Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States

 ,
, 

Abstract
1. Introduction
2. Methods
2.1. Materials
2.2. Cell Culture and Transfection
2.3. SRB Assay for Cell Proliferation, Mitochondrial Membrane Potential, Cellular ATP Levels Determination
2.4. Xenograft Experiments
2.5. IHC and Immunofluorescence (IF), Immunoblotting and TUNEL Assays
2.6. RNA Preparation and Quantitative Real-Time PCR (q-RT-PCR)
2.7. Statistics
3. Results
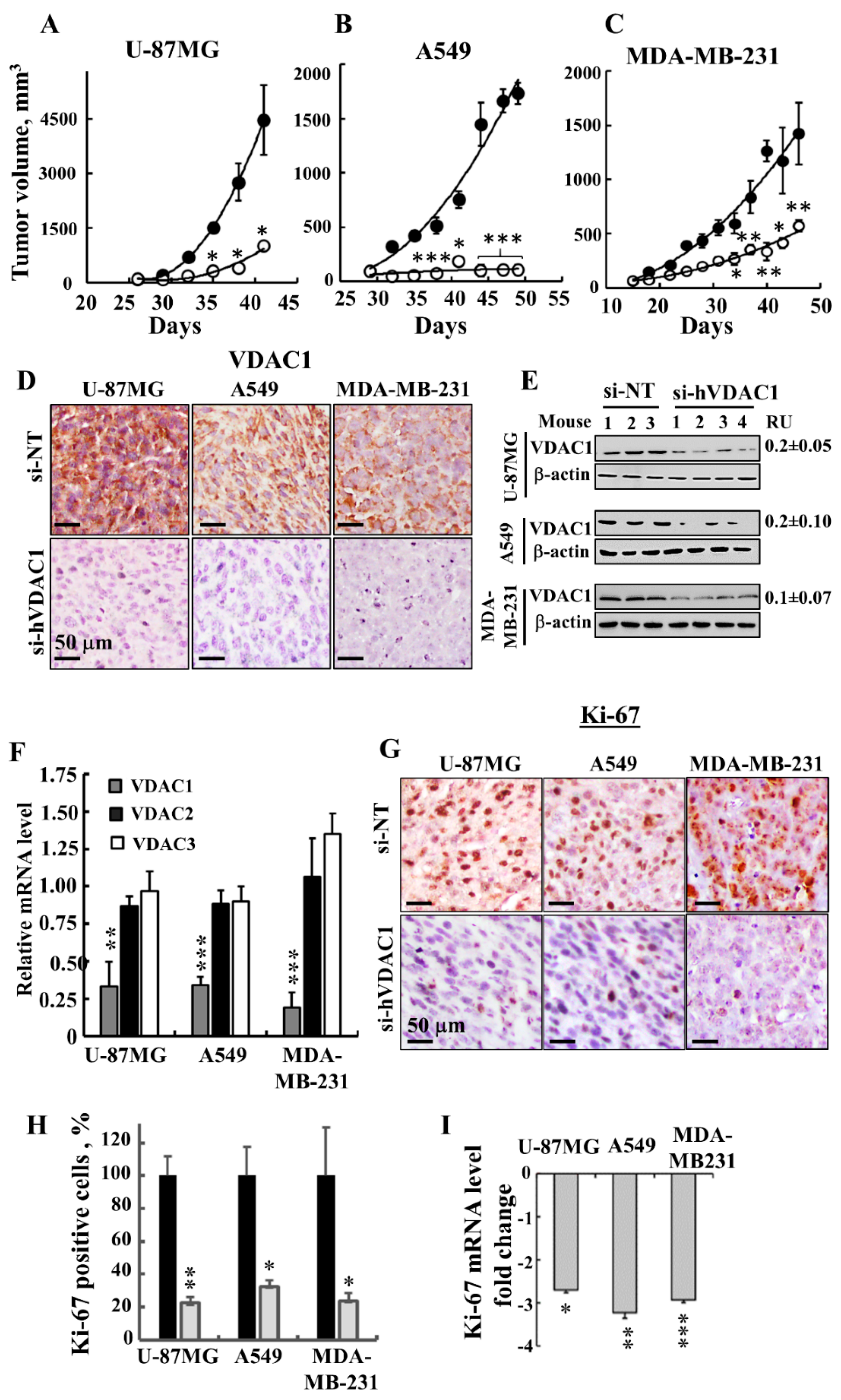
3.1. Silencing VDAC1 Expression Inhibits Cancer Cell Growth and Tumour Development
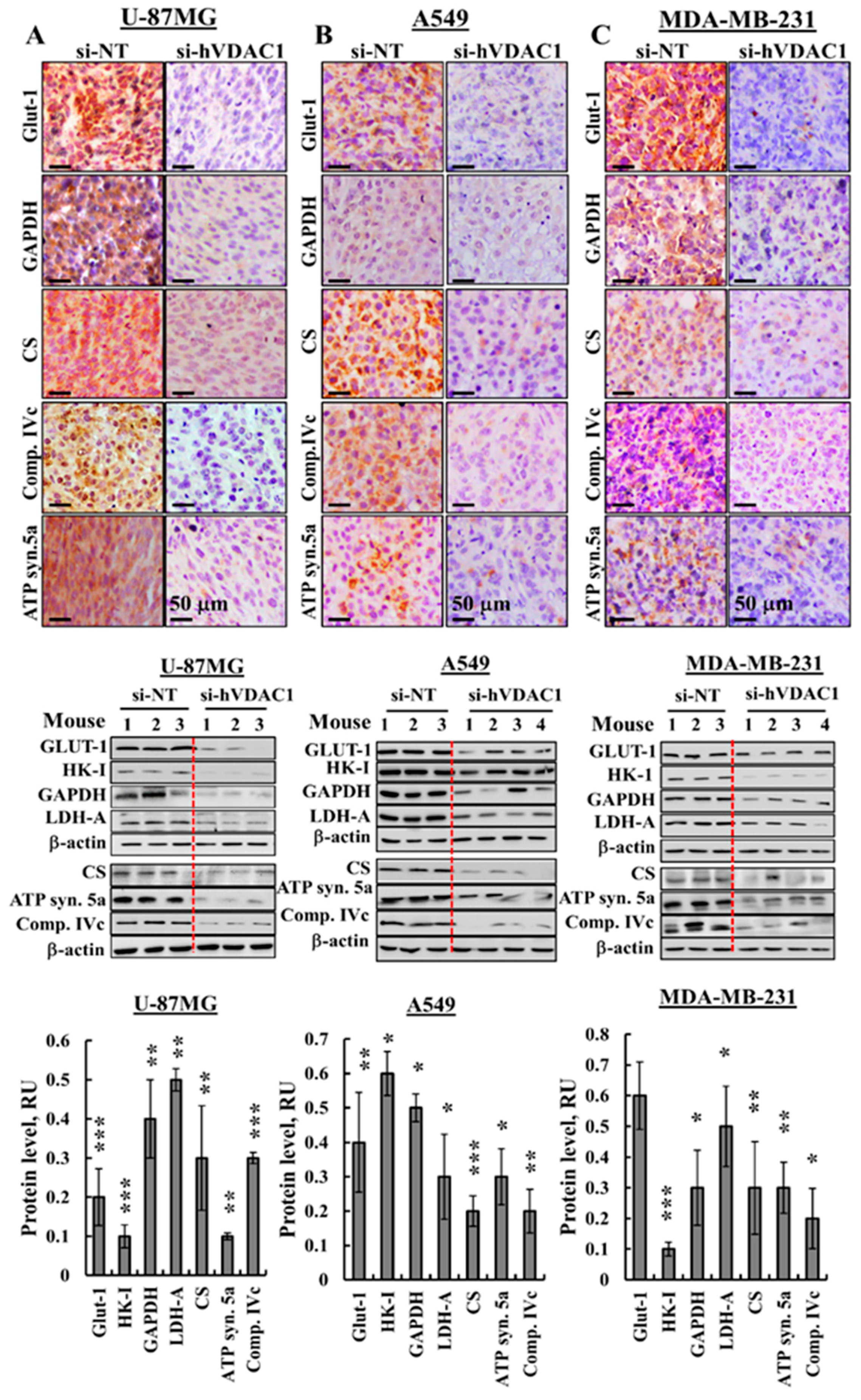
3.2. Reprogrammed Cancer Cell Metabolism is Reversed by VDAC1 Depletion
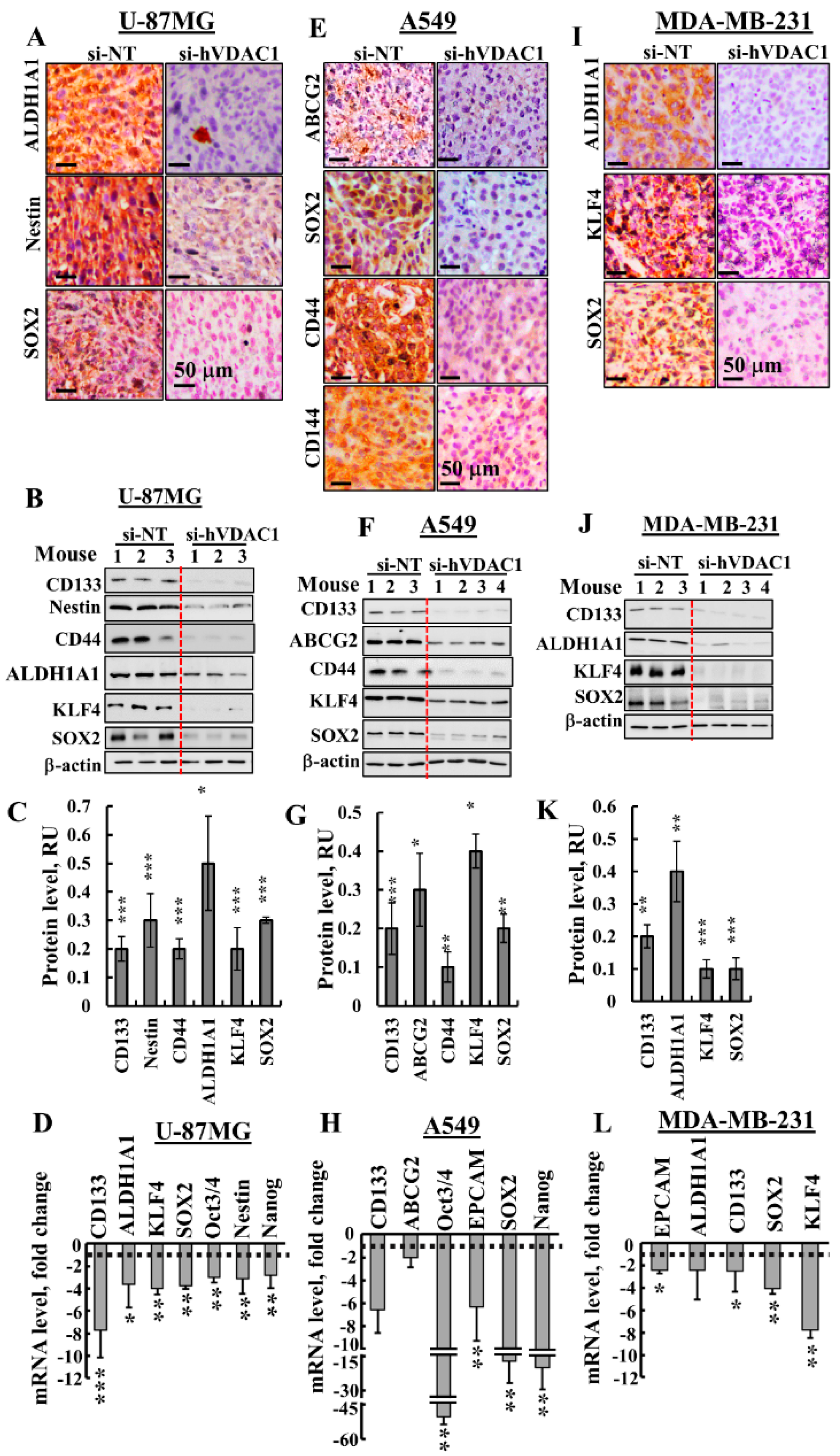
3.3. Treating Tumours with si-hVDAC1 Eliminates CSCs
3.4. Differentiation of Cancer Cells in GBM, Lung Cancer and Breast Cancer Tumours
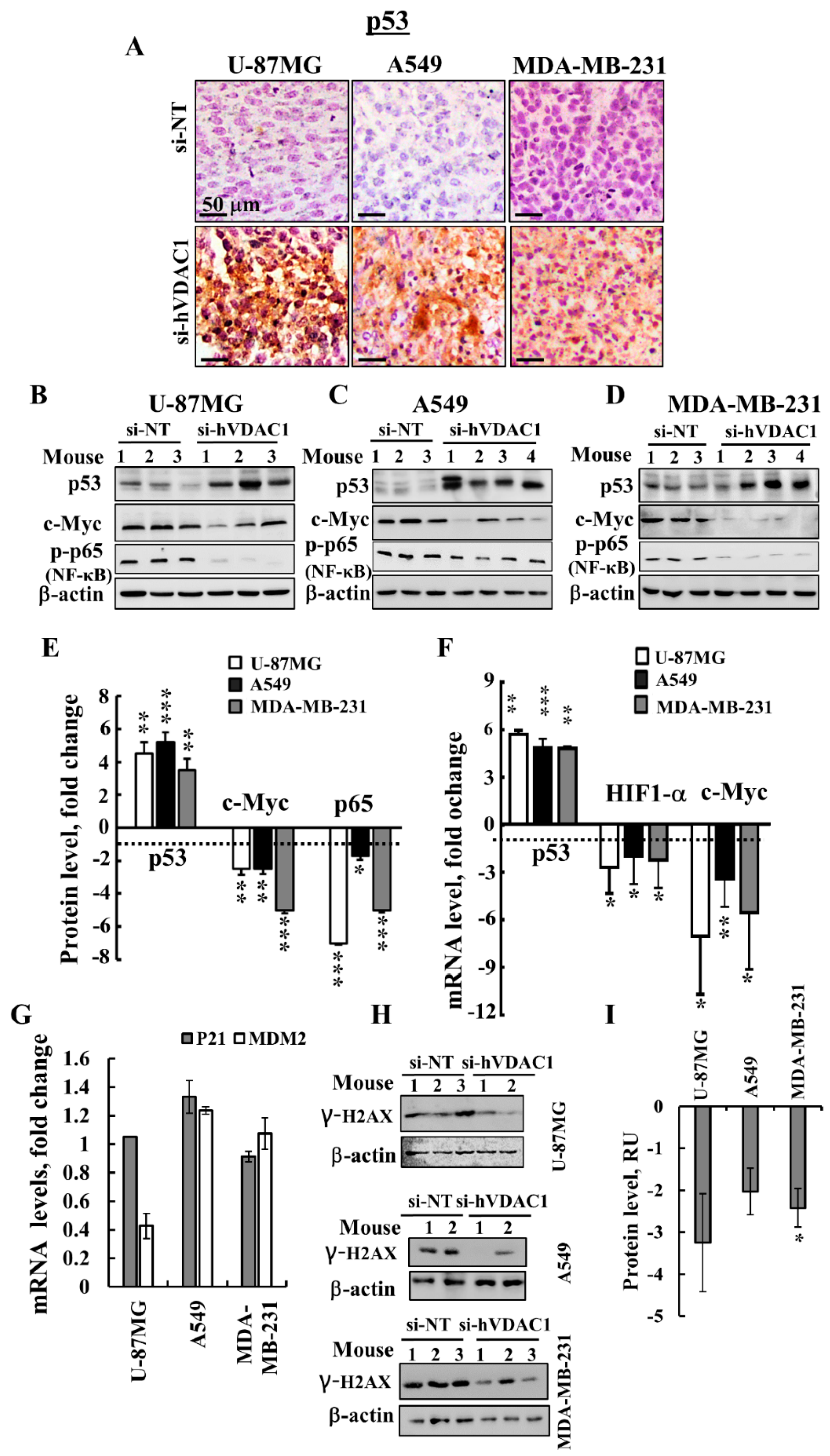
3.5. VDAC1 Depletion Alters the Expression of a Master Transcription Factor (TF)
4. Discussion
4.1. Reprogrammed Cancer Cell Metabolism is Reversed by VDAC1 Depletion, Resulting in Inhibited Cell Proliferation and Tumour Growth
4.2. Alterations in the Expression of TFs are Involved in si-hVDAC1-Induced Reprogramming of Cancer Cells
4.3. si-hVDAC1 Reduces CSC Levels and Induces Differentiation
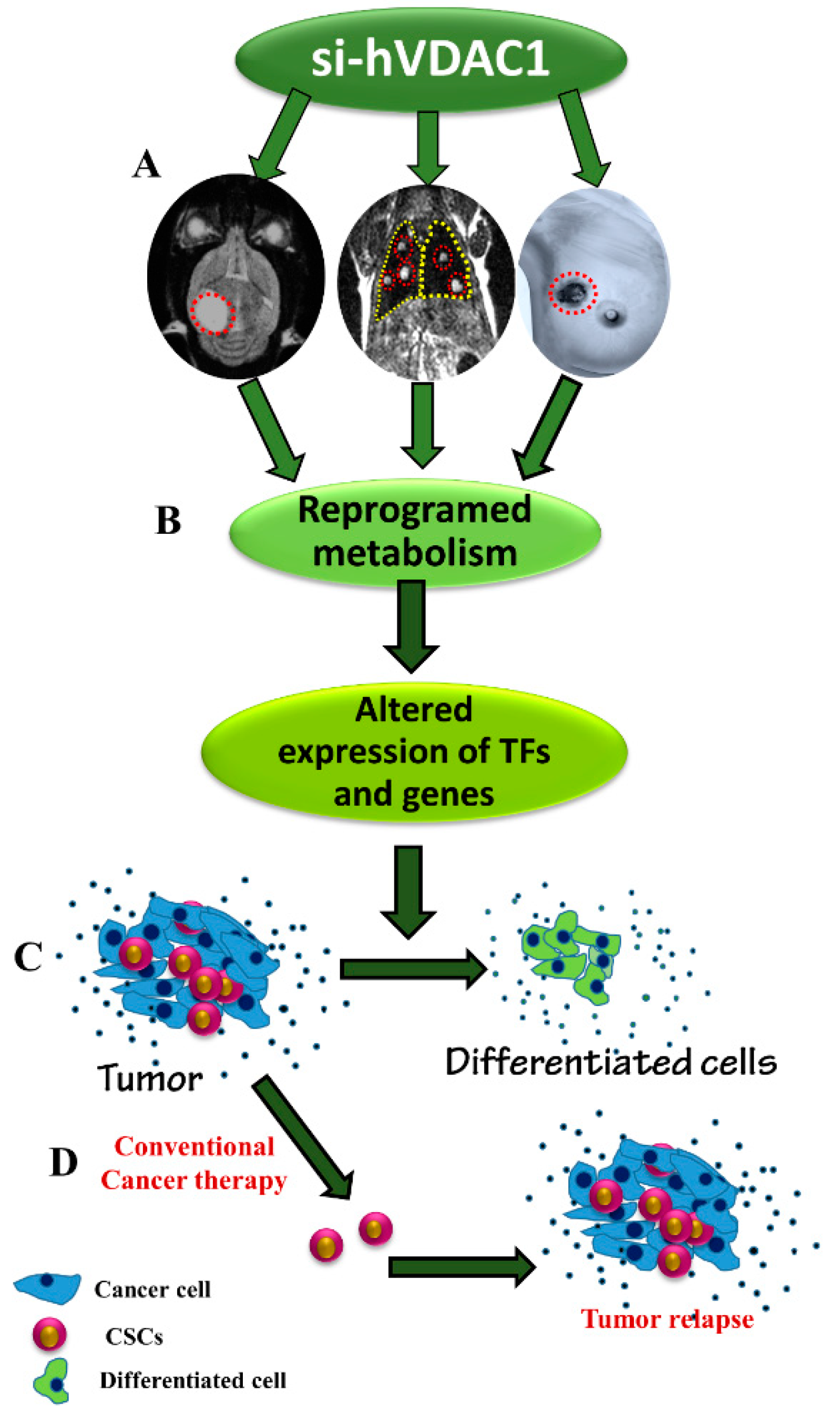
