A Phase I/II Study Targeting Angiogenesis Using Bevacizumab Combined with Chemotherapy and a Histone Deacetylase Inhibitor (Valproic Acid) in Advanced Sarcomas

 ,
,  ,
, 

Abstract
1. Introduction
2. Results
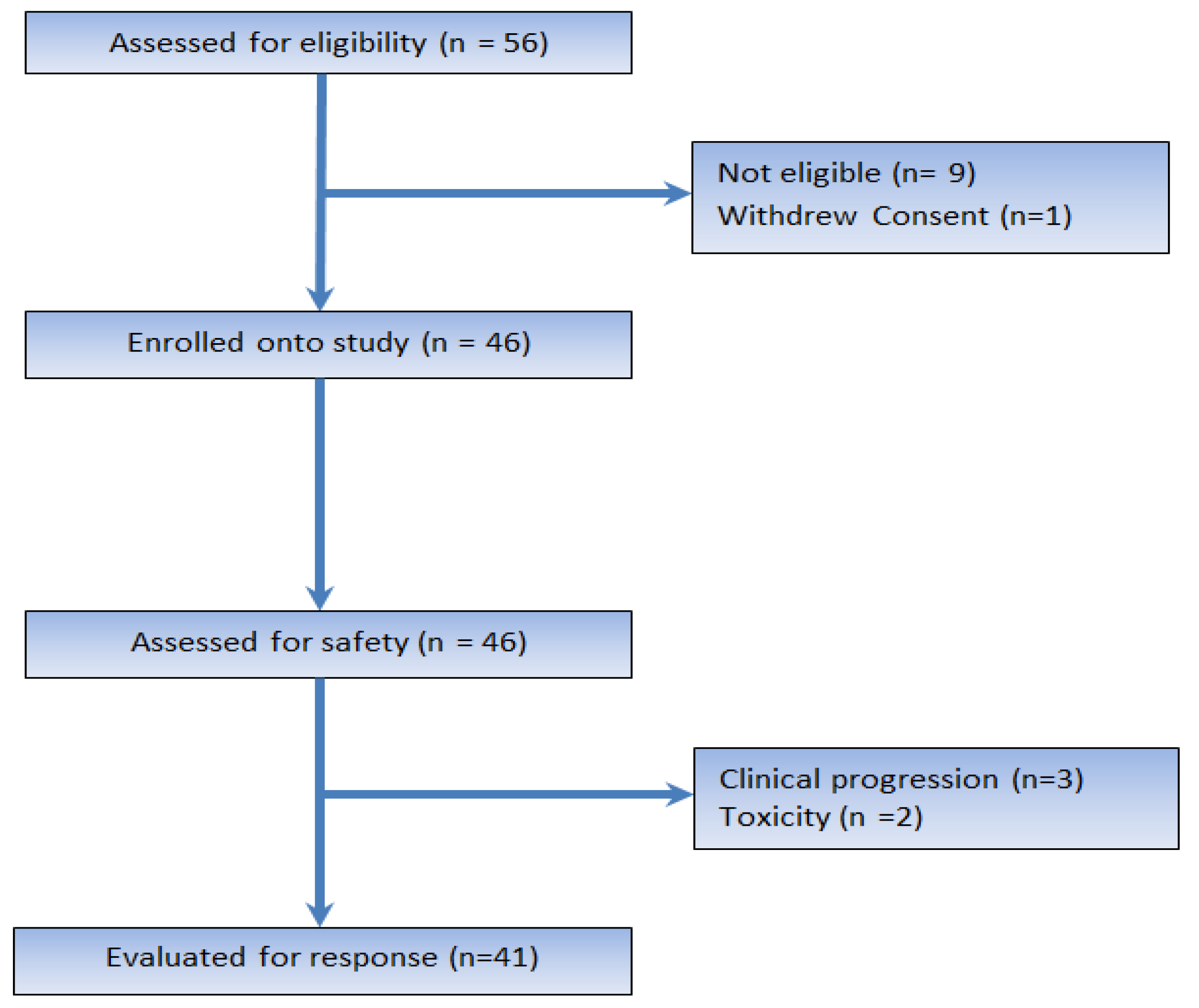
2.1. Patient Characteristics and Disposition
2.2. Safety and Toxicity
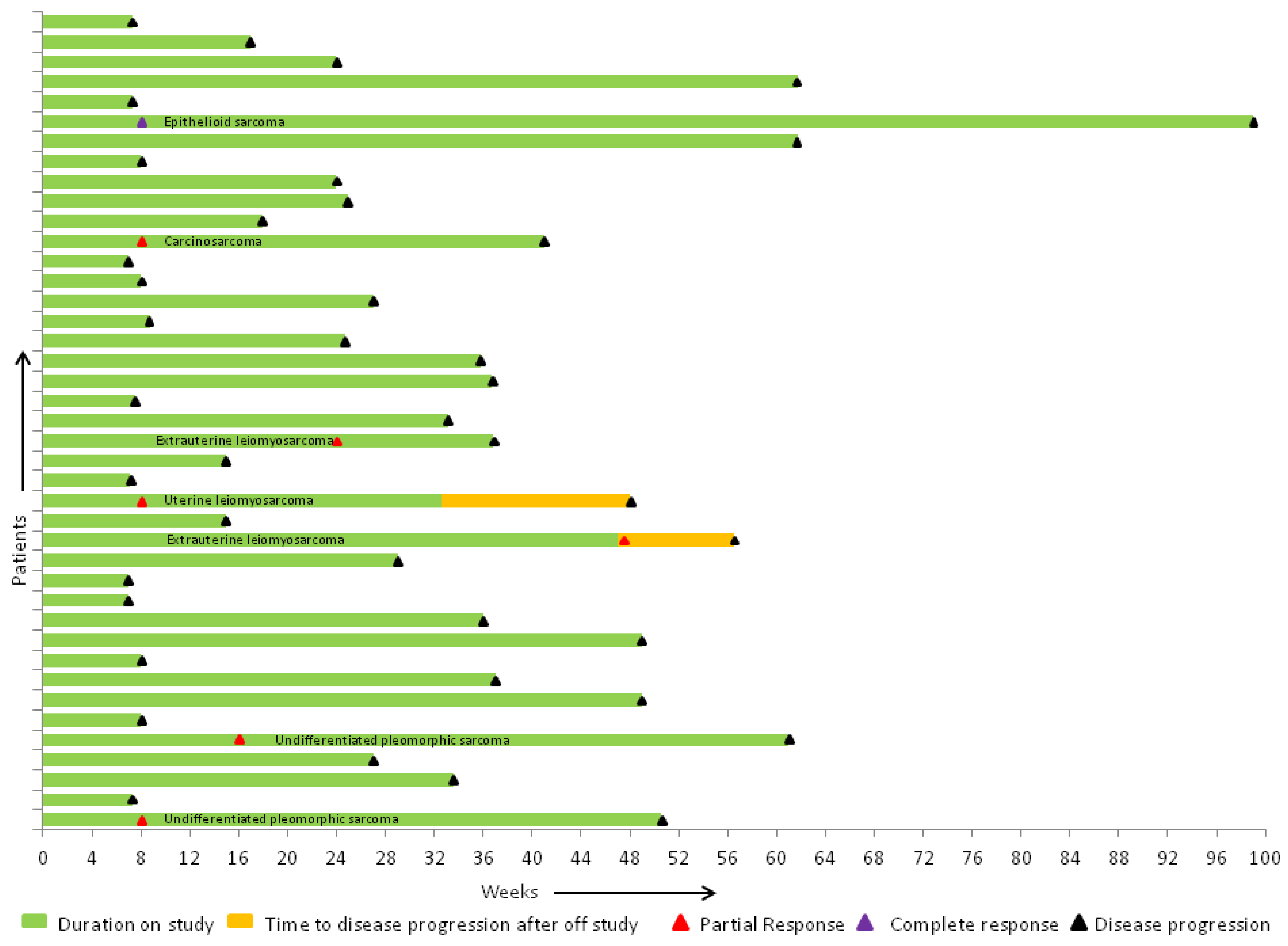
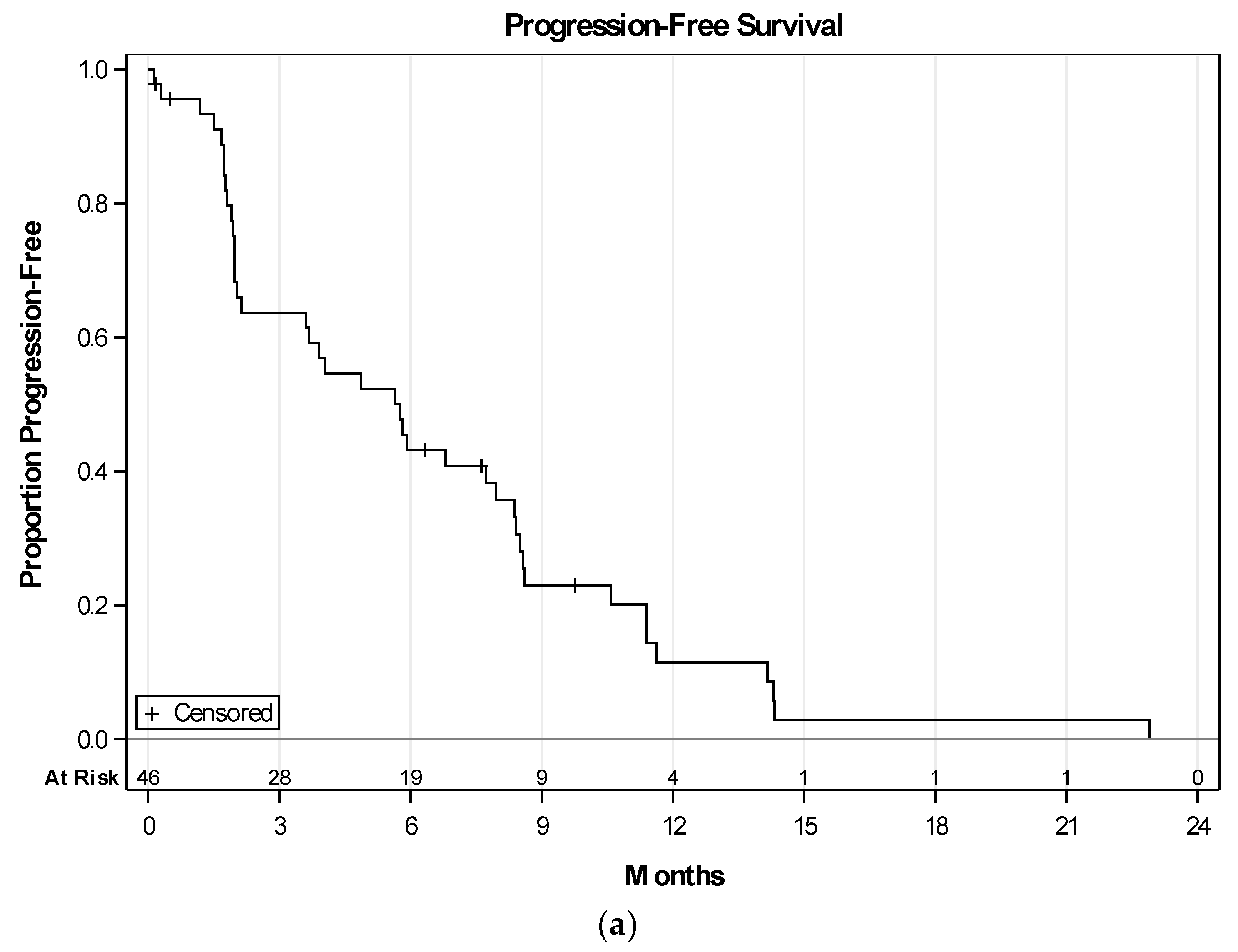
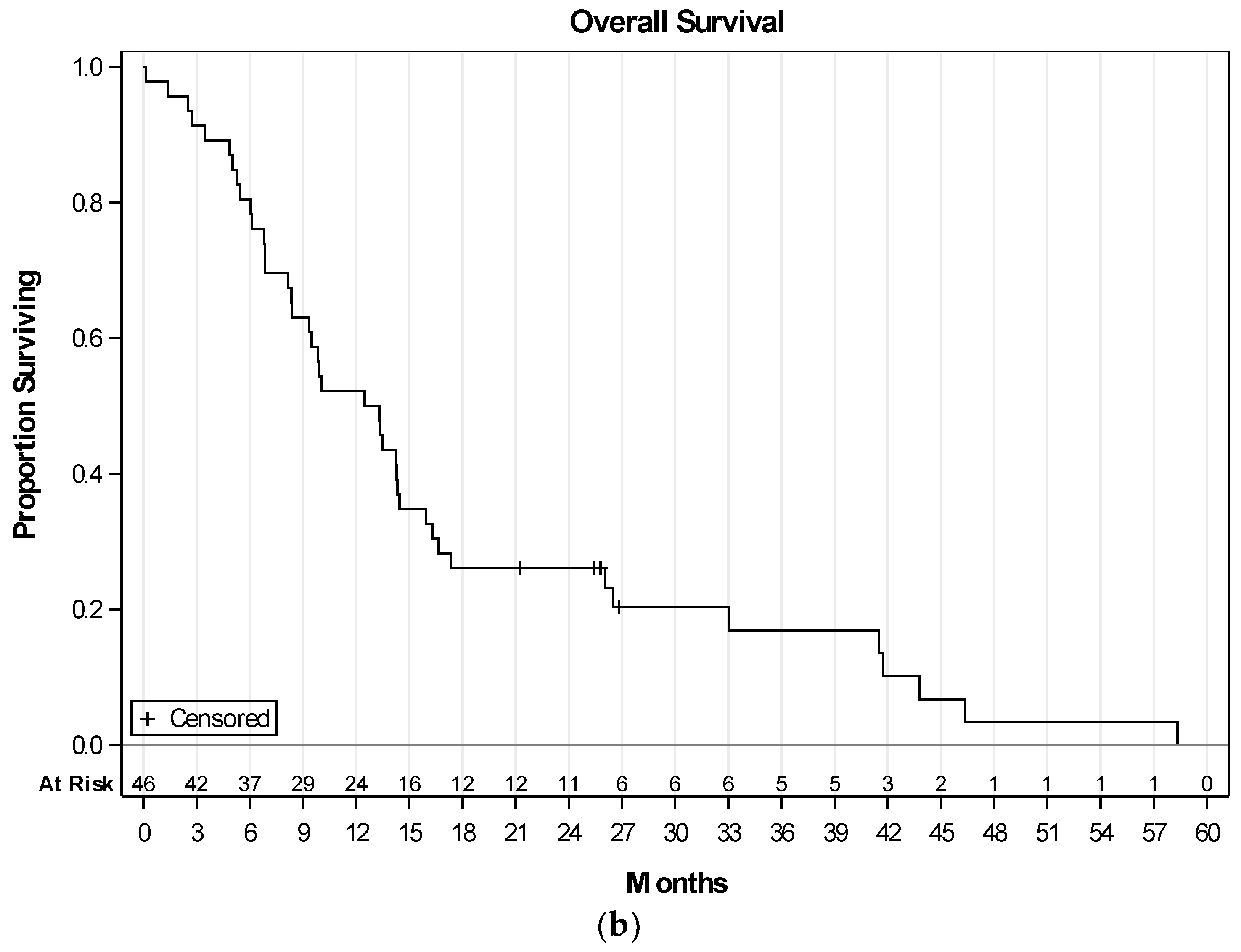
2.3. Efficacy of Therapy
3. Discussion
4. Materials and Methods
4.1. Patient Eligibility
4.2. Therapy
4.3. Study Parameters
4.4. Analysis Samples
4.5. Statistical Considerations and Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Potti, A.; Ganti, A.K.; Tendulkar, K.; Sholes, K.; Chitajallu, S.; Koch, M.; Kargas, S. Determination of vascular endothelial growth factor (VEGF) overexpression in soft tissue sarcomas and the role of overexpression in leiomyosarcoma. J. Cancer Res. Clin. Oncol. 2004, 130, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Kuhnen, C.; Lehnhardt, M.; Tolnay, E.; Muehlberger, T.; Vogt, P.M.; Muller, K.M. Patterns of expression and secretion of vascular endothelial growth factor in malignant soft-tissue tumours. J. Cancer Res. Clin. Oncol. 2000, 126, 219–225. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, D.R.; Anderson, S.E.; Albritton, K.; Yamada, J.; Riedel, E.; Scheu, K.; Schwartz, G.K.; Chen, H.; Maki, R.G. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. J. Clin. Oncol. 2005, 23, 7135–7142. [Google Scholar] [CrossRef] [PubMed]
