Immunotherapy, Radiotherapy, and Hyperthermia: A Combined Therapeutic Approach in Pancreatic Cancer Treatment

 , ,
, ,
Abstract
1. Introduction:
2. Pancreatic Cancer Tumor Microenvironment (PC-TME)
3. Specific and Integrated Immunotherapy
3.1. Immunotherapy for Pancreatic Cancer employing Checkpoint Inhibitors
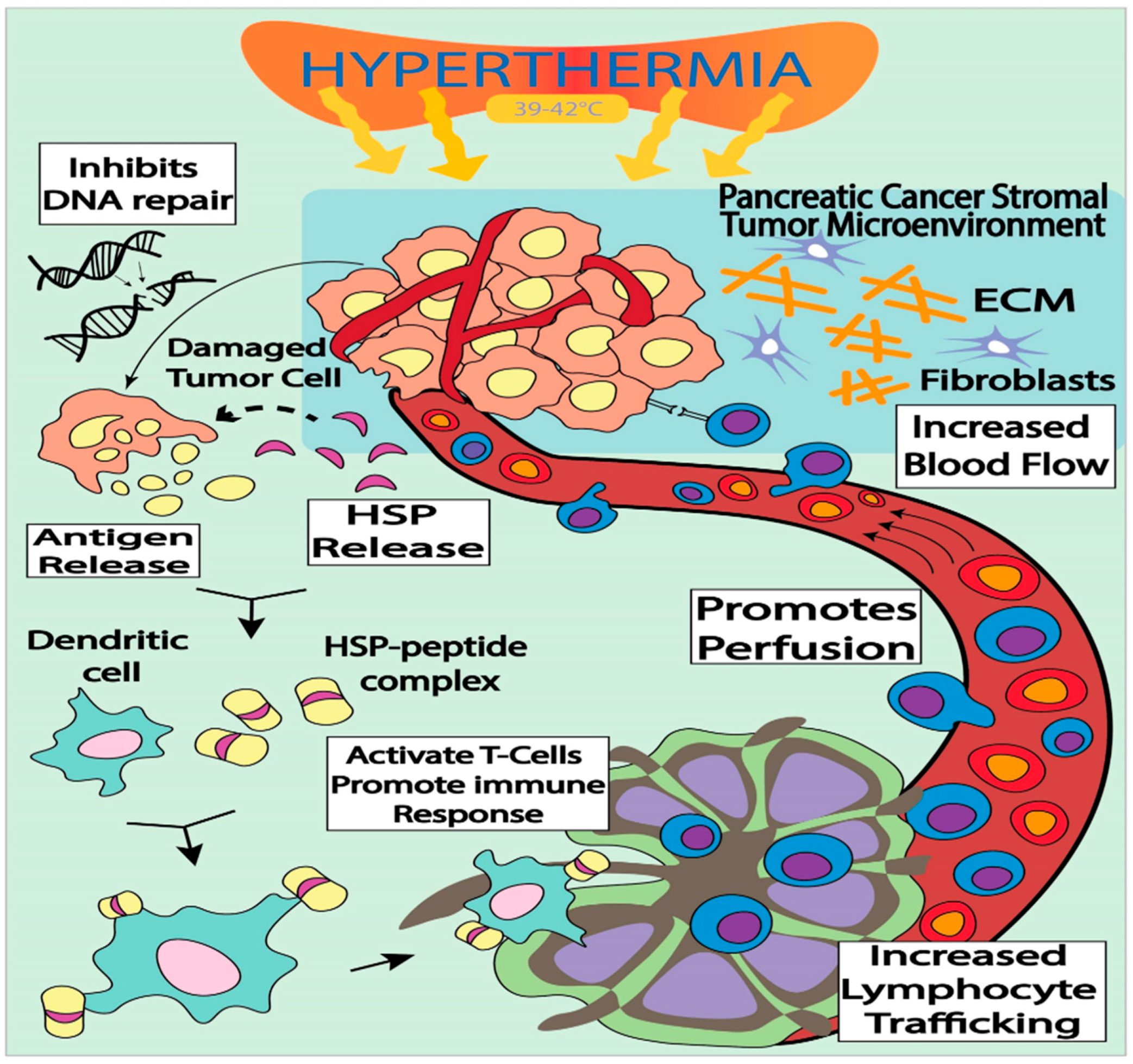
3.2. Immune-Modulation by targeted Hyperthermia as a Treatment Modality for Pancreatic Cancer