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wenger, J.B.; Chun, S.Y.; Dang, D.T.; Luesch, H.; Dang, L.H. Combination therapy targeting cancer metabolism. Med. Hypotheses 2011, 76, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Dmitriev, A.A.; Lakunina, V.A.; Kirpiy, A.A.; Kudryavtseva, A.V. Targeting VDAC-bound hexokinase II: A promising approach for concomitant anti-cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1221–1233. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef]
- Koren, I.; Raviv, Z.; Shoshan-Barmatz, V. Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol. Ther. 2010, 9, 1046–1052. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Majeed, R.H.A.; Qurishi, Y.; Qazi, A.K.; Hussain, A.; Ahmed, M.; Najar, R.A.; Bhat, J.A.; Singh, S.K.; Saxena, A.K. Therapeutic Targeting of Cancer Cell Metabolism: Role of Metabolic Enzymes, Oncogenes and Tumor Suppressor Genes. J. Cancer Sci. Ther. 2012, 4, 281–291. [Google Scholar] [CrossRef]
- Jeon, S.M.; Hay, N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch. Pharm. Res. 2015, 38, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Steinberg, G.R.; Singh, G.; Tsakiridis, T. AMP-activated protein kinase (AMPK) beyond metabolism: A novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 2014, 15, 156–169. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar]
- Xavier, J.M.; Morgado, A.L.; Sola, S.; Rodrigues, C.M. Mitochondrial translocation of p53 modulates neuronal fate by preventing differentiation-induced mitochondrial stress. Antioxid. Redox Signal. 2014, 21, 1009–1024. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Wu, S.; Li, J.; Fu, W.; He, W.; Liu, X.; Slutsky, A.S.; Zhang, H.; Li, Y. Human alveolar epithelial type II cells in primary culture. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.J. Biology of alveolar type II cells. Respirology 2006, 11, S12–S15. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kotton, D.N.; Fine, A. Lung stem cells. Cell Tissue Res. 2008, 331, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Hogan, B.L. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 493–512. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Lin, V.C.; Jin, R.; Tan, P.H.; Aw, S.E.; Woon, C.T.; Bay, B.H. Progesterone induces cellular differentiation in MDA-MB-231 breast cancer cells transfected with progesterone receptor complementary DNA. Am. J. Pathol. 2003, 162, 1781–1787. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. CMLS 2008, 65, 3981–3999. [Google Scholar] [CrossRef]
- El-Deiry, W.S. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Momand, J.; Wu, H.H.; Dasgupta, G. MDM2—Master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Krelin, Y.; Arif, T.; Jeger, R.; Shoshan-Barmatz, V. A New Role for the Mitochondrial Pro-apoptotic Protein SMAC/Diablo in Phospholipid Synthesis Associated with Tumorigenesis. Mol. Ther. 2018, 26, 680–694. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pedini, F.; Mollinari, C.; Condorelli, G.; Bonci, D.; Bez, A.; Colombo, A.; Parati, E.; Peschle, C.; De Maria, R. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J. Exp. Med. 2004, 200, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Krelin, Y.; Zhang, L.; Kang, T.B.; Appel, E.; Kovalenko, A.; Wallach, D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008, 15, 1350–1355. [Google Scholar] [CrossRef]
