Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model




Abstract
1. Introduction
2. Results
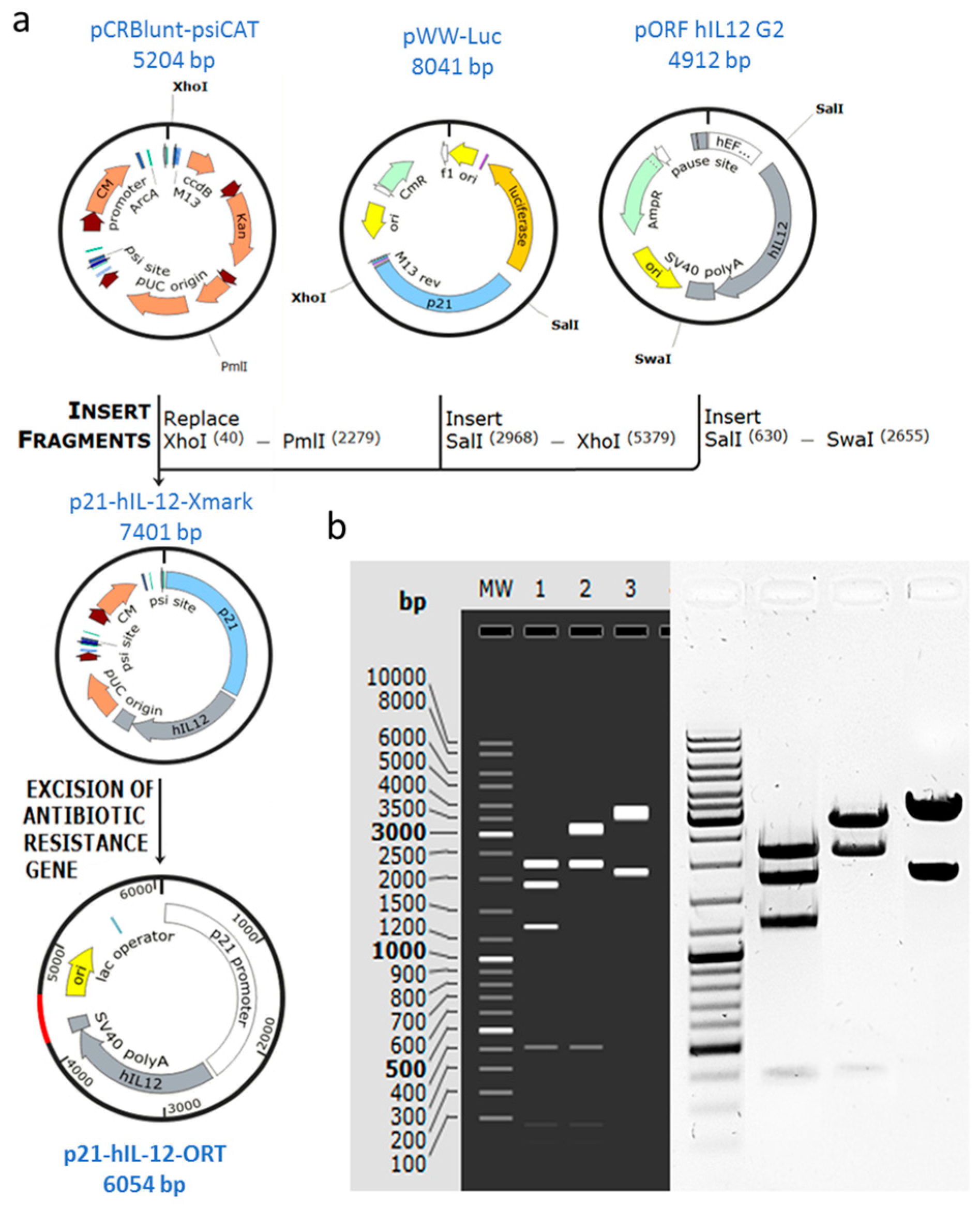
2.1. Confirmation of Successful Construction of the Plasmid
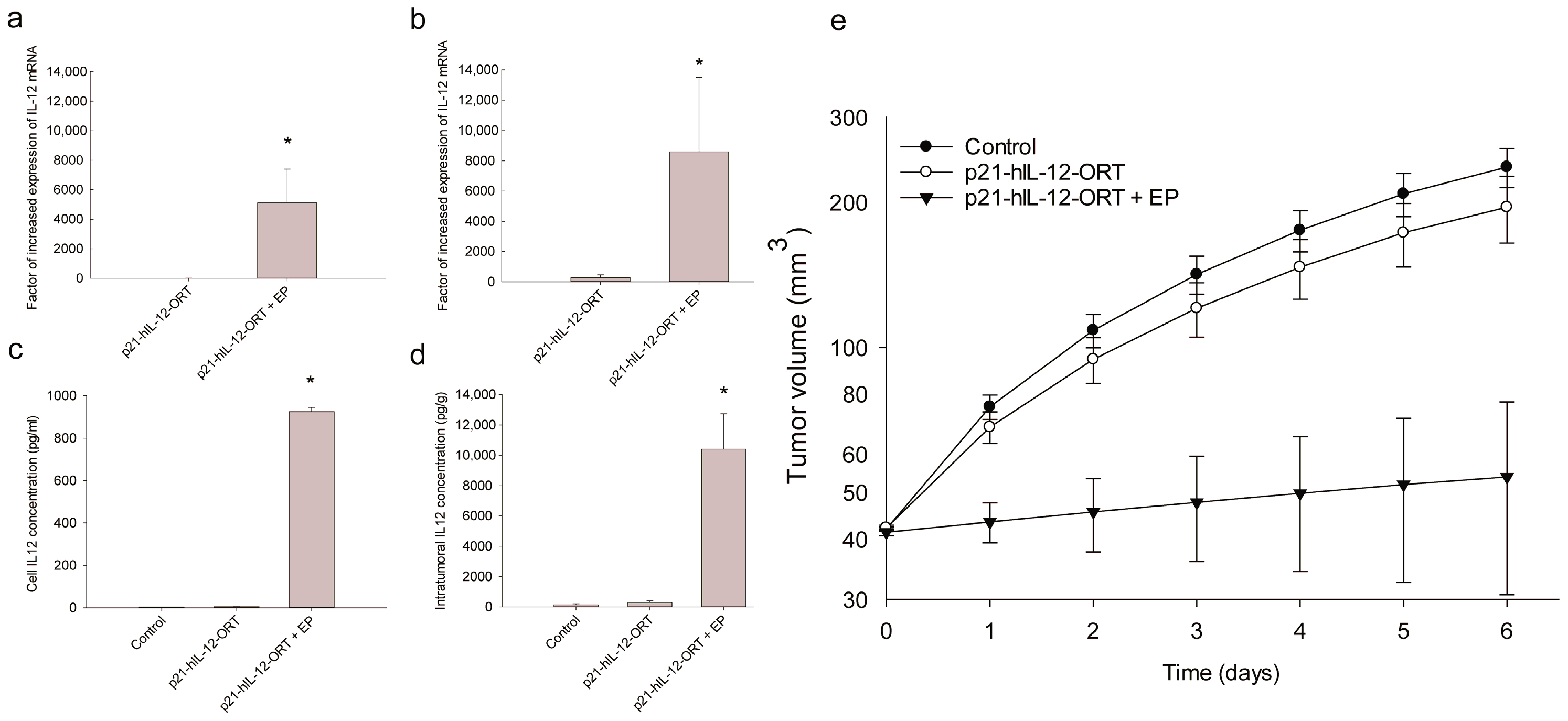
2.2. Expression Profile after GET In Vitro and In Vivo
2.3. Antitumor Effectiveness
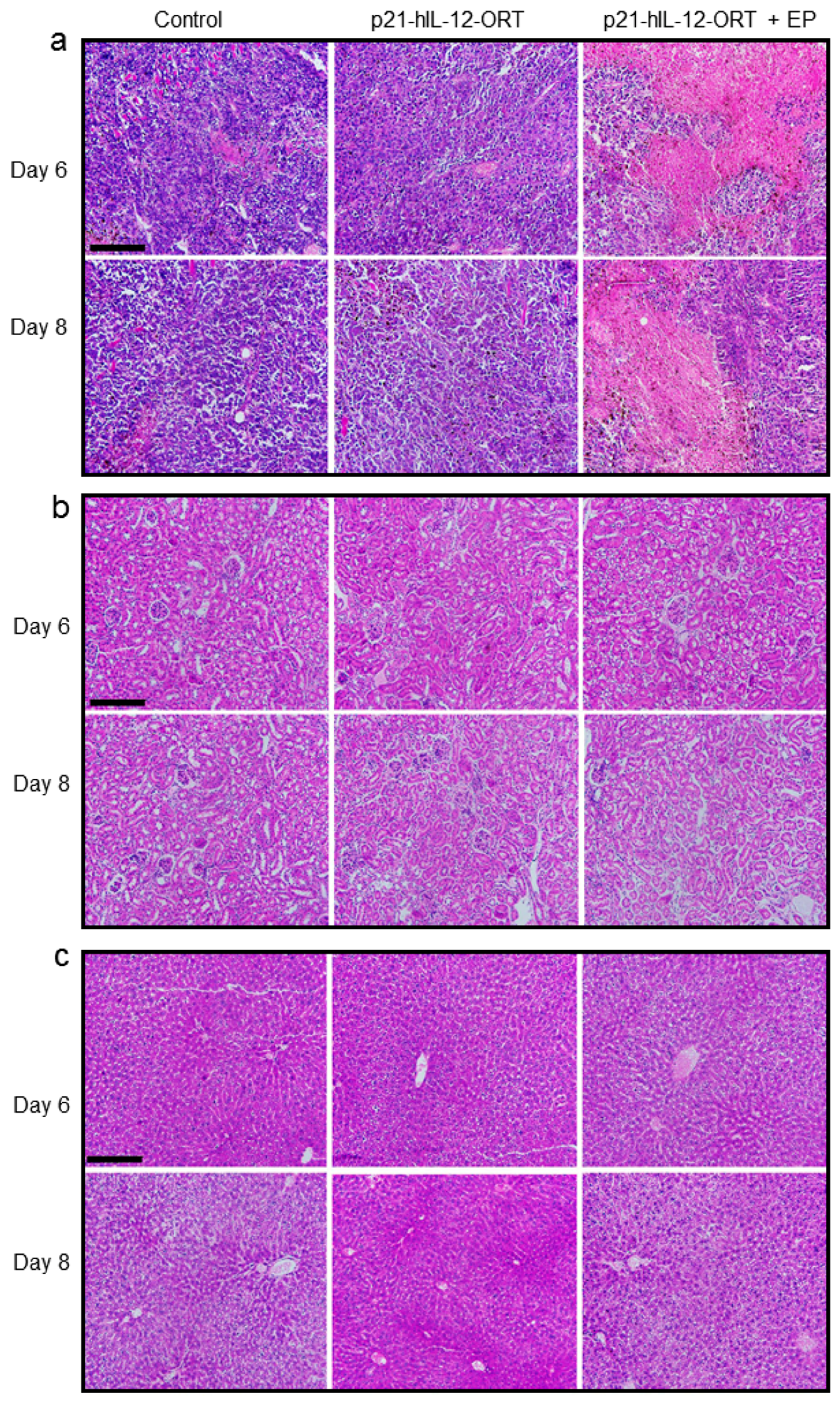
2.4. Toxicity
2.4.1. Histological Changes
2.4.2. Blood Hematology
2.4.3. Blood Chemistry
3. Discussion
4. Materials and Methods
4.1. Construction of the p21-hIL-12-ORT Plasmid
4.2. Production and Purification of the Plasmid DNA
4.3. Cells
4.4. Animals and Tumors
4.5. In Vitro GET
4.6. In Vivo GET
4.7. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Blood Chemistry and Hematology
4.10. Antitumor Effectiveness
4.11. Histology
4.12. Statistical Analysis
5. Patents
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lasek, W.; Zagozdzon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. IL-12 deaths: Explanation and a puzzle. Science 1995, 270, 908. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar] [PubMed]
- Hernandez-Alcoceba, R.; Poutou, J.; Ballesteros-Briones, M.C.; Smerdou, C. Gene therapy approaches against cancer using in vivo and ex vivo gene transfer of interleukin-12. Immunotherapy 2016, 8, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Cemazar, M.; Jarm, T.; Sersa, G. Cancer Electrogene Therapy with Interleukin-12. Curr. Gene Ther. 2010, 10, 300–311. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. ClinicalTrail.Gov. Available online: https://clinicaltrials.gov/ (accessed on 28 December 2017).