- Verschraegen, C.F.; Arias-Pulido, H.; Lee, S.J.; Movva, S.; Cerilli, L.A.; Eberhardt, S.; Schmit, B.; Quinn, R.; Muller, C.Y.; Rabinowitz, I.; et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: The Axtell regimen. Ann. Oncol. 2012, 23, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Agulnik, M.; Yarber, J.L.; Okuno, S.H.; von Mehren, M.; Jovanovic, B.D.; Brockstein, B.E.; Evens, A.M.; Benjamin, R.S. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann. Oncol. 2013, 24, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005, 65, 8961–8967. [Google Scholar] [CrossRef] [PubMed]
- Glasspool, R.M.; Teodoridis, J.M.; Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 2006, 94, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Cress, W.D.; Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 2000, 184, 1–16. [Google Scholar] [CrossRef]
- Timmermann, S.; Lehrmann, H.; Polesskaya, A.; Harel-Bellan, A. Histone acetylation and disease. Cell. Mol. Life Sci. 2001, 58, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, U.; Hoelzer, D. Histone acetylation modifiers in the pathogenesis of malignant disease. Mol. Med. 2000, 6, 623–644. [Google Scholar] [PubMed]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Michaelis, U.R.; Fleming, I.; Suhan, T.; Cinatl, J.; Blaheta, R.A.; Hoffmann, K.; Kotchetkov, R.; Busse, R.; Nau, H.; et al. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol. Pharmacol. 2004, 65, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.Z.; Kato, Y.; Shabbeer, S.; Wei, Y.; Verheul, H.M.; Salumbides, B.; Sanni, T.; Atadja, P.