3.3. PC Immunotherapy with RT as a Treatment Modality
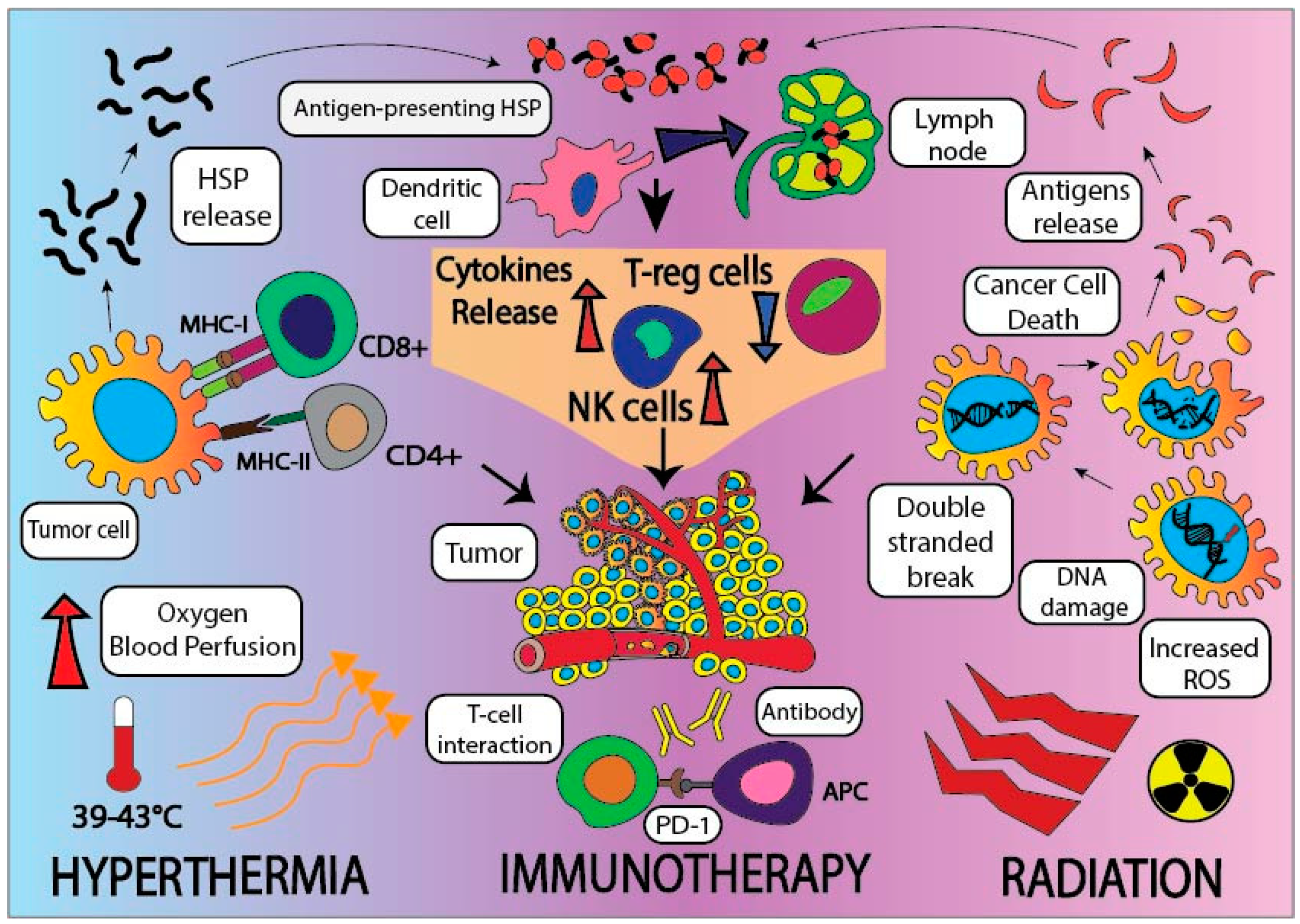
3.4. Employing Tripartite Modalities, including Radiation, Hyperthermia, and Immune Checkpoint Inhibitors, to Treat Pancreatic Cancer
4. Vaccines against Pancreatic Cancer Neoantigens, and its Success in Immunotherapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2015. Available online: https://seer.cancer.gov/csr/1975_2015/ (accessed on 28 November 2018).
