The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Inflammation Is a Clinical Concern for Myeloid Malignancies
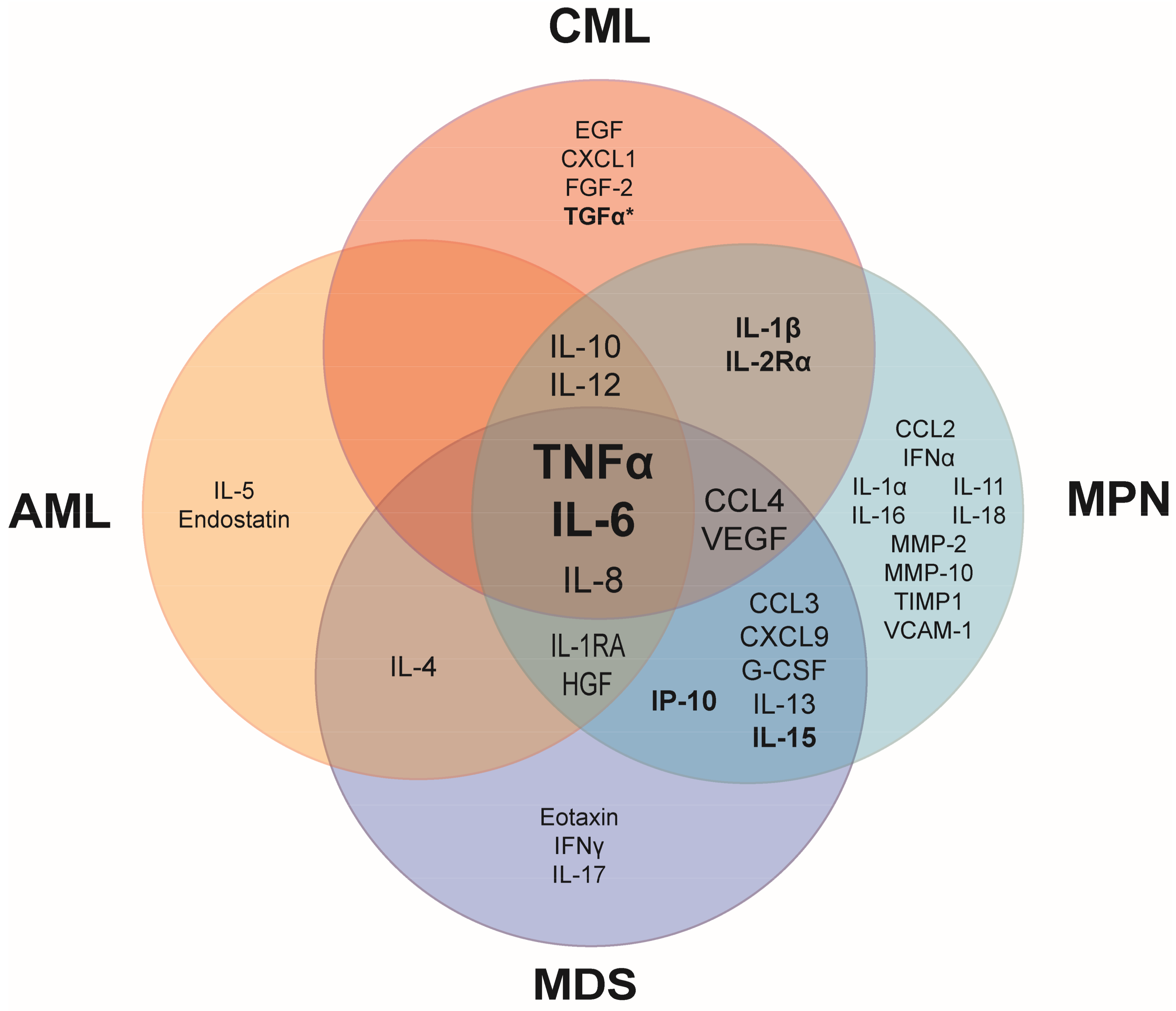
2.1. Chronic Inflammation Is a Shared Characteristic in Myeloid Malignancies
2.2. Autoinflammatory and Autoimmune Diseases in Hematological Malignancies
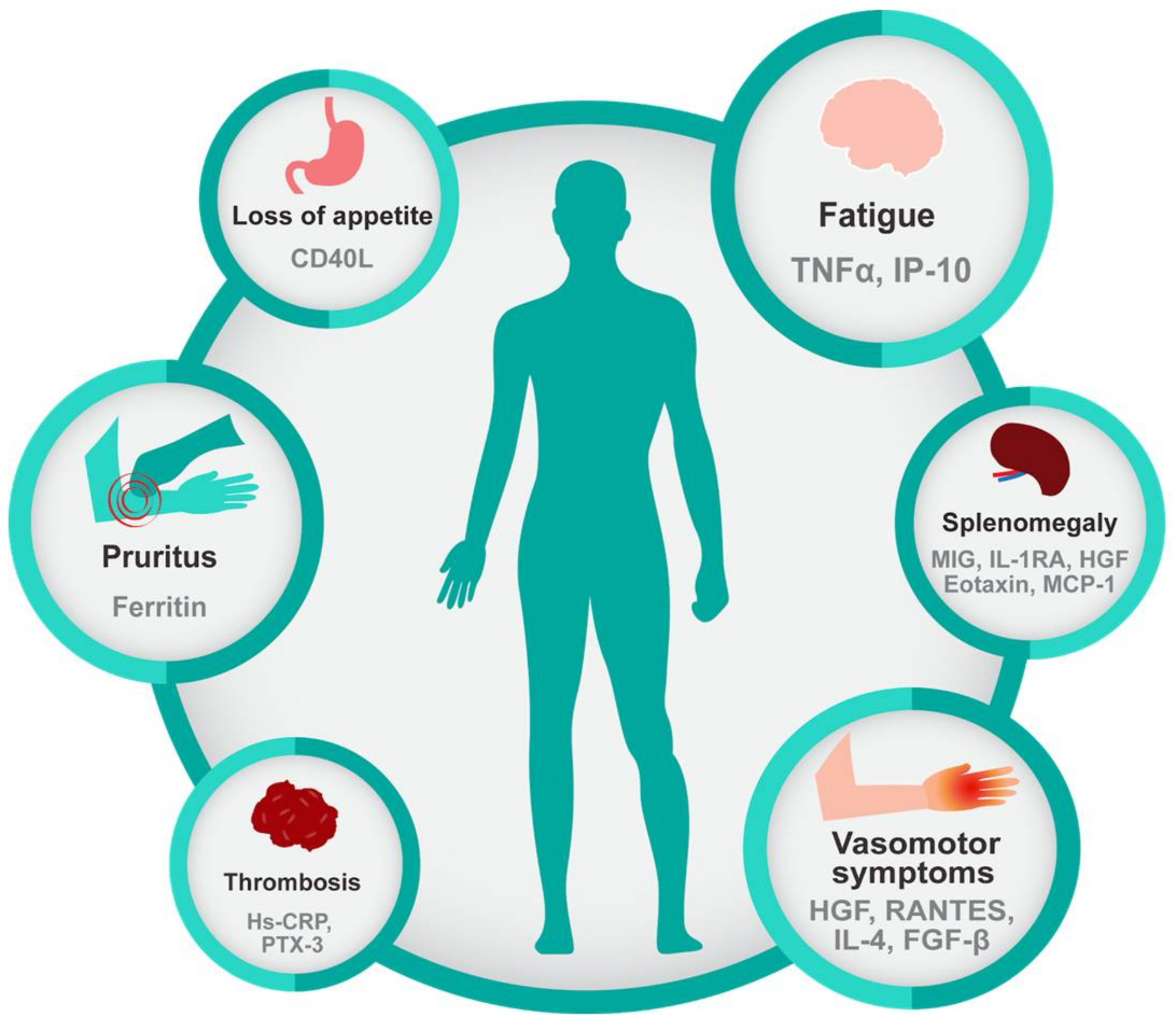
2.3. Inflammation Exacerbates Symptom Burden in Myeloid Malignancies
2.4. Inflammatory Diseases and Cancers Share Therapies
3. Inflammation Drives the Onset/Progression of Myeloid Malignancies
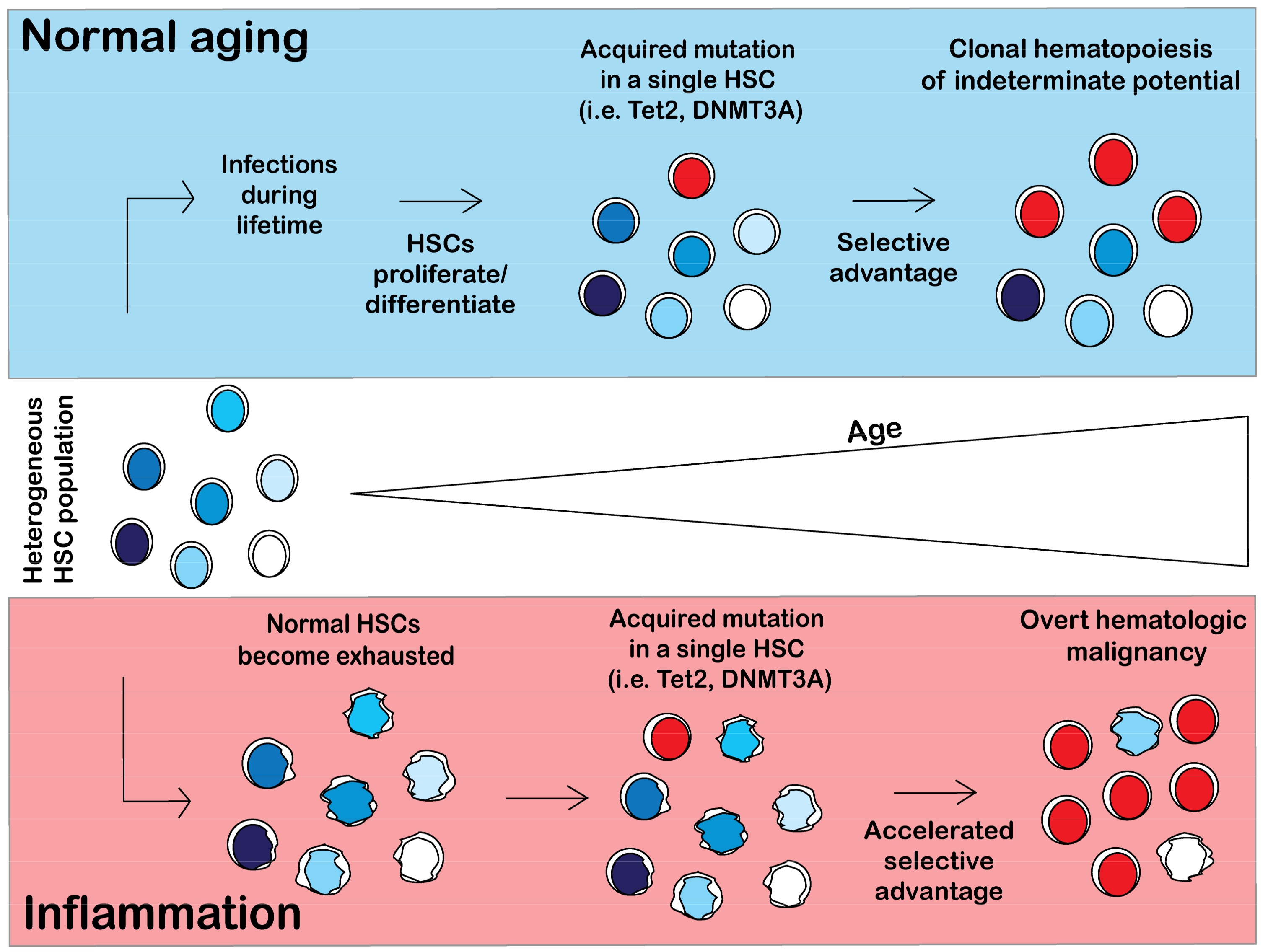
3.1. Inflammation Promotes Clonal Hematopoiesis
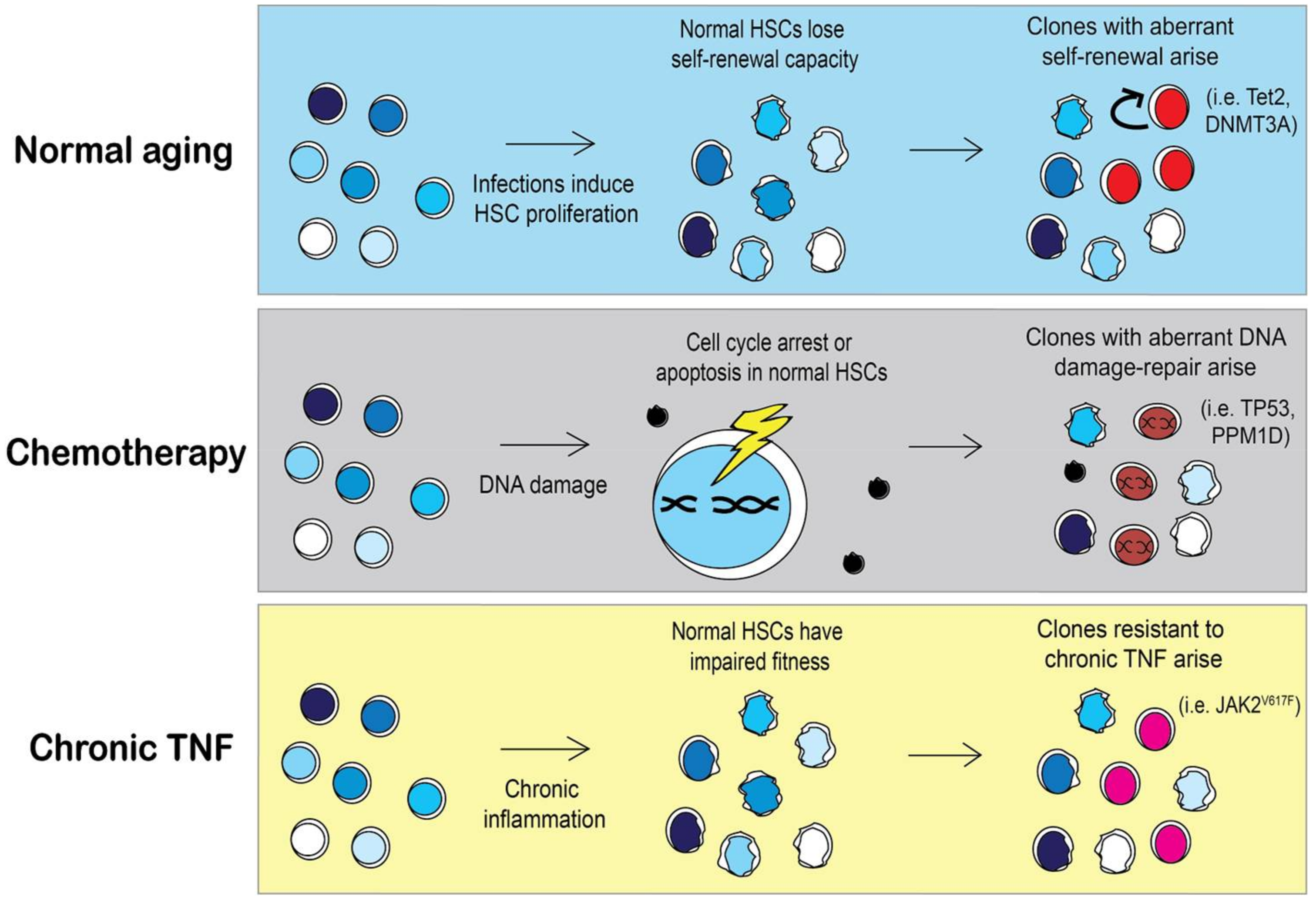
3.2. Aging of Hematopoietic Stem Cells Is Mediated by Inflammation
3.3. Selective Pressures Will Influence the Outgrowth of Specific Clones
3.3.1. Chemotherapy and Other DNA Damaging Agents as a Selective Pressure
3.3.2. Lifestyle Pressures That Influence Inflammation and Hematopoiesis
3.4. The Bone Marrow Microenvironment and Inflammation
4. Future Prospects in the Clinic and on the Bench
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Bergua, J.M.; Campos, C.; Gayoso, I.; Arcos, M.J.; Banas, H.; Morgado, S.; Casado, J.G.; Solana, R.; Tarazona, R. Cytokine profiles in acute myeloid leukemia patients at diagnosis: Survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine 2013, 61, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Nievergall, E.; Reynolds, J.; Kok, C.H.; Watkins, D.B.; Biondo, M.; Busfield, S.J.; Vairo, G.; Fuller, K.; Erber, W.N.; Sadras, T.; et al. TGF-alpha and IL-6 plasma levels selectively identify CML patients who fail to achieve an early molecular response or progress in the first year of therapy. Leukemia 2016, 30, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene 2011, 30, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Scheinberg, P.; Wu, C.O.; Samsel, L.; Nunez, O.; Prince, C.; Ganetzky, R.D.; McCoy, J.P., Jr.; Maciejewski, J.P.; Young, N.S. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica 2011, 96, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Fung, F.Y.; Li, M.; Breunis, H.; Timilshina, N.; Minden, M.D.; Alibhai, S.M. Correlation between cytokine levels and changes in fatigue and quality of life in patients with acute myeloid leukemia. Leuk. Res. 2013, 37, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.A.; Albitar, M.; Estey, E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005, 104, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Gao, X.; Wang, J.; Sun, Y.; Shekhar, S. Cytokines in autoimmune disease. Mediat. Inflamm. 2017, 2017, 5089815. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom Smedby, K.; Vajdic, C.M.; Falster, M.; Engels, E.A.; Martinez-Maza, O.; Turner, J.; Hjalgrim, H.; Vineis, P.; Seniori Costantini, A.; Bracci, P.M.; et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: A pooled analysis within the InterLymph Consortium. Blood 2008, 111, 4029–4038. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; Pfeiffer, R.M.; Landgren, O.; Gadalla, S.; Berndt, S.I.; Engels, E.A. Risks of myeloid malignancies in patients with autoimmune conditions. Br. J. Cancer 2009, 100, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, S.Y.; Bjorkholm, M.; Hultcrantz, M.; Derolf, A.R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Bjorn, M.E. MPNs as inflammatory diseases: The evidence, consequences, and perspectives. Mediat. Inflamm. 2015, 2015, 102476. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, S.Y.; Landgren, O.; Samuelsson, J.; Björkholm, M.; Goldin, L.R. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica 2010, 95, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.L.; Hasselbalch, H.C. Antecedent cardiovascular disease and autoimmunity in philadelphia-negative chronic myeloproliferative neoplasms. Leuk. Res. 2016, 41, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Hermouet, S.; Vilaine, M. The JAK2 46/1 haplotype: A marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica 2011, 96, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, E.; Lascu, E.; Wang, Y.L.; Gjoni, S.; Cross, N.C.; Baumann, R.; Tam, K.; Scherl, E.; Longman, R.S.; Silver, R.T. The JAK2V617F mutation seen in myeloproliferative neoplasms (MPNs) occurs in patients with inflammatory bowel disease: Implications of a pilot study. Int. J. Clin. Med. 2013, 4, 10–15. [Google Scholar] [CrossRef]
- Newton-Cheh, C.; Johnson, T.; Gateva, V.; Tobin, M.D.; Bochud, M.; Coin, L.; Najjar, S.S.; Zhao, J.H.; Heath, S.C.; Eyheramendy, S.; et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009, 41, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, F.; De Martinis, M.; Ginaldi, L. An update on autoinflammatory diseases. Curr. Med. Chem. 2014, 21, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Brenner, R.; Ben-Zvi, I.; Shinar, Y.; Liphshitz, I.; Silverman, B.; Peled, N.; Levy, C.; Ben-Chetrit, E.; Livneh, A.; Kivity, S. Familial mediterranean fever and incidence of cancer: An analysis of 8534 israeli patients with 258,803 person-years. Arthritis Rheumatol. 2018, 70, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Pasquet, F.; Pavic, M.; Ninet, J.; Hot, A. [autoimmune diseases and cancers. Part i: Cancers complicating autoimmune diseases and their treatment]. Rev. Med. Intern. 2014, 35, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Vergara-Lluri, M.E.; Piatek, C.I.; Pullarkat, V.; Siddiqi, I.N.; O’Connell, C.; Feinstein, D.I.; Brynes, R.K. Autoimmune myelofibrosis: An update on morphologic features in 29 cases and review of the literature. Hum. Pathol. 2014, 45, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R. The role of cytokines in cancer-related fatigue. Cancer 2001, 92, 1684–1688. [Google Scholar] [CrossRef]
