Protein Biomarkers for Early Detection of Pancreatic Ductal Adenocarcinoma: Progress and Challenges



Abstract
1. Unmet Needs for Early Detection of Pancreatic Ductal Adenocarcinoma
2. Development of Molecular Biomarkers
“The age-adjusted incidence of pancreatic cancer in subjects ≥50 years of age is 38/100,000. If a test with 99% sensitivity and 99% specificity for pancreatic cancer is used to screen 100,000 subjects ≥50 years of age, the test would identify nearly all pancreatic cancers in the population screened (n = 37). However, the test would also falsely identify nearly a 1000 subjects as having pancreatic cancer.”[12]
3. Recent Advances in Protein Biomarkers for Early Detection of PDAC
| Cohen 2017 [10]: Found that combining ctDNA testing for KRAS mutations in combination with 4 plasma proteins (CA19-9, CEA, HGF, OPN) outperformed CA19-9 alone in discriminating PDAC from normal controls, chronic pancreatitis, and other benign pancreatic diseases. A disappointing 64% sensitivity begs the question as to whether including additional biomarkers or optimizing the assays may improve performance. Evaluation in an independent test set is desirable. |
| Capello 2017 [21]: Began with 17 protein biomarker candidates from previous studies and validated several that can distinguish PDAC from controls and chronic pancreatitis (TIMP1, LRG1, REG3A, IGFBP2, COL18A1, TNFRSF1A), finding TIMP1 + LRG1 + CA19-9 outperformed CA19-9 alone. Evaluation in an independent test set is desirable. |
| Kaur 2017 [26]: Performed an in-depth analysis of the literature and observed that MUC5AC has favorable biomarker properties: secretion, high over-expression in PanIN, and numerous epitopes. They developed an in-house ELISA against MUC5AC, finding that MUC5AC + CA19-9 outperforms CA19-9 alone in two, large independent validation cohorts. |
| Kim 2017 [27]: Began with a state-of-the-art cell reprogramming model of PDAC and sophisticated systems biology network analysis to nominate thrombo-spondin-2 (THBS2); when combined with CA19-9 it outperformed CA19-9 in discriminating PDAC from controls and chronic pancreatitis. The two biomarker panel was evaluated in a discovery phase and two validation phases, however, the number of Stage I/II patients was 0 in the discovery phase, and 7 Stage I and 34 Stage II in validation set A. |
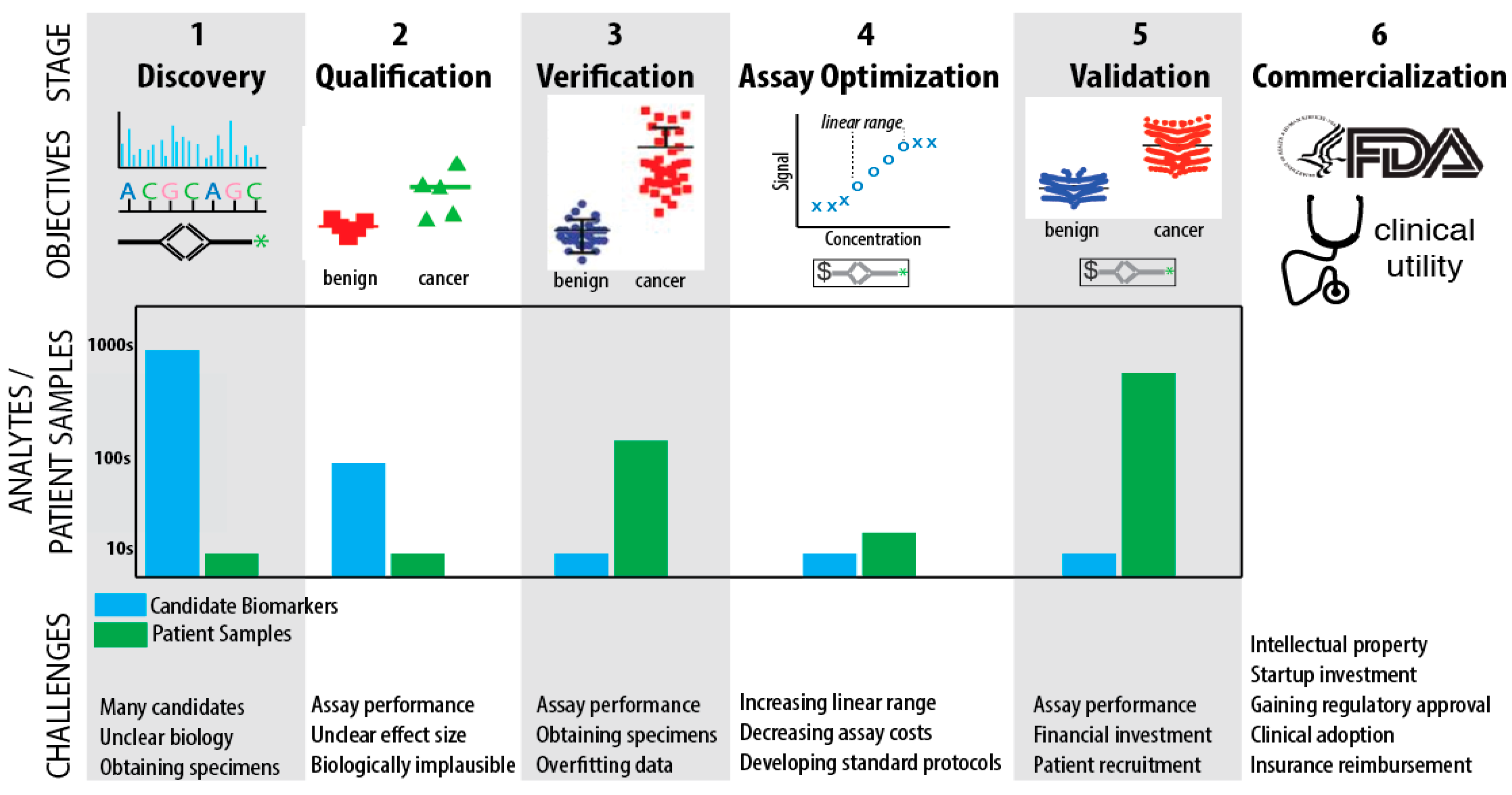
4. Biomarker Challenges
4.1. Challenges in Discovery, Qualification, and Verification
4.2. Challenges in Assay Optimization, Validation, and Commercialization
5. Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- SEER. Cancer Stat Facts: National Cancer Institute. 2017. Available online: https://seer.cancer.gov/statfacts/ (accessed on 29 November 2017).