- The European Agency for the Evaluation of Medicinal Products. Note for Guidance on the Aspects of Gene Transfer Medicinal Products. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000873.jsp&mid=WC0b01ac058002956b (accessed on 28 December 2017).
- Mignon, C.; Sodoyer, R.; Werle, B. Antibiotic-free selection in biotherapeutics: Now and forever. Pathogens 2015, 4, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Vandermeulen, G.; Marie, C.; Scherman, D.; Préat, V. New generation of plasmid backbones devoid of antibiotic resistance marker for gene therapy trials. Mol. Ther. 2011, 19, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Kamensek, U.; Tesic, N.; Sersa, G.; Cemazar, M. Constructing clinically applicable plasmids for cancer gene therapy. In 1st World Congress on Electroporation and Pulsed Electric Fields in Biology, Medicine and Food & Environmental Technologies; IFMBE Proceedings; Springer: Singapore, 2016; Volume 53, pp. 313–316. [Google Scholar]
- McCarthy, O.; Worthington, J.; Barrett, E.; Cosimo, E.; Boyd, M.; Mairs, R.J.; Ward, C.; McKeown, S.R.; Hirst, D.G.; Robson, T.; et al. p(21(WAF1))-mediated transcriptional targeting of inducible nitric oxide synthase gene therapy sensitizes tumours to fractionated radiotherapy. Gene Ther. 2007, 14, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.; McCarthy, H.O.; Barrett, E.; Adams, C.; Robson, T.; Hirst, D.G. Use of the radiation-inducible WAF1 promoter to drive iNOS gene therapy as a novel anti-cancer treatment. J. Gene Med. 2004, 6, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Robson, T.; Hirst, D.G. Transcriptional targeting in cancer gene therapy. J. Biomed. Biotechnol. 2003, 2003, 110–137. [Google Scholar] [CrossRef] [PubMed]
- Kamensek, U.; Sersa, G.; Cemazar, M. Evaluation of p21 promoter for interleukin 12 radiation induced transcriptional targeting in a mouse tumor model. Mol. Cancer 2013, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Cranenburgh, R.M. Operator-repressor titration: Stable plasmid maintenance without selectable marker genes. In Minicircle and Miniplasmid DNA Vectors: The Future of Nonviral and Viral Gene Transfer; Schleef, M., Ed.; Wiley Online Library, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 7–21. [Google Scholar] [CrossRef]
- Bosnjak, M.; Prosen, L.; Dolinsek, T.; Blagus, T.; Markelc, B.; Cemazar, M.; Bouquet, C.; Sersa, G. Biological properties of melanoma and endothelial cells after plasmid AMEP gene electrotransfer depend on integrin quantity on cells. J. Membr. Biol. 2013, 246, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Spanggaard, I.; Snoj, M.; Cavalcanti, A.; Bouquet, C.; Sersa, G.; Robert, C.; Cemazar, M.; Dam, E.; Vasseur, B.; Attali, P.; et al. Gene electrotransfer of plasmid antiangiogenic metargidin peptide (AMEP) in disseminated melanoma: Safety and efficacy results of a phase I first-in-man study. Hum. Gene Ther. Clin. Dev. 2013, 24, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ogden, G.R. An overview of the cell cycle arrest protein, p21(WAF1). Oral Oncol. 2000, 36, 3–7. [Google Scholar] [CrossRef]