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Wu, C.-Y.; Yeh, Y.-C.; Shyr, Y.-M.; Wu, Y.-Y.; Kuo, C.-Y.; Hung, Y.-P.; Chen, M.-H.; Lee, W.-P.; Luo, J.-C.; et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: A randomized, open-label, prospective trial. Oncotarget 2015, 6, 18162–18173. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Askan, G.; Hu, Z.I.; O’Reilly, E.M. Immunotherapy in pancreatic ductal adenocarcinoma: An emerging entity? Ann. Oncol. 2017, 28, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Neesse, P.; Michl, K.K.; Frese, C.; Feig, N.; Cook, M.A.; Jacobetz, M.P.; Lolkema, M.; Buchholz, K.P.; Olive, T.M.; Gress, D.A. Tuveson, Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Jain, R.K. Tumor microvasculature and microenvironment: Targets for anti-angiogenesis and normalization. Microvasc. Res. 2007, 74, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-F.; Qin, Y.; Li, D.-G.; Kerr, D. Retrospective Clinical Study of Advanced Pancreatic Cancer Treated with Chemotherapy and Abdominal Hyperthermia. J. Glob. Oncol. 2017, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; Vriend, L.E.M.; Krawczyk, P.M.; Horsman, M.R.; Franken, N.A.P.; Crezee, J. Targeting therapy-resistant cancer stem cells by hyperthermia. Int. J. Hyperth. 2017, 33, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Masunaga, S.; Nagata, K.; Suzuki, M.; Kashino, G.; Kinashi, Y.; Ono, K. Inhibition of repair of radiation-induced damage by mild temperature hyperthermia, referring to the effect on quiescent cell populations. Radiat. Med. 2007, 25, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, G.; Szasz, O.; Szasz, A. Oncothermia: A new paradigm and promising method in cancer therapies. Acupunct. Electrother. Res. 2013, 38, 161–197. [Google Scholar] [CrossRef] [PubMed]
- Aliru, M.L.; Schoenhals, J.E.; Venkatesulu, B.P.; Anderson, C.C.; Barsoumian, H.B.; Younes, A.I.; Mahadevan, L.S.K.; Soeung, M.; Aziz, K.E.; Welsh, J.W.; et al. Radiation therapy and immunotherapy: What is the optimal timing or sequencing? Immunotherapy 2018, 10, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Amedei, E.; Niccolai, M.; Benagiano, C.; Della Bella, F.; Cianchi, P.; Bechi, A.; Taddei, L.; Bencini, M.; Farsi, P.; Cappello, D.; et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol. Immunother. 2013, 62, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Coleman, C.N.; Formenti, S.C. Radiotherapy: Changing the Game in Immunotherapy. Trends Cancer 2016, 2, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Wang, S.; Nadella, S.; Jablonski, S.A.; Weiner, L.M. Cholecystokinin receptor antagonist alters pancreatic cancer microenvironment and increases efficacy of immune checkpoint antibody therapy in mice. Cancer Immunol. Immunother. 2018, 67, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, C.; Zhang, X.; Hu, Q.; Zhang, Y.; Liu, Q.; Wen, D.; Milligan, J.; Bellotti, A.; Huang, L.; et al. A melanin-mediated cancer immunotherapy patch. Sci. Immunol. 2017, 2, e5692. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Yu, J.; Wen, D.; Kahkoska, A.R.; Gu, Z. Polymeric microneedles for transdermal protein delivery. Adv. Drug Deliv. Rev. 2018, 127, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.; Drake, C. Immunotherapy earns its spot in the ranks of cancer therapy. J. Exp. Med. 2012, 209, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Kunk, P.R.; Bauer, T.W.; Slingluff, C.L.; Rahma, O.E. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J. Immunother. Cancer 2016, 4, e14. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity. 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Lund, A.W. Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer 2012, 12, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Tsukumo, S.-I.; Yasutomo, K. Regulation of CD8+ T Cells and Antitumor Immunity by Notch Signaling. Front. Immunol. 2018, 9, e101. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Brunet, L.R.; Hagemann, T.; Gaya, A.; Mudan, S.; Marabelle, A. Have lessons from past failures brought us closer to the success of immunotherapy in metastatic pancreatic cancer? Oncoimmunology 2016, 5, e1112942. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015, 4, e1008824. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W. Effect of local hyperthermia on blood flow and microenvironment: A review. Cancer Res. 1984, 44, 4721–4730. [Google Scholar]