- Fernando, P.; Kelly, J.F.; Balazsi, K.; Slack, R.S.; Megeney, L.A. Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 11025–11030. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep. 2012, 13, 322–330. [Google Scholar] [CrossRef]
- Murray, T.V.; McMahon, J.M.; Howley, B.A.; Stanley, A.; Ritter, T.; Mohr, A.; Zwacka, R.; Fearnhead, H.O. A non-apoptotic role for caspase-9 in muscle differentiation. J. Cell Sci. 2008, 121, 3786–3793. [Google Scholar] [CrossRef]
- Nur, E.K.A.; Gross, S.R.; Pan, Z.; Balklava, Z.; Ma, J.; Liu, L.F. Nuclear translocation of cytochrome c during apoptosis. J. Biol. Chem. 2004, 279, 24911–24914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Aviles, E.R., Jr.; Fujikawa, D.G. Nuclear translocation of mitochondrial cytochrome c, lysosomal cathepsins B and D, and three other death-promoting proteins within the first 60 minutes of generalized seizures. J. Neurosci. Res. 2010, 88, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.J.; Kitagawa, H.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol. Metab. TEM 2013, 24, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Labagnara, M.; Friedman, M.; Kwasnicki, A.; Murali, R. Glioblastoma: Molecular pathways, stem cells and therapeutic targets. Cancers 2015, 7, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell. Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Marin-Hernandez, A.; Saavedra, E.; Pardo, J.P.; Ralph, S.J.; Rodriguez-Enriquez, S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 2014, 50, 10–23. [Google Scholar] [CrossRef]