- Sum, C.H.; Wettig, S.; Slavcev, R.A. Impact of DNA vector topology on non-viral gene therapeutic safety and efficacy. Curr. Gene Ther. 2014, 14, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Kamensek, U.; Sersa, G.; Vidic, S.; Tevz, G.; Kranjc, S.; Cemazar, M. Irradiation, cisplatin and 5-azacytidine up-regulate cytomegalovirus promoter in tumors and muscles: Implementation of noninvasive fluorescence imaging. Mol. Imaging Biol. 2011, 13, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.R.; Harkins, R.N.; Wang, P.Y.; Qian, H.S.; Liu, P.X.; Rubanyi, G.M. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 2004, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Tahara, H. Cytokine gene-mediated immunotherapy: Current status and future perspectives. Cancer Sci. 2009, 100, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Sersa, G.; Teissie, J.; Cemazar, M.; Signori, E.; Kamensek, U.; Marshall, G.; Miklavcic, D. Electrochemotherapy of tumors as in situ vaccination boosted by immunogene electrotransfer. Cancer Immunol. Immunother. 2015, 64, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.M.; Wells, D.J. Electroporation for gene transfer to skeletal muscles—Current status. Biodrugs 2004, 18, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.A.; Hyde, S.C.; Nunez-Alonso, G.; Bazzani, R.P.; Harding-Smith, R.; Pringle, I.A.; Lawton, A.E.; Abdullah, S.; Roberts, T.C.; McCormick, D.; et al. The use of CpG-free plasmids to mediate persistent gene expression following repeated aerosol delivery of pDNA/PEI complexes. Biomaterials 2012, 33, 5618–5627. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Kobiyama, K.; Takeshita, F.; Ishii, K.J. Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front. Cell. Infect. Microbiol. 2013, 2, 168. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.C.; Coppola, D. Electrically mediated delivery of vector plasmid DNA elicits an antitumor effect. Gene Ther. 2002, 9, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, M.; Jesenko, T.; Kamensek, U.; Sersa, G.; Lavrencak, J.; Heller, L.; Cemazar, M. Electrotransfer of different control plasmids elicits different antitumor effectiveness in B16.F10 melanoma. Cancers 2018, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Kamensek, U.; Rols, M.-P.; Cemazar, M.; Golzio, M. Visualization of nonspecific antitumor effectiveness and vascular effects of gene electro-transfer to tumors. Curr. Gene Ther. 2016, 16, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Lampreht Tratar, U.; Loiacono, L.; Cemazar, M.; Kamensek, U.; Fazio, V.M.; Sersa, G.; Signori, E. Gene electrotransfer of plasmid-encoding IL-12 recruits the M1 acrophages and antigen-presenting cells inducing the eradication of aggressive B16F10 murine melanoma. Mediat. Inflamm. 2017, 2017, 5285890. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Daud, A. Plasmid IL-12 electroporation in melanoma. Hum. Vaccin. Immunother. 2012, 8, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Imboden, M.; Shi, F.; Pugh, T.D.; Freud, A.G.; Thom, N.J.; Hank, J.A.; Hao, Z.; Staelin, S.T.; Sondel, P.M.; Mahvi, D.M. Safety of interleukin-12 gene therapy against cancer: A murine biodistribution and toxicity study. Hum. Gene Ther. 2003, 14, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.; Merkler, K.; Westover, J.; Cruz, Y.; Coppola, D.; Benson, K.; Daud, A.; Heller, R. Evaluation of toxicity following electrically mediated interleukin-12 gene delivery in a B16 mouse melanoma model. Clin. Cancer Res. 2006, 12, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Tolmachov, O. Designing plasmid vectors. Methods Mol. Biol. 2009, 542, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Vosberg, H.P. Molecular cloning of DNA. An introduction into techniques and problems. Hum. Genet. 1977, 40, 1–72. [Google Scholar] [CrossRef] [PubMed]