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors: The hydroxamic acid derivative LBH589. Clin. Cancer Res. 2006, 12, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Rossig, L.; Li, H.; Fisslthaler, B.; Urbich, C.; Fleming, I.; Forstermann, U.; Zeiher, A.M.; Dimmeler, S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 2002, 91, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Brodie, M.J.; Dichter, M.A. Antiepileptic drugs. N. Engl. J. Med. 1996, 334, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, M.; Gallardo-Rincon, D.; Arce, C.; Cetina, L.; Aguilar-Ponce, J.L.; Arrieta, O.; Gonzalez-Fierro, A.; Chavez-Blanco, A.; de la Cruz-Hernandez, E.; Camargo, M.F.; et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann. Oncol. 2007, 18, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Maki, R.G.; Wathen, J.K.; Patel, S.R.; Priebat, D.A.; Okuno, S.H.; Samuels, B.; Fanucchi, M.; Harmon, D.C.; Schuetze, S.M.; Reinke, D.; et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: Results of sarcoma alliance for research through collaboration study 002 [corrected]. J. Clin. Oncol. 2007, 25, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Blanco, A.; Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Cetina, L.; Candelaria, M.; Cantu, D.; Gonzalez-Fierro, A.; Garcia-Lopez, P.; Zambrano, P.; et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study. Mol. Cancer 2005, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Hensley, M.L.; Miller, A.; O’Malley, D.M.; Mannel, R.S.; Behbakht, K.; Bakkum-Gamez, J.N.; Michael, H. Randomized