- Rios, M.; Foretz, M.; Viollet, B.; Prieto, A.; Fraga, M.; Costoya, J.A.; Senaris, R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013, 73, 2628–2638. [Google Scholar] [CrossRef]
- Suzuki, K.; Matsubara, H. Recent advances in p53 research and cancer treatment. J. Biomed. Biotechnol. 2011, 2011, 978312. [Google Scholar] [CrossRef]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell. Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M.; Weiler, M.; Batchelor, T.; Yung, A.W.; Platten, M. Pathway inhibition: Emerging molecular targets for treating glioblastoma. Neuro Oncol. 2011, 13, 566–579. [Google Scholar] [CrossRef]
- Mendez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.C.; Newcomb, E.W.; Zagzag, D. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Chaudhary, N.; Pfluger, P.T. Metabolic benefits from Sirt1 and Sirt1 activators. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 431–437. [Google Scholar] [CrossRef]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef]
- Simmons, G.E., Jr.; Pruitt, W.M.; Pruitt, K. Diverse roles of SIRT1 in cancer biology and lipid metabolism. Int. J. Mol. Sci. 2015, 16, 950–965. [Google Scholar] [CrossRef]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Bertucci, F.; Cabaud, O.; Wicinski, J.; Finetti, P.; Josselin, E.; Adelaide, J.; Nguyen, T.T.; Monville, F.; et al. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013, 73, 7290–7300. [Google Scholar] [CrossRef]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef]