- Myers, J.S. Proinflammatory cytokines and sickness behavior: Implications for depression and cancer-related symptoms. Oncol. Nurs. Forum 2008, 35, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.L.; Dueck, A.C.; Scherber, R.M.; Mesa, R.A. Impact of inflammation on myeloproliferative neoplasm symptom development. Mediat. Inflamm. 2015, 2015, 284706. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to mpn pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Manitta, V.; Zordan, R.; Cole-Sinclair, M.; Nandurkar, H.; Philip, J. The symptom burden of patients with hematological malignancy: A cross-sectional observational study. J. Pain Symptom Manag. 2011, 42, 432–442. [Google Scholar] [CrossRef] [PubMed]
- McFarland, D.C.; Shaffer, K.M.; Polizzi, H.; Mascarenhas, J.; Kremyanskaya, M.; Holland, J.; Hoffman, R. Prevalence of physical problems detected by the distress thermometer and problem list in patients with myeloproliferative disorders. J. Natl. Compr. Cancer Netw. 2017, 15, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Bjorn, M.E.; Hasselbalch, H.C. Minimal residual disease or cure in MPNs? Rationales and perspectives on combination therapy with interferon-alpha2 and ruxolitinib. Expert Rev. Hematol. 2017, 10, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.T.; Kiladjian, J.J.; Hasselbalch, H.C. Interferon and the treatment of polycythemia vera, essential thrombocythemia and myelofibrosis. Expert Rev. Hematol. 2013, 6, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Bruedigam, C.; Poveromo, L.; Heidel, F.H.; Purdon, A.; Vu, T.; Austin, R.; Heckl, D.; Breyfogle, L.J.; Kuhn, C.P.; et al. Depletion of JAK2V617F myeloproliferative neoplasm-propagating stem cells by interferon-alpha in a murine model of polycythemia vera. Blood 2013, 121, 3692–3702. [Google Scholar] [CrossRef] [PubMed]
- Cassinat, B.; Verger, E.; Kiladjian, J.J. Interferon alfa therapy in calr-mutated essential thrombocythemia. N. Engl. J. Med. 2014, 371, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Cassinat, B.; Chevret, S.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Grandchamp, B.; Chomienne, C.; Fenaux, P. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008, 112, 3065–3072. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Kantarjian, H.; Manshouri, T.; Luthra, R.; Estrov, Z.; Pierce, S.; Richie, M.A.; Borthakur, G.; Konopleva, M.; Cortes, J.; et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J. Clin. Oncol. 2009, 27, 5418–5424. [Google Scholar] [CrossRef] [PubMed]