- Yagawa, Y.; Tanigawa, K.; Kobayashi, Y.; Yamamoto, M. Cancer immunity and therapy using hyperthermia with immunotherapy, radiotherapy, chemotherapy, and surgery. J. Cancer Meta. Treat 2017, 3, e218. [Google Scholar] [CrossRef]
- Binder, R.J.; Srivastava, P.K. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat. Immunol. 2005, 6, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Udono, H.; Blachere, N.; Li, Z. Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics 1994, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Tsan, M.-F.; Gao, B. Heat shock proteins and immune system. J. Leukoc. Biol. 2009, 85, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.M.; Lee, S.C.; Andrade Filho, P.A.; Lord, C.A.; Jie, H.-B.; Davidson, H.C.; Lopez-Albaitero, A.; Gibson, S.P.; Gooding, W.E.; Ferrone, S.; et al. Cetuximab-Activated Natural Killer and Dendritic Cells Collaborate to Trigger Tumor Antigen-Specific T-cell Immunity in Head and Neck Cancer Patients. Clin. Cancer Res. 2013, 19, 1858–1872. [Google Scholar] [CrossRef] [PubMed]
- Breloer, M.; Dorner, B.; Moré, S.H.; Roderian, T.; Fleischer, B.; von Bonin, A. Heat shock proteins as “danger signals”: Eukaryotic Hsp60 enhances and accelerates antigen-specific IFN-γ production in T cells. Eur. J. Immunol. 2001, 31, 2051–2059. [Google Scholar] [CrossRef]
- Skitzki, J.J.; Repasky, E.A.; Evans, S.S. Hyperthermia as an immunotherapy strategy for cancer. Curr. Opin. Investig. Drugs. 2009, 10, 550–558. [Google Scholar] [PubMed]
- Schildkopf, P.; Ott, O.J.; Frey, B.; Wadepohl, M.; Sauer, R.; Fietkau, R.; Gaipl, U.S. Biological rationales and clinical applications of temperature controlled hyperthermia—implications for multimodal cancer treatments. Curr. Med. Chem. 2010, 17, 3045–3057. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; June, C.H. Adoptive T Cell Transfer for Cancer Immunotherapy in the Era of Synthetic Biology. Immunity 2013, 39, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.; Péter, M.; Liebisch, G.; Horváth, I.; Török, Z.; Nagy, E.; Maslyanko, A.; Benkő, S.; Schmitz, G.; Harwood, J.L.; et al. Lipidomics reveals membrane lipid remodelling and release of potential lipid mediators during early stress responses in a murine melanoma cell line. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids. 2010, 1801, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, R.; Oda, T.; Hashimoto, S.; Kurokawa, T.; Inagaki, Y.; Shimomura, O.; Ohara, Y.; Yamada, K.; Akashi, Y.; Enomoto, T.; et al. Cetuximab delivery and antitumor effects are enhanced by mild hyperthermia in a xenograft mouse model of pancreatic cancer. Cancer Sci. 2016, 107, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Mallory, M.; Gogineni, E.; Jones, G.C.; Greer, L.; Ii, C.B.S. Therapeutic hyperthermia: The old, the new, and the upcoming. Crit. Rev. Oncol./Hematol. 2016, 97, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J.; Roizin-Towle, L. Biological effects of heat. Cancer Res. 1984, 44, 4708–4713. [Google Scholar]
- Ostberg, J.R.; Gellin, C.; Patel, R.; Repasky, E.A. Regulatory Potential of Fever-Range Whole Body Hyperthermia on Langerhans Cells and Lymphocytes in an Antigen-Dependent Cellular Immune Response. J. Immunol. 2001, 167, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Hasegawa, T.; Miyazawa, K.; Yoshida, K.; Takeda, T.; Takeda, H.; Takahashi, T.; Yamamoto, I.; Kobayashi, S. Hyperthemia enhances activated lymphocytes and molecular target therapy. Gan. Kagaku Ryoho. 2011, 38, 1924–1926. [Google Scholar]
- Datta, N.R.; Pestalozzi, B.; Clavien, P.-A.; Siebenhüner, A.; Puric, E.; Khan, S.; Mamot, C.; Riesterer, O.; Knuchel, J.; Reiner, C.S.; et al. Members of the H.T. members of the HEATPAC Trial Group, “HEATPAC”—A phase II randomized study of concurrent thermochemoradiotherapy versus chemoradiotherapy alone in locally advanced pancreatic cancer. Radiat. Oncol. 2017, 12, e183. [Google Scholar] [CrossRef] [PubMed]