- Wang, L.; Park, P.; Zhang, H.; La Marca, F.; Lin, C.Y. Prospective identification of tumorigenic osteosarcoma cancer stem cells in OS99-1 cells based on high aldehyde dehydrogenase activity. Int. J. Cancer 2011, 128, 294–303. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef]
- Schonberg, D.L.; Lubelski, D.; Miller, T.E.; Rich, J.N. Brain tumor stem cells: Molecular characteristics and their impact on therapy. Mol. Aspects Med. 2014, 39, 82–101. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, J.D.; Barr, M.; Fennell, D.; Richard, D.; Reynolds, J.; O’Leary, J.; O’Byrne, K. The cancer stem-cell hypothesis: Its emerging role in lung cancer biology and its relevance for future therapy. J. Thorac. Oncol. 2012, 7, 1880–1890. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef]
- La Porta, C.A.; Zapperi, S. Human breast and melanoma cancer stem cells biomarkers. Cancer Lett. 2013, 338, 69–73. [Google Scholar] [CrossRef]
- Yoshida, G.J.; Saya, H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016, 107, 5–11. [Google Scholar] [CrossRef]
- Cunningham, J.J.; Ulbright, T.M.; Pera, M.F.; Looijenga, L.H. Lessons from human teratomas to guide development of safe stem cell therapies. Nat. Biotechnol. 2012, 30, 849–857. [Google Scholar] [CrossRef]
- Ugorski, M.; Dziegiel, P.; Suchanski, J. Podoplanin—A small glycoprotein with many faces. Am. J. Cancer Res. 2016, 6, 370–386. [Google Scholar]
- Astarita, J.L.; Acton, S.E.; Turley, S.J. Podoplanin: Emerging functions in development, the immune system, and cancer. Front. Immunol. 2012, 3, 283. [Google Scholar] [CrossRef] [PubMed]