- Utke Rank, C.; Weis Bjerrum, O.; Larsen, T.S.; Kjaer, L.; de Stricker, K.; Riley, C.H.; Hasselbalch, H.C. Minimal residual disease after long-term interferon-alpha2 treatment: A report on hematological, molecular and histomorphological response patterns in 10 patients with essential thrombocythemia and polycythemia vera. Leuk. Lymphoma 2016, 57, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, M.; Silver, R.T.; Barel, A.; Orazi, A. Recombinant interferon-alpha in myelofibrosis reduces bone marrow fibrosis, improves its morphology and is associated with clinical response. Mod. Pathol. 2015, 28, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, H.; Zagrijtschuk, O.; Buxhofer-Ausch, V.; Thaler, J.; Schloegl, E.; Gastl, G.A.; Wolf, D.; Kralovics, R.; Gisslinger, B.; Strecker, K.; et al. Ropeginterferon alfa-2b, a novel ifnalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 2015, 126, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, R.M.; Dueck, A.C.; Geyer, H.L.; Kiladjian, J.J.; Slot, S.; Zweegman, S.; te Boekhorst, P.A.; Commandeur, S.; Schouten, H.C.; Sackmann, F.; et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: Prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J. Clin. Oncol. 2012, 30, 4098–4103. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Schwager, S.; Radia, D.; Cheville, A.; Hussein, K.; Niblack, J.; Pardanani, A.D.; Steensma, D.P.; Litzow, M.R.; Rivera, C.E.; et al. The myelofibrosis symptom assessment form (MFSAF): An evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk. Res. 2009, 33, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma cytokines in polycythemia vera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am. J. Hematol. 2012, 87, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Xing, S.; Li, Z.; Fu, X.; Li, Q.; Krantz, S.B.; Zhao, Z.J. Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 2005, 280, 22788–22792. [Google Scholar] [CrossRef] [PubMed]
- Mackay-Wiggan, J.; Jabbari, A.; Nguyen, N.; Cerise, J.E.; Clark, C.; Ulerio, G.; Furniss, M.; Vaughan, R.; Christiano, A.M.; Clynes, R. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight 2016, 1, e89790. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.; Armstrong, A.W. JAK inhibitors: Treatment efficacy and safety profile in patients with psoriasis. J. Immunol. Res. 2014, 2014, 283617. [Google Scholar] [CrossRef] [PubMed]