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Lennon, A.M.; Wolfgang, C.L.; Canto, M.I.; Klein, A.P.; Herman, J.M.; Goggins, M.; Fishman, E.K.; Kamel, I.; Weiss, M.J.; Diaz, L.A.; et al. The early detection of pancreatic cancer: What will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res. 2014, 74, 3381–33819. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, S.; Egawa, S.; Fukuyama, S.; Motoi, F.; Sunamura, M.; Isaji, S.; Imaizumi, T.; Okada, S.; Kato, H.; Suda, K.; et al. Pancreatic Cancer Registry in Japan: 20 years of experience. Pancreas 2004, 28, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Chari, S.T.; Kelly, K.; Hollingsworth, M.A.; Thayer, S.P.; Ahlquist, D.A.; Andersen, D.K.; Batra, S.K.; Brentnall, T.A.; Canto, M.; Cleeter, D.F.; et al. Early detection of sporadic pancreatic cancer: Summative review. Pancreas 2015, 44, 693–712. [Google Scholar] [CrossRef] [PubMed]
- Paulovich, A.G.; Whiteaker, J.R.; Hoofnagle, A.N.; Wang, P. The interface between biomarker discovery and clinical validation: The tar pit of the protein biomarker pipeline. Proteom. Clin. Appl. 2008, 2, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Borrebaeck, C.A. Precision diagnostics: Moving towards protein biomarker signatures of clinical utility in cancer. Nat. Rev. Cancer 2017, 17, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.S.; Feng, Z.; Janes, H.; Bossuyt, P.M.; Potter, J.D. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: Standards for study design. J. Natl. Cancer Inst. 2008, 100, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef] [PubMed]
- Pannala, R.; Basu, A.; Petersen, G.M.; Chari, S.T. New-onset diabetes: A potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 2009, 10, 88–95. [Google Scholar] [CrossRef]
- Groselj, U.; Tansek, M.Z.; Battelino, T. Fifty years of phenylketonuria newborn screening—A great success for many, but what about the rest? Mol. Genet. Metab. 2014, 113, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Prosser, L.A.; Grosse, S.D.; Kemper, A.R.; Tarini, B.A.; Perrin, J.M. Decision analysis, economic evaluation, and newborn screening: Challenges and opportunities. Genet. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.J. Decision analysis for the evaluation of diagnostic tests, prediction models and molecular markers. Am. Stat. 2008, 62, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Ghatnekar, O.; Andersson, R.; Svensson, M.; Persson, U.; Ringdahl, U.; Zeilon, P.; Borrebaeck, C.A. Modelling the benefits of early diagnosis of pancreatic cancer using a biomarker signature. Int. J. Cancer 2013, 133, 2392–2397. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.; Ibrahim, I.; Ponce, C.G.; Slater, E.P.; Matthai, E.; Carrato, A.; Earl, J.; Robbers, K.; van Mil, A.M.; Potjer, T.; et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies From Three European Expert Centers. J. Clin. Oncol. 