- Beeharry, M.K.; Liu, W.-T.; Yao, X.-X.; Yan, M.; Zhu, Z.-G. A critical analysis of the cytoreductive surgery with hyperthermic intraperitoneal chemotherapy combo in the clinical management of advanced gastric cancer: An effective multimodality approach with scope for improvement. Transl. Gastroenterol. Hepatol. 2016, 1, e77. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Kokura, S.; Okayama, T.; Ishikawa, T.; Takagi, T.; Handa, O.; Naito, Y.; Yoshikawa, T. Effect of hyperthermia combined with gemcitabine on apoptotic cell death in cultured human pancreatic cancer cell lines. Int. J. Hyperth. 2009, 25, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Maebayashi, T.; Ishibashi, N.; Aizawa, T.; Sakaguchi, M.; Sato, T.; Kawamori, J.; Tanaka, Y. Treatment outcomes of concurrent hyperthermia and chemoradiotherapy for pancreatic cancer: Insights into the significance of hyperthermia treatment. Oncol. Lett. 2017, 13, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Thind, K.; Padrnos, L.J.; Ramanathan, R.K.; Borad, M.J. Immunotherapy in pancreatic cancer treatment: A new frontier. Therap. Adv. Gastroenterol. 2017, 10, 168–194. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Wang, X.; Soh, H.; Seyedin, S.; Cortez, M.A.; Krishnan, S.; Massarelli, E.; Hong, D.; Naing, A.; Diab, A.; et al. Combining Radiation and Immunotherapy: A New Systemic Therapy for Solid Tumors? Cancer Immunol. Res. 2014, 2, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining Radiotherapy and Cancer Immunotherapy: A Paradigm Shift. JNCI J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, e51. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti-CTLA-4 Antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Demaria, S.; Schiff, P.B.; Chachoua, A.; Formenti, S.C. An Abscopal Response to Radiation and Ipilimumab in a Patient with Metastatic Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2013, 1, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.G.; Bourhis, J.; Coukos, G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J. Clin. 2017, 67, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: Using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.H.; Byrne, K.T.; Vonderheide, R.H. Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer 2018, 4, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T. Hyperthermia Combined with Chemotherapy: Pancreatic Cancer. In Hyperthermic Oncology from Bench to Bedside; Springer: Singapore, 2016; pp. 275–285. [Google Scholar]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Apetoh, L. Radiotherapy and Immunogenic Cell Death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.-L. Modulating both tumor cell death and innate immunity is essential for improving radiation therapy effectiveness. Front. Immunol. 2017, 8, e613. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Regulatory T cells and immune tolerance to tumors. Immunol. Res. 2010, 46, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Nguy, S.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Daley, D.; Barilla, R.; et al. Radiation therapy induces macrophages to suppress t-cell responses against pancreatic tumors in mice. Gastroenterology 2016, 150, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Azad, S.; Yin Lim, Z.; D’Costa, K.; Jones, A.; Diana, O.J.; Sansom, P.; Kruger, S.; Liu, W.G.; McKenna, O.; Dushek, R.J.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Laheru, D.; Jaffee, E.M. Immunotherapy for pancreatic cancer—Science driving clinical progress. Nat. Rev. Cancer 2005, 5, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, A.D.; Broome, P.; Topuzov, E.G.; Garin, A.M.; Pulay, I.; Humphreys, J.; Whitehead, A.; Takhar, A.; Rowlands, B.J.; Beckingham, I.J. An international multicenter randomized controlled trial of G17DT in patients with pancreatic cancer. Pancreas 2012, 41, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V2.