- Spoerl, S.; Mathew, N.R.; Bscheider, M.; Schmitt-Graeff, A.; Chen, S.; Mueller, T.; Verbeek, M.; Fischer, J.; Otten, V.; Schmickl, M.; et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014, 123, 3832–3842. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Hashimoto, D.; Hayase, E.; Ogasawara, R.; Ohigashi, H.; Ara, T.; Yokoyama, E.; Ebata, K.; Matsuoka, S.; Hill, G.; et al. Ruxolitinib protects skin stem cells and maintains skin homeostasis in murine graft-versus-host disease. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: Current and future prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W.; Study, A.I. Tofacitinib, an oral janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.O.; Talpaz, M.; Gupta, V.; Foltz, L.M.; Savona, M.R.; Paquette, R.; Turner, A.R.; Coughlin, P.; Winton, E.; Burn, T.C.; et al. Primary analysis of a phase ii open-label trial of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Haematologica 2017, 102, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Mesa, R.A.; Li, C.Y.; Gray, L.; Tefferi, A. Etanercept, a soluble tumor necrosis factor receptor, palliates constitutional symptoms in patients with myelofibrosis with myeloid metaplasia: Results of a pilot study. Blood 2002, 99, 2252–2254. [Google Scholar] [CrossRef] [PubMed]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: Whether to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, S.U.; Kjaer, L.; Skov, V.; Bjorn, M.E.; Andersen, C.L.; Bjerrum, O.W.; Brochmann, N.; El Fassi, D.; Kruse, T.A.; Larsen, T.S.; et al. Safety and efficacy of combination therapy of interferon-alpha2+JAK1-2 inhibitor in the philadelphia-negative chronic myeloproliferative neoplasms. Preliminary results from the danish combi-trial—An open label, single arm, non-randomized multicenter phase ii study. Blood 2015, 126, 824. [Google Scholar]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 2017, 21, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Gillis, N.K.; Ball, M.; Zhang, Q.; Ma, Z.; Zhao, Y.; Yoder, S.J.; Balasis, M.E.; Mesa, T.E.; Sallman, D.A.; Lancet, J.E.; et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol. 2017, 18, 112–121. [Google Scholar] [CrossRef]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef] [PubMed]
- Rundberg Nilsson, A.; Soneji, S.; Adolfsson, S.; Bryder, D.; Pronk, C.J. Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS ONE 2016, 11, e0158369. [Google Scholar] [CrossRef] [PubMed]
- Linton, P.J.; Dorshkind, K. Age-related changes in lymphocyte development and function. Nat. Immunol. 2004, 5, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef] [PubMed]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Astle, C.M. Loss of stem cell repopulating ability upon transplantation. Effects of donor age, cell number, and transplantation procedure. J. Exp. Med. 1982, 156, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Esplin, B.L.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Nie, L.; Zhang, Q.; Humphrey, M.B.; Yang, Q.; Borghesi, L.A.; Kincade, P.W. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J. Immunol. 2011, 186, 5367–5375. [Google Scholar] [CrossRef] [PubMed]