2016, 34, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.L.; Roberts, N.J.; Jones, S.; Wheelan, S.J.; Papadopoulos, N.; Vogelstein, B.; Kinzler, K.W.; Hruban, R.H.; Klein, A.P.; Eshleman, J.R. Familial and sporadic pancreatic cancer share the same molecular pathogenesis. Fam. Cancer 2015, 14, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Partyka, K.; Maupin, K.A.; Hurley, M.; Andrews, P.; Kaul, K.; Moser, A.J.; Zeh, H.; Brand, R.E.; Haab, B.B. Identification of blood-protein carriers of the CA 19-9 antigen and characterization of prevalence in pancreatic diseases. Proteomics 2011, 11, 3665–3674. [Google Scholar] [CrossRef] [PubMed]
- Capello, M.; Bantis, L.E.; Scelo, G.; Zhao, Y.; Li, P.; Dhillon, D.S.; Patel, N.J.; Kundnani, D.L.; Wang, H.; Abbruzzese, J.L.; et al. Sequential Validation of Blood-Based Protein Biomarker Candidates for Early-Stage Pancreatic Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Bunger, S.; Laubert, T.; Roblick, U.J.; Habermann, J.K. Serum biomarkers for improved diagnostic of pancreatic cancer: A current overview. J. Cancer Res. Clin. Oncol. 2011, 137, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Loosen, S.H.; Neumann, U.P.; Trautwein, C.; Roderburg, C.; Luedde, T. Current and future biomarkers for pancreatic adenocarcinoma. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Gallego, J.; Lopez, C.; Pazo-Cid, R.; Lopez-Rios, F.; Carrato, A. Biomarkers in pancreatic ductal adenocarcinoma. Clin. Transl. Oncol. 2017, 19, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, C.; Zhou, T.; Liu, X.; Liu, X.; Li, X.; Chen, D. Role of exosomal proteins in cancer diagnosis. Mol. Cancer 2017, 16, 145. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Smith, L.M.; Patel, A.; Menning, M.; Watley, D.C.; Malik, S.S.; Krishn, S.R.; Mallya, K.; Aithal, A.; Sasson, A.R.; et al. A Combination of MUC5AC and CA19-9 Improves the Diagnosis of Pancreatic Cancer: A Multicenter Study. Am. J. Gastroenterol. 2017, 112, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bamlet, W.R.; Oberg, A.L.; Chaffee, K.G.; Donahue, G.; Cao, X.J.; Chari, S.; Garcia, B.A.; Petersen, G.M.; Zaret, K.S. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Sci. Transl. Med. 2017, 9, eaah5583. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Firpo, M.A.; Scaife, C.L.; Adler, D.G.; Emerson, L.L.; Boucher, K.M.; Mulvihill, S.J. Serum osteopontin and tissue inhibitor of metalloproteinase 1 as diagnostic and prognostic biomarkers for pancreatic adenocarcinoma. Pancreas 2013, 42, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Radon, T.P.; Massat, N.J.; Jones, R.; Alrawashdeh, W.; Dumartin, L.; Ennis, D.; Duffy, S.W.; Kocher, H.M.; Pereira, S.P.; Guarner Posthumous, L.; et al. Identification of a Three-Biomarker Panel in Urine for Early Detection of Pancreatic Adenocarcinoma. Clin. Cancer Res. 2015, 21, 3512–3521. [Google Scholar] [CrossRef] [PubMed]