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with gvax pancreas prime and listeria monocytogenes–expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Robbins, P.F. Cancer immunotherapy targeting neoantigens. Semin. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-Q.; Buchsbaum, D.J. Monoclonal antibodies in the treatment of pancreatic cancer. Immunotherapy 2009, 1, 223–229. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; Stashwick, C.; Plesa, G.; Tanyi, J.L. Overcoming barriers of car T-cell therapy in patients with mesothelin-expressing cancers. Immunotherapy 2017, 9, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Andersson, R.; Bauden, M.; Hammes, S.; Holdenrieder, S.; Ansari, D. Immune checkpoint therapy for pancreatic cancer. World J. Gastroenterol. 2016, 22, e9457. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Trials | Control Arm | Experimental Arm | Type of Hyperthermia |
|---|---|---|---|
| NCT01077427 Phase III | Adjuvant Gemcitabine and Capecitabine | Adjuvant Gemcitabine Cisplatin and regional hyperthermia | Regional hyperthermia |
| NCT02439593 Phase II | Chemo radiotherapy | Thermo chemo radiotherapy | Regional hyperthermia |
| NCT03251365 Phase II and III | Adjuvant chemotherapy Gemcitabine | HIPEC gemcitabine | Hyperthermic intraabdominal chemotherapy |
| NCT02862015 Phase II | Folfirinox or gemcitabine-based chemotherapy | Oncothermia | Whole body hyperthermia |
| NCT02973217 | Standard chemotherapy | Specific form of thermotherapy—Immuno Stimulating interstitial laser thermotherapy | Thermotherapy |
| Trials | Control Arm | Experimental Arm | Immunotherapy |
|---|---|---|---|
| NCT02405585 Phase II | Neoadjuvant chemotherapy followed by SBRT with Gemcitabine in borderline resectable pancreatic cancer | Neoadjuvant chemotherapy with Immunotherapy Algenpantucel-L Followed by SBRT with Gemcitabine | Algenpantucel-L |
| NCT01959672 Phase II | Neoadjuvant chemotherapy (gemcitabine, leucovorin, 5FU) followed by SBRT with Nelfinavir | Add Oregovomab with chemotherapy | Oregovomab (Chin) |
| NCT01072981 Phase III | Post-Surgery, adjuvant Gemcitabine, or 5FU chemo radiation | Post-Surgery, adjuvant Gemcitabine, or 5FU chemo radiation with Algenpantucel-L | Algenpantucel-L |
| NCT01903083 Phase I | No control | Chemo immunotherapy followed by assessment for surgery | Tadalafil |
| NCT02648282 Phase II | Chemotherapy with radiation therapy | GVAX vaccine and Pembrolizumab along with chemo radiation therapy | GVAX and Pembrolizumab |
| NCT03104439 Phase II | Radiation therapy | Nivolumab and Ipilimumab with radiation therapy | Nivolumab and Ipilimumab |
| NCT02305186 Phase II | Neoadjuvant chemoradiation | Neoadjuvant chemoradiation with Pembrolizumab | Pembrolizumab |
| NCT01342224 Phase I | No control | Vaccination with chemotherapy followed by radiation therapy | Tadalafil and Vaccination |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, J.; Shukla, H.D.; Soman, S.; Samanta, S.; Singh, P.; Kamlapurkar, S.; Saeed, A.; Amin, N.P.; Vujaskovic, Z. Immunotherapy, Radiotherapy, and Hyperthermia: A Combined Therapeutic Approach in Pancreatic Cancer Treatment. Cancers 2018, 10, 469. https://doi.org/10.3390/cancers10120469
Mahmood J, Shukla HD, Soman S, Samanta S, Singh P, Kamlapurkar S, Saeed A, Amin NP, Vujaskovic Z. Immunotherapy, Radiotherapy, and Hyperthermia: A Combined Therapeutic Approach in Pancreatic Cancer Treatment. Cancers. 2018; 10(12):469. https://doi.org/10.3390/cancers10120469
Chicago/Turabian StyleMahmood, Javed, Hem D. Shukla, Sandrine Soman, Santanu Samanta, Prerna Singh, Shriya Kamlapurkar, Ali Saeed, Neha P. Amin, and Zeljko Vujaskovic. 2018. "Immunotherapy, Radiotherapy, and Hyperthermia: A Combined Therapeutic Approach in Pancreatic Cancer Treatment" Cancers 10, no. 12: 469. https://doi.org/10.3390/cancers10120469
APA StyleMahmood, J., Shukla, H. D., Soman, S., Samanta, S., Singh, P., Kamlapurkar, S., Saeed, A., Amin, N. P., & Vujaskovic, Z. (2018). Immunotherapy, Radiotherapy, and Hyperthermia: A Combined Therapeutic Approach in Pancreatic Cancer Treatment. Cancers, 10(12), 469. https://doi.org/10.3390/cancers10120469