- Matatall, K.A.; Jeong, M.; Chen, S.; Sun, D.; Chen, F.; Mo, Q.; Kimmel, M.; King, K.Y. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. 2016, 17, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.D.; et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 2017, 21, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ling, F.; Wang, H.C.; Sun, X.H. Chronic TLR signaling impairs the long-term repopulating potential of hematopoietic stem cells of wild type but not Id1 deficient mice. PLoS ONE 2013, 8, e55552. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, H.; Rodriguez, S.; Wang, L.; Wang, S.; Serezani, H.; Kapur, R.; Cardoso, A.A.; Carlesso, N. Sepsis induces hematopoietic stem cell exhaustion and myelosuppression through distinct contributions of TRIF and MYD88. Stem Cell Rep. 2016, 6, 940–956. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.M.; Fisher, D.S.; Murray, A.W. The speed of evolution and maintenance of variation in asexual populations. Curr. Biol. 2007, 17, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFΑ facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Dreyling, M.; Schiegnitz, E.; Haferlach, T.; Hasford, J.; Unterhalt, M.; Hiddemann, W. Moderate increase of secondary hematologic malignancies after myeloablative radiochemotherapy and autologous stem-cell transplantation in patients with indolent lymphoma: Results of a prospective randomized trial of the german low grade lymphoma study group. J. Clin. Oncol. 2004, 22, 4926–4933. [Google Scholar] [PubMed]
- Frick, M.; Galan-Sousa, J.; Hoyer, K.; Chan, W.; Jäger, M.; Yoshida, K.; Seemann, R.; Nörenberg, D.; Waldhüter, N.; Fleischer-Notter, H.; et al. Clonal hematopoiesis: Cell of origin, lineage repartition and dynamic evolution during chemotherapy. Blood 2017, 130, 632. [Google Scholar]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. Dnmt3a mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Bjorkholm, M.; Hultcrantz, M.; Derolf, A.R. Leukemic transformation in myeloproliferative neoplasms: Therapy-related or unrelated? Best Pract Res Clin Haematol 2014, 27, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Kantor, E.D.; Lampe, J.W.; Kratz, M.; White, E. Lifestyle factors and inflammation: Associations by body mass index. PLoS ONE 2013, 8, e67833. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate decision of mesenchymal stem cells: Adipocytes or osteoblasts? Cell Death Differ. 