- Faca, V.M.; Song, K.S.; Wang, H.; Zhang, Q.; Krasnoselsky, A.L.; Newcomb, L.F.; Plentz, R.R.; Gurumurthy, S.; Redston, M.S.; Pitteri, S.J.; et al. A mouse to human search for plasma proteome changes associated with pancreatic tumor development. PLoS Med. 2008, 5, e123. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Ashfaq, R.; Maitra, A.; Adsay, N.V.; Shen-Ong, G.L.; Berg, K.; Hollingsworth, M.A.; Cameron, J.L.; Yeo, C.J.; Kern, S.E.; et al. Highly expressed genes in pancreatic ductal adenocarcinomas: A comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003, 63, 8614–8622. [Google Scholar] [PubMed]
- Debernardi, S.; Massat, N.J.; Radon, T.P.; Sangaralingam, A.; Banissi, A.; Ennis, D.P.; Dowe, T.; Chelala, C.; Pereira, S.P.; Kocher, H.M.; et al. Noninvasive urinary miRNA biomarkers for early detection of pancreatic adenocarcinoma. Am. J. Cancer Res. 2015, 5, 3455–3466. [Google Scholar] [PubMed]
- Ioannidis, J.P. Why Most Clinical Research Is Not Useful. PLoS Med. 2016, 13, e1002049. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar]
- O’Brien, D.P.; Sandanayake, N.S.; Jenkinson, C.; Gentry-Maharaj, A.; Apostolidou, S.; Fourkala, E.O.; Camuzeaux, S.; Blyuss, O.; Gunu, R.; Dawnay, A.; et al. Serum CA19-9 is significantly upregulated up to 2 years before diagnosis with pancreatic cancer: Implications for early disease detection. Clin. Cancer Res. 2015, 21, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Hanash, S. A call for a fresh new look at the plasma proteome. Proteom. Clin. Appl. 2012, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Harsha, H.C.; Kandasamy, K.; Ranganathan, P.; Rani, S.; Ramabadran, S.; Gollapudi, S.; Balakrishnan, L.; Dwivedi, S.B.; Telikicherla, D.; Selvan, L.D.N.; et al. A compendium of potential biomarkers of pancreatic cancer. PLoS Med. 2009, 6, e1000046. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M. Statistical challenges in the development and evaluation of marker-based clinical tests. BMC Med. 2012, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Ressom, H.W.; Wang, A.; Xuan, J.; Liu, M.C.; Gehan, E.A.; Wang, Y. The properties of high-dimensional data spaces: Implications for exploring gene and protein expression data. Nat. Rev. Cancer 2008, 8, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E. Why your new cancer biomarker may never work: Recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012, 72, 6097–6101. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P. Biomarker failures. Clin. Chem. 2013, 59, 202–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mayo Clinic Prospective Resource for Biomarker Validation and Early Detection of Pancreatic Cancer. Mayo Clinic: Rochester, MN, USA, 2017. Available online: http://www.mayo.edu/research/centers-programs/cancer-research/research-programs/gastrointestinal-cancer-program/mayo-clinic-pancreatic-cancer-spore/core-resources/tissue-core (accessed on 29 November 2017).