phase III trial of gemcitabine plus docetaxel plus bevacizumab or placebo as first-line treatment for metastatic uterine leiomyosarcoma: An NRG Oncology/Gynecologic Oncology Group study. J. Clin. Oncol. 2015, 33, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Lee, H.K.; Kang, C.S.; Park, W.S.; Lee, J.Y.; Shim, S.I. p53 gene mutations and p53 protein expression in human soft tissue sarcomas. Arch. Pathol. Lab. Med. 1997, 121, 395–399. [Google Scholar] [PubMed]
- Schneider-Stock, R.; Radig, K.; Oda, Y.; Mellin, W.; Rys, J.; Niezabitowski, A.; Roessner, A. p53 gene mutations in soft-tissue sarcomas-correlations with p53 immunohistochemistry and DNA ploidy. J. Cancer Res. Clin. Oncol. 1997, 123, 211–218. [Google Scholar] [PubMed]
- Taubert, H.; Meye, A.; Wurl, P. Soft tissue sarcomas and p53 mutations. Mol. Med. 1998, 4, 365–372. [Google Scholar] [PubMed]
- Zhang, L.; Yu, D.; Hu, M.; Xiong, S.; Lang, A.; Ellis, L.M.; Pollock, R.E. Wild-type p53 suppresses angiogenesis in human leiomyosarcoma and synovial sarcoma by transcriptional suppression of vascular endothelial growth factor expression. Cancer Res. 2000, 60, 3655–3661. [Google Scholar] [PubMed]
- Koehler, K.; Liebner, D.; Chen, J.L. TP53 mutational status is predictive of pazopanib response in advanced sarcomas. Ann. Oncol. 2016, 27, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Brachova, P.; Thiel, K.W.; Leslie, K.K. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 19257–19275. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.B.; Nesslinger, N.J.; Deitch, A.D.; Gumerlock, P.H.; deVere White, R.W. Complex functions of mutant p53 alleles from human prostate cancer. Prostate 2002, 51, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.; Hart, S.N.; Lima, J.F.; Kipp, B.R.; Klebig, M.; Winters, J.L.; Szabo, C.; Zhang, L.; Eckloff, B.W.; Petersen, G.M.; et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013, 145, 1098–1109.e1. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Level | N (%) |
|---|---|---|
| Gender | Female | 30 (65.2) |
| Male | 16 (34.8) | |
| Tumor Histology | Angiosarcoma | 4 (8.7) |
| Carcinosarcoma | 4 (8.7) | |
| Epithelioid Sarcoma | 2 (4.3) | |
| Extrauterine leiomyosarcoma | 9 (19.6) | |
| Undifferentiated uterine sarcoma | 2 (4.3) | |
| Liposarcoma | 4 (8.7) | |
| MPNST 1 | 2 (4.3) | |
| Rhabdomyosarcoma | 1 (2.2) | |
| Malignant solitary fibrous tumor | 1 (2.2) | |
| Synovial sarcoma | 2 (4.3) | |
| Undifferentiated Pleomorphic sarcoma | 7 (15.2) | |
| Uterine Leiomyosarcoma | 8 (17.4) | |
| 2 ECOG | 0 | 21 (45.7) |
| 1 | 24 (52.2) | |
| 2 | 1 (2.2) | |
| Prior chemotherapy | No | 12 (26.1) |
| Yes | 34 (73.9) | |
| Prior lines of chemotherapy | 1 | 15 (32.6) |
| 2 | 8 (17.4) | |
| 3 | 8 (17.4) | |
| 4 | 1 (2.2) | |
| 5 | 1 (2.2) | |
| 6 | 1 (2.2) | |
| Prior Gemcitabine + Docetaxel | No | 32 (69.6) |
| Yes | 14 (30.4) | |
| Prior radiation | No | 30 (65.2) |
| Yes | 16 (34.8) | |
| Prior surgery | No | 2 (4.3) |
| Yes | 44 (95.7) |
| Toxicity | Grade | ||||
|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | Total | |