2016, 23, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Krings, A.; Rahman, S.; Huang, S.; Lu, Y.; Czernik, P.J.; Lecka-Czernik, B. Bone marrow fat has brown adipose tissue characteristics, which are attenuated with aging and diabetes. Bone 2012, 50, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Styner, M.; Thompson, W.R.; Galior, K.; Uzer, G.; Wu, X.; Kadari, S.; Case, N.; Xie, Z.; Sen, B.; Romaine, A.; et al. Bone marrow fat accumulation accelerated by high fat diet is suppressed by exercise. Bone 2014, 64, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Govindarajah, V.; Goddard, B.; Hinge, A.; Muench, D.E.; Filippi, M.-D.; Aronow, B.; Cancelas, J.A.; Salomonis, N.; Leighton Grimes, H.; et al. Obesity alters the long-term fitness of the hematopoietic stem cell compartment through modulation ofgfi1expression. J. Exp. Med. 2017, 215, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, L.; Jin, W.; Ma, X.; Ma, Y.; Dong, F.; Zhu, H.; Li, J.; Wang, K. Influence of body mass index on incidence and prognosis of acute myeloid leukemia and acute promyelocytic leukemia: A meta-analysis. Sci. Rep. 2017, 7, 17998. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.D.; Thompson, C.A.; Wang, A.H.; Vierkant, R.A.; Habermann, T.M.; Ross, J.A.; Mesa, R.A.; Virnig, B.A.; Cerhan, J.R. Anthropometric, medical history and lifestyle risk factors for myeloproliferative neoplasms in the iowa women’s health study cohort. Int. J. Cancer 2014, 134, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Deschler, B.; Lubbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood 2005, 105, 2664–2670. [Google Scholar] [CrossRef] [PubMed]
- Menegaux, F.; Ripert, M.; Hemon, D.; Clavel, J. Maternal alcohol and coffee drinking, parental smoking and childhood leukaemia: A french population-based case-control study. Paediatr. Perinat. Epidemiol. 2007, 21, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Bjork, J.; Albin, M.; Mauritzson, N.; Stromberg, U.; Johansson, B.; Hagmar, L. Smoking and myelodysplastic syndromes. Epidemiology 2000, 11, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Elbaek, M.V.; Sorensen, A.L.; Hasselbalch, H.C. Chronic inflammation and autoimmunity as risk factors for the development of chronic myelomonocytic leukemia? Leuk. Lymphoma 2016, 57, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk. Res. 2015, 39, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Lindholm Sorensen, A.; Hasselbalch, H.C. Smoking and philadelphia-negative chronic myeloproliferative neoplasms. Eur. J. Haematol. 2016, 97, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [PubMed]
- Wu, H.-J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Lane, E.R.; Zisman, T.L.; Suskind, D.L. The microbiota in inflammatory bowel disease: Current and therapeutic insights. J. Inflamm. Res. 2017, 10, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Katz-Agranov, N.; Zandman-Goddard, G. The microbiome and systemic lupus erythematosus. Immunol. Res. 2017, 65, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Takeda, K. Role of gut microbiota in rheumatoid arthritis. J. Clin. Med. Res. 2017, 6. [Google Scholar] [CrossRef]