- Sakamoto, H.; Harada, S.; Nishioka, N.; Maeda, K.; Kurihara, T.; Sakamoto, T.; Higuchi, K.; Kitano, M.; Takeyama, Y.; Kogire, M.; et al. A Social Program for the Early Detection of Pancreatic Cancer: The Kishiwada Katsuragi Project. Oncology 2017, 93, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lami, G.; Biagini, M.R.; Galli, A. Endoscopic ultrasonography for surveillance of individuals at high risk for pancreatic cancer. World J. Gastrointest. Endosc. 2014, 6, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Potjer, T.P.; Mertens, B.J.; Nicolardi, S.; van der Burgt, Y.E.; Bonsing, B.A.; Mesker, W.E.; Tollenaar, R.A.; Vasen, H.F. Application of a Serum Protein Signature for Pancreatic Cancer to Separate Cases from Controls in a Pancreatic Surveillance Cohort. Transl. Oncol. 2016, 9, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Nepom, G.T.; Lonial, S. Academic, Foundation, and Industry Collaboration in Finding New Therapies. N. Engl. J. Med. 2017, 376, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.D.; Alexander, B.M.; Chandra, A. Innovation Incentives and Biomarkers. Clin. Pharmacol. Ther. 2018, 103, 34–36. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | n (Training; val1; val2) Training Stage I/II Val. A Stage I/II Val. B Stage I/II Val. C Stage I/II | Biomarkers(s) | Cancer vs. Control AUC, SN, SP | Cancer vs. CP AUC, SN, SP |
|---|---|---|---|---|
| Poruk 2013 [28] | 220; 0; 0 ^ 7/35 Stage I/II na na | CA19-9 | 92, 84, 88 *,^ | * |
| OPN | 72, na, na *,^ | * | ||
| TIMP1 | 77, na, na *,^ | * | ||
| CA19-9 + OPN + TIMP1 | na, 87, 91 *,^ | * | ||
| Radon [29]∇ | 134; 58 4/14 Stage I/II Unclear | LYVE1 | 84, 68, 92; na, na, na | 73, 77, 62; na, na, na |
| REG1A | 75, 75, 69; na, na, na | 72, 75, 70; na, na, na | ||
| TFF1 | 70, 79, 53; na, na, na | 65, 77, 56; na, na, na | ||
| panel + creatine + age | 90, 82, 89; 93, 80, 77 | 83, 86, 67; 85, 100, 50 | ||
| panel + plasma CA19.9 | na, na, na; 97, 88, 96 | na, na, na; 87, 75, 94 | ||
| Kaur 2017 [26] | 346; 94; 321 70 Stage I/II Unclear Unclear | MUC5AC | 84,70,83; 70,68,73; 74,65,83 § | * |
| CA19-9 | 57,48,67 §; na, na, na; na, na, na | * | ||
| MUC5AC + CA19-9 | 86,72,85 §; na, na, na; na, na, na | * | ||
| Capello 2017 [21] | 121; 30; 142; 35~ Unclear 10 Stage I/II 42 Stage I/II 21 Resectable | CA19-9 | 88, 73, 23~ | 82, 29, 24~ |
| TIMP1 | 81, 41, 50~ | 73, 22, 33~ | ||
| LRG1 | 85, 43, 25~ | 68, 11, 12~ | ||
| TIMP1 + LRG1 + CA19-9 (“OR” rule) | 96, 85, 67~ | 89, 45, 54~ | ||
| Kim 2017 [27] | 20; 189; 537 0 Stage I/II 7/34 Stage I/II 4/37 Stage I/II | CA19-9 | 85, 69, 100; 58, 78, 99 | 77, na, na; 82, na, na |
| THBS2 | 84, 33,96; 58, 94, 75 | 73, na, na; 73, na, na | ||
| CA19-9 + THBS2 | 96, 74, 96; 76, 88, 93 | 84, na, na; 87, na, na | ||
| Cohen 2017 [10] | na; 403; 0 na 29/102 Stage I/II | ctDNA | na, 30, na | na, na, na |
| CA19-9 | na, 49, na | na, na, na | ||
| CEA + HGF + OPN | na, 18, na | na, na, na | ||
| ctDNA + CA19-9 | na, 60, na | na, na, na | ||
| ctDNA + CEA + HGF + OPN | na, 42, na | na, na, na | ||
| CA19-9 + CEA + HGF + OPN | na, 54, na | na, na, na | ||
| ctDNA+CA19-9+CE+ HGF +OPN | na, 64, 99.5 | na, na, na |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root, A.; Allen, P.; Tempst, P.; Yu, K. Protein Biomarkers for Early Detection of Pancreatic Ductal Adenocarcinoma: Progress and Challenges. Cancers 2018, 10, 67. https://doi.org/10.3390/cancers10030067
Root A, Allen P, Tempst P, Yu K. Protein Biomarkers for Early Detection of Pancreatic Ductal Adenocarcinoma: Progress and Challenges. Cancers. 2018; 10(3):67. https://doi.org/10.3390/cancers10030067
Chicago/Turabian StyleRoot, Alex, Peter Allen, Paul Tempst, and Kenneth Yu. 2018. "Protein Biomarkers for Early Detection of Pancreatic Ductal Adenocarcinoma: Progress and Challenges" Cancers 10, no. 3: 67. https://doi.org/10.3390/cancers10030067
APA StyleRoot, A., Allen, P., Tempst, P., & Yu, K. (2018). Protein Biomarkers for Early Detection of Pancreatic Ductal Adenocarcinoma: Progress and Challenges. Cancers, 10(3), 67. https://doi.org/10.3390/cancers10030067