- Martin-Villar, E.; Borda-d’Agua, B.; Carrasco-Ramirez, P.; Renart, J.; Parsons, M.; Quintanilla, M.; Jones, G.E. Podoplanin mediates ECM degradation by squamous carcinoma cells through control of invadopodia stability. Oncogene 2015, 34, 4531–4544. [Google Scholar] [CrossRef] [PubMed]
- Radler, P.D.; Wehde, B.L.; Wagner, K.U. Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol. Cell. Endocrinol. 2017, 451, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Krelin, Y.; Shoshan-Barmatz, V. Reducing VDAC1 expression induces a non-apoptotic role for pro-apoptotic proteins in cancer cell differentiation. Biochim. Biophys. Acta 2016, 1857, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.X.; Najbauer, J.; Woo, C.C.; Dashtipour, K.; Ribak, C.E.; Leon, M. Expression of active caspase-3 in mitotic and postmitotic cells of the rat forebrain. J. Comp. Neurol. 2001, 433, 4–22. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fold of Change (p-Value) | Function |
|---|---|---|
| CDK1- Cyclin-dependent kinase 1 [HGNC:1722] | −7.5 (0.0081) | Essential for the G1/S transition in cell cycle progression and leads to preparation for S phase entry |
| CDK2- Cyclin-dependent kinase 2 [HGNC:1771] | −4.33 (0.0080) | Involved in the activation of proliferative TF and interacts with p21 |
| CDK4- Cyclin-dependent kinase 4 [HGNC:1773] | −2.18 (0.016) | In neural stem cells, proposed to inhibit neurogenesis and expand the population of basal progenitors by shortening the duration of G1 |