- Farinha, P.; Gascoyne, R.D. Helicobacter pylori and malt lymphoma. Gastroenterology 2005, 128, 1579–1605. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, A.; Yáñez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014, 15, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; Housseau, F.; Sears, C.L. Sporadic colorectal cancer: Microbial contributors to disease prevention, development and therapy. Br. J. Cancer 2016, 115, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Pitmon, E.; Wang, K. Microbiome, inflammation and colorectal cancer. Semin. Immunol. 2017, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Manz, M.G. Impact of inflammation on early hematopoiesis and the microenvironment. Int. J. Hematol. 2017, 106, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Hoermann, G.; Greiner, G.; Valent, P. Cytokine regulation of microenvironmental cells in myeloproliferative neoplasms. Mediat. Inflamm. 2015, 2015, 869242. [Google Scholar] [CrossRef] [PubMed]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdiles, M.C. Inflammation as a keystone of bone marrow stroma alterations in primary myelofibrosis. Mediat. Inflamm. 2015, 2015, 415024. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Sajid, Z.; Pedersen, R.K.; Gudmand-Hoeyer, J.; Ellervik, C.; Skov, V.; Kjaer, L.; Pallisgaard, N.; Kruse, T.A.; Thomassen, M.; et al. Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PLoS ONE 2017, 12, e0183620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fleischman, A.G.; Wodarz, D.; Komarova, N.L. Determining the role of inflammation in the selection of JAK2 mutant cells in myeloproliferative neoplasms. J. Theor. Biol. 2017, 425, 43–52. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Craver, B.M.; El Alaoui, K.; Scherber, R.M.; Fleischman, A.G. The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies. Cancers 2018, 10, 104. https://doi.org/10.3390/cancers10040104
Craver BM, El Alaoui K, Scherber RM, Fleischman AG. The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies. Cancers. 2018; 10(4):104. https://doi.org/10.3390/cancers10040104
Chicago/Turabian StyleCraver, Brianna M., Kenza El Alaoui, Robyn M. Scherber, and Angela G. Fleischman. 2018. "The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies" Cancers 10, no. 4: 104. https://doi.org/10.3390/cancers10040104
APA StyleCraver, B. M., El Alaoui, K., Scherber, R. M., & Fleischman, A. G. (2018). The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies. Cancers, 10(4), 104. https://doi.org/10.3390/cancers10040104