| Anemia | 1 | 1 | 1 | 0 | 3 |
| Leukopenia | 1 | 1 | 1 | 0 | 3 |
| Neutropenia | 0 | 1 | 0 | 0 | 1 |
| Thrombocytopenia | 1 | 1 | 1 | 0 | 3 |
| Hypertension | 5 | 4 | 0 | 0 | 9 |
| Elevated INR | 0 | 1 | 0 | 0 | 1 |
| Fatigue | 5 | 0 | 0 | 0 | 5 |
| Alopecia | 3 | 0 | 0 | 0 | 3 |
| Diarrhea | 2 | 0 | 0 | 0 | 2 |
| Heartburn | 3 | 0 | 0 | 0 | 3 |
| Mucositis | 2 | 0 | 0 | 0 | 2 |
| Nausea | 2 | 0 | 0 | 0 | 2 |
| Taste Alteration | 1 | 0 | 0 | 0 | 1 |
| Vomiting | 1 | 0 | 0 | 0 | 1 |
| Hemorrhage | 2 | 1 | 0 | 0 | 3 |
| Leg edema | 1 | 0 | 0 | 0 | 1 |
| Hyponatremia | 0 | 7 | 0 | 0 | 7 |
| Liver Enzyme Elevation | 25 | 5 | 0 | 0 | 30 |
| Confusion | 2 | 2 | 0 | 0 | 4 |
| Dizziness | 2 | 1 | 0 | 0 | 3 |
| Memory Impairment | 1 | 0 | 0 | 0 | 1 |
| Neurology Other | 4 | 1 | 0 | 0 | 5 |
| Psychosis-Hallucination | 0 | 1 | 0 | 0 | 1 |
| Somnolence | 2 | 0 | 0 | 0 | 2 |
| Oral pain | 1 | 0 | 0 | 0 | 1 |
| Headache | 0 | 1 | 0 | 0 | 1 |
| Myalgia | 2 | 0 | 0 | 0 | 2 |
| Voice Changes-Hoarseness | 1 | 0 | 0 | 0 | 1 |
| Voice Changes-Slurred Speech | 1 | 0 | 0 | 0 | 1 |
| Urinary incontinence | 0 | 1 | 0 | 0 | 1 |
| Total | 71 | 29 | 3 | 0 | 103 |
| Best Response | |||||
|---|---|---|---|---|---|
| CR N = 1 | PR N = 6 | SD N = 21 | PD N = 13 | ||
| Diagnosis | Angiosarcoma | 0 (0) | 0 (0) | 3 (100.0) | 0 (0) |
| Carcinosarcoma | 0 (0) | 1 (33.3) | 2 (66.7) | 0 (0) | |
| Epithelioid sarcoma | 1 (50) | 0 (0) | 1 (50) | 0 (0) | |
| Extra Uterine Leiomyosarcoma | 0 (0) | 2 (22.2) | 5 (55.6) | 2 (22.2) | |