| Cyclin B1- G2/mitotic-specific cyclin-B1 [HGNC:1579] | −4.62 (0.0019) | Involved in regulating G2/M phase of the cell cycle. Contributes to the switch-like all or none behaviour of the cell in deciding to commit to mitosis |
| Cyclin B2- G2/mitotic-specific cyclin-B2 [HGNC:1580] | −5.51 (0.0127) | Play a key role in transforming growth factor beta-mediated cell cycle control |
| Cyclin D3- G1/S-specific cyclin-D3 [HGNC:1585] | 3.64 (0.015) | Postulated to carry out cell cycle-independent functions in a range of terminally differentiated cells |
| CDK 5, regulatory subunit 1 (p35) [HGNC:1775] | 6.12 (0.0035) | Cyclin 5 complex that is essential for neuronal synaptic activity |
| CDK 5, regulatory subunit 2 (p39) [HGNC:1776] | 5.72 (0.016) | Involved in the activation of CDK5/TPKII |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. https://doi.org/10.3390/cancers10120499
Arif T, Paul A, Krelin Y, Shteinfer-Kuzmine A, Shoshan-Barmatz V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers. 2018; 10(12):499. https://doi.org/10.3390/cancers10120499
Chicago/Turabian StyleArif, Tasleem, Avijit Paul, Yakov Krelin, Anna Shteinfer-Kuzmine, and Varda Shoshan-Barmatz. 2018. "Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States" Cancers 10, no. 12: 499. https://doi.org/10.3390/cancers10120499
APA StyleArif, T., Paul, A., Krelin, Y., Shteinfer-Kuzmine, A., & Shoshan-Barmatz, V. (2018). Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers, 10(12), 499. https://doi.org/10.3390/cancers10120499