1BBL | Human pancreatic xenografts in nude mice | Peritumoral injection | Pre-clinical [126] |
| Human adenovirus 5 (Delta-24-RGDOX) | 24-bp deletion in E1A, RGD-4C motif insertion in fiber, insertion of OX40L | Glioma | OV: intratumoral; anti-PD-L1: intratumoral | Pre-clinical [124] |
| NDV (NDV-ICOSL) | Insertion of ICOSL | Melanoma | OV: Intratumoral; anti-CTLA-4: Intraperitoneal | Pre-clinical [127] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.; Avci, N.G.; Shin, D.H.; Martinez-Velez, N.; Jiang, H. Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses. Cancers 2018, 10, 171. https://doi.org/10.3390/cancers10060171
Nguyen T, Avci NG, Shin DH, Martinez-Velez N, Jiang H. Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses. Cancers. 2018; 10(6):171. https://doi.org/10.3390/cancers10060171
Chicago/Turabian StyleNguyen, Teresa, Naze G. Avci, Dong Ho Shin, Naiara Martinez-Velez, and Hong Jiang. 2018. "Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses" Cancers 10, no. 6: 171. https://doi.org/10.3390/cancers10060171
APA StyleNguyen, T., Avci, N. G., Shin, D. H., Martinez-Velez, N., & Jiang, H. (2018). Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses. Cancers, 10(6), 171. https://doi.org/10.3390/cancers10060171