- Cemazar, M.; Sersa, G.; Wilson, J.; Tozer, G.M.; Hart, S.L.; Grosel, A.; Dachs, G.U. Effective gene transfer to solid tumors using different nonviral gene delivery techniques: Electroporation, liposomes, and integrin-targeted vector. Cancer Gene Ther. 2002, 9, 399–406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cemazar, M.; Pavlin, D.; Kranjc, S.; Grosel, A.; Mesojednik, S.; Sersa, G. Sequence and time dependence of transfection efficiency of electrically-assisted gene delivery to tumors in mice. Curr. Drug Deliv. 2006, 3, 77–81. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Test | Normals | Units | Day 8 (AM ± SE) | ||
|---|---|---|---|---|---|
| Ctrl | p21-hIL-12-ORT | p21-hIL-12-ORT + EP | |||
| WBC | 3.2–12.7 | ×109/L | 6.1 ± 1.6 | 6.4 ± 1.4 | 6.2 ± 0.8 |
| RBC | 7.0–10.1 | ×1012/L | 4.1 ± 1.0 * | 5.4 ± 1.7 | 8.1 ± 1.2 |
| HGB | 118–149 | g/L | 59.6 ± 13.4 ** | 76.5 ± 26.4 + | 121.0 ± 17.6 |
| HCT | 36.7–46.8 | L/L | 23.2 ± 4.8 ++ | 31.8 ± 8.7 | 46.3 ± 5.1 |
| MCV | 42.2–59.2 | fL | 57.5 ± 3.2 | 62.7 ± 5.0 | 60.5 ± 4.2 |
| MCH | 13.8–18.4 | pg | 14.6 ± 0.5 | 13.7 ± 1.5 | 14.9 ± 0.2 |
| MCHC | 310–347 | g/L | 256.4 ± 8.7 | 221.0 ± 26.1 | 252.1 ± 14.0 |
| CHCM | 307–340 | g/L | 238.0 ± 11.1 | 231.0 ± 11.3 | 230.8 ± 12.2 |
| CH | 13.8–18.4 | pg | 13.5 ± 0.3 | 14.0 ± 0.4 | 13.6 ± 0.2 |
| RDW | 11.7–15.1 | % | 17.0 ± 2.7 | 20.0 ± 2.6 | 16.7 ± 1.8 |
| HDW | 18–26 | g/L | 23.2 ± 2.1 | 27.3 ± 3.8 | 24.0 ± 2.7 |
| PLT | 766–1657 | ×109/L | 1001.4 ± 118.8 | 923.8 ± 87.7 | 1067.4 ± 101.8 |
| MPV | 5.0–8.0 | fL | 7.4 ± 0.4 | 7.6 ± 0.6 | 8.2 ± 0.8 |
| % NEUT | 6.8–31.1 | % | 27.1 ± 6.7 | 19.7 ± 5.4 | 9.5 ± 0.9 |
| % LYMPH | 60.2–95.0 | % | 67.7 ± 6.9 | 74.7 ± 5.3 | 83.0 ± 2.3 |
| % MONO | 0–4.3 | % | 2.1 ± 0.2 | 1.7 ± 0.3 | 2.2 ± 0.5 |
| % EOS | 0.2–5.9 | % | 1.7 ± 0.4 | 2.0 ± 0.7 | 4.3 ± 1.4 |
| % BASO | 0–1.0 | % | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.0 |
| % LUC | 0–3.2 | % | 1.3 ± 0.2 | 1.8 ± 0.9 | 0.8 ± 0.1 |
| # NEUT | 0.5–2.0 | ×109/L | 1.3 ± 0.2 | 1.2 ± 0.4 | 0.6 ± 0.1 |
| # LYMPH | 3.8–8.9 | ×109/L | 4.6 ± 1.4 | 5.0 ± 1.4 | 5.2 ± 0.8 |
| # MONO | 0–0.3 | ×109/L | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| # EOS | 0–0.4 | ×109/L | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.2 ± 0.1 |
| # BASO | 0–0.1 | ×109/L | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 |
| # LUC | 0–0.3 | ×109/L | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| Test | Normals | Units | Day 8 (AM ± SE) | ||
|---|---|---|---|---|---|
| Ctrl | p21-hIL-12-ORT | p21-hIL-12-ORT + EP | |||
| Creatine | 0.2–0.9 | mg/dL | 0.4 ± 0.0 | 0.4 ± 0.0 | 0.4 ± 0.0 |
| TP2 | 3.5–7.2 | g/dL | 4.7 ± 0.2 | 4.2 ± 0.3 | 5.0 ± 0.3 |
| Albumin | 2.5–3 | g/dL | 3.2 ± 0.3 | 3.0 ± 0.2 | 3.4 ± 0.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamensek, U.; Tesic, N.; Sersa, G.; Cemazar, M. Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model. Cancers 2018, 10, 60. https://doi.org/10.3390/cancers10030060
Kamensek U, Tesic N, Sersa G, Cemazar M. Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model. Cancers. 2018; 10(3):60. https://doi.org/10.3390/cancers10030060
Chicago/Turabian StyleKamensek, Urska, Natasa Tesic, Gregor Sersa, and Maja Cemazar. 2018. "Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model" Cancers 10, no. 3: 60. https://doi.org/10.3390/cancers10030060
APA StyleKamensek, U., Tesic, N., Sersa, G., & Cemazar, M. (2018). Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model. Cancers, 10(3), 60. https://doi.org/10.3390/cancers10030060