| High Grade/Undifferentiated Uterine Sarcoma | 0 (0) | 0 (0) | 1 (50.0) | 1 (50.0) | |
| Liposarcoma | 0 (0) | 0 (0) | 0 (0) | 4 (100.0) | |
| MPNST | 0 (0) | 0 (0) | 0 (0) | 1 (100.0) | |
| Rhabdomyosarcoma | 0 (0) | 0 (0) | 1 (100.0) | 0 (0) | |
| SFT | 0 (0) | 0 (0) | 1 (100.0) | 0 (0) | |
| Synovial | 0 (0) | 0 (0) | 1 (50.0) | 1 (50.0) | |
| Undifferentiated Pleomorphic/Spindle Cell Sarcoma | 0 (0) | 2 (40.0) | 1 (20.0) | 2 (40.0) | |
| Uterine Leiomyosarcoma | 0 (0) | 1 (12.5) | 5 (62.5) | 2 (25.0) | |
| Leiomyosarcomas | 0 (0) | 3 (17.7) | 10 (58.8) | 4 (23.5) | |
| Prior chemotherapy | No | 1 (9.1) | 2 (18.2) | 6 (54.5) | 2 (18.2) |
| Yes | 0 (0) | 4 (12.9) | 15 (48.4) | 12 (38.7) | |
| Prior Gemcitabine + Docetaxel | No | 1 (3.4) | 5 (17.2) | 14 (48.3) | 9 (31.0) |
| Yes | 0 (0) | 1 (7.7) | 7 (53.8) | 5 (38.5) | |
| Gain of function p53 mutation | No | 1 (2.9) | 5 (14.7) | 17 (50.0) | 11 (32.4) |
| Yes | 0 (0) | 0 (0) | 2 (66.7) | 1 (33.3) | |
| Histology | TP53 Mutation | Variant | Genomic Position | Allele Frequency |
|---|---|---|---|---|
| Pleomorphic Sarcoma | GOF | * exon7:c.G743A:p.R248Q [33] exon5:c.C451A:p.P151T | chr7: 7577538 chr7: 7578479 | 33% 31% |
| Uterine leiomyosarcoma | GOF | * exon5:c.G524A:p.R175H [33] | chr7: 7578406 | 90% |
| Retroperitoneal leiomyosarcoma | GOF | * exon8:c.844C > G; p.R282G [34,35] | chr7: 7577094 | 72% |
| Uterine leiomyosarcoma | LOF | exon6:c.C574T:p.Q192X | chr7: 7578275 | 84% |
| Uterine Leiomyosarcoma | LOF | exon10:c.C1024T:p.R342X | chr7: 7574003 | 95% |
| Carcinosarcoma | LOF | exon2:c.52delA:p.T18fs | chr7: 7579860 | 38% |
| Extra-uterine Leiomyosarcoma | LOF | exon4:c.205delG:p.A69fs | chr7: 7579481 | 56% |
| Extra-uterine leiomyosarcoma | LOF | exon8:c.785delG:p.G262fs | chr7: 7577152 | 59% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monga, V.; Swami, U.; Tanas, M.; Bossler, A.; Mott, S.L.; Smith, B.J.; Milhem, M. A Phase I/II Study Targeting Angiogenesis Using Bevacizumab Combined with Chemotherapy and a Histone Deacetylase Inhibitor (Valproic Acid) in Advanced Sarcomas. Cancers 2018, 10, 53. https://doi.org/10.3390/cancers10020053
Monga V, Swami U, Tanas M, Bossler A, Mott SL, Smith BJ, Milhem M. A Phase I/II Study Targeting Angiogenesis Using Bevacizumab Combined with Chemotherapy and a Histone Deacetylase Inhibitor (Valproic Acid) in Advanced Sarcomas. Cancers. 2018; 10(2):53. https://doi.org/10.3390/cancers10020053
Chicago/Turabian StyleMonga, Varun, Umang Swami, Munir Tanas, Aaron Bossler, Sarah L. Mott, Brian J. Smith, and Mohammed Milhem. 2018. "A Phase I/II Study Targeting Angiogenesis Using Bevacizumab Combined with Chemotherapy and a Histone Deacetylase Inhibitor (Valproic Acid) in Advanced Sarcomas" Cancers 10, no. 2: 53. https://doi.org/10.3390/cancers10020053
APA StyleMonga, V., Swami, U., Tanas, M., Bossler, A., Mott, S. L., Smith, B. J., & Milhem, M. (2018). A Phase I/II Study Targeting Angiogenesis Using Bevacizumab Combined with Chemotherapy and a Histone Deacetylase Inhibitor (Valproic Acid) in Advanced Sarcomas. Cancers, 10(2), 53. https://doi.org/10.3390/cancers10020053