Renal Cell Carcinoma Is Abrogated by p53 Stabilization through Transglutaminase 2 Inhibition

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Expression of TGase 2 Is Increased in RCC Clinical Samples and Is Associated with Survival of RCC Patients
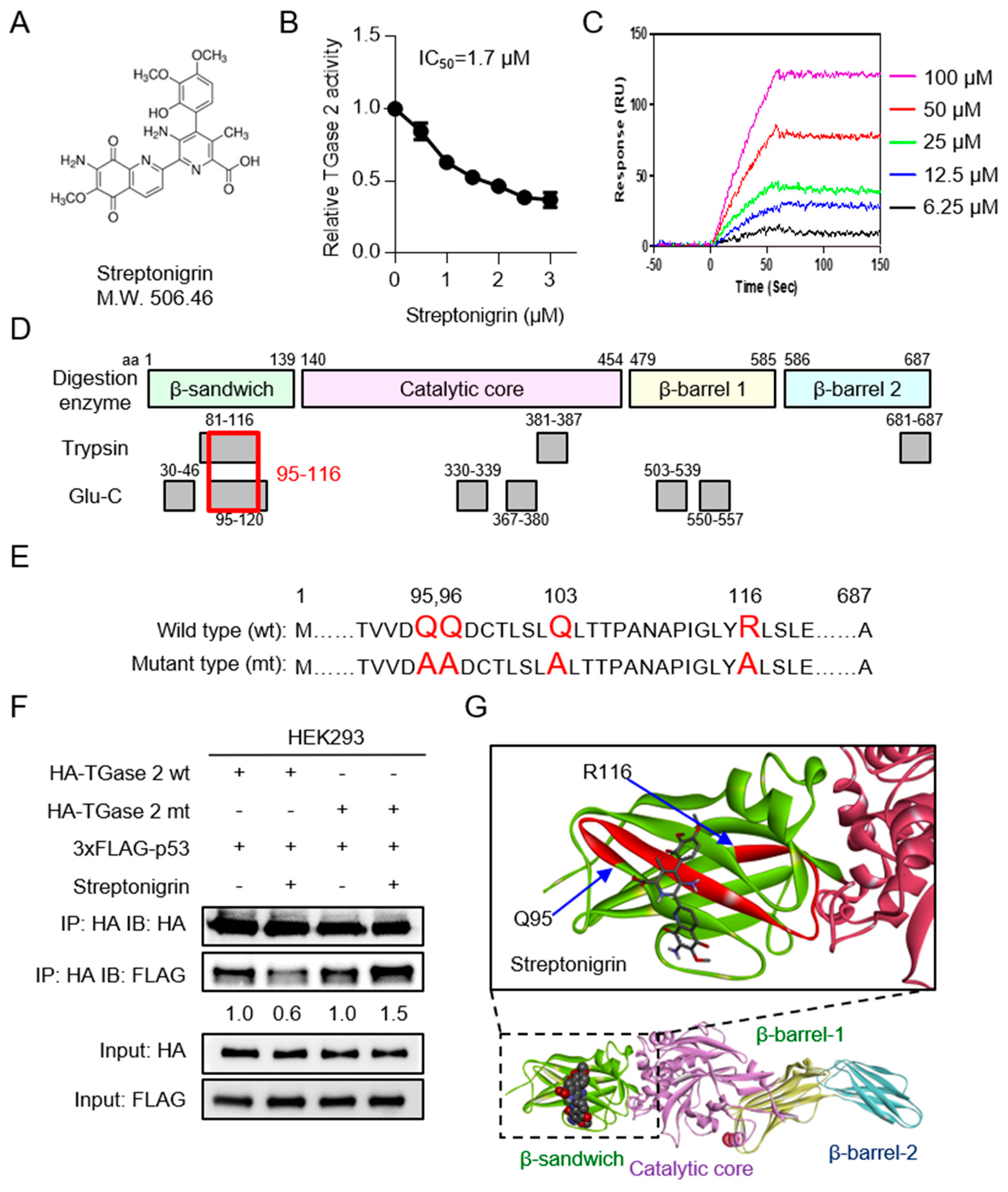
2.2. Streptonigrin Inhibits TGase 2 by Interacting with the β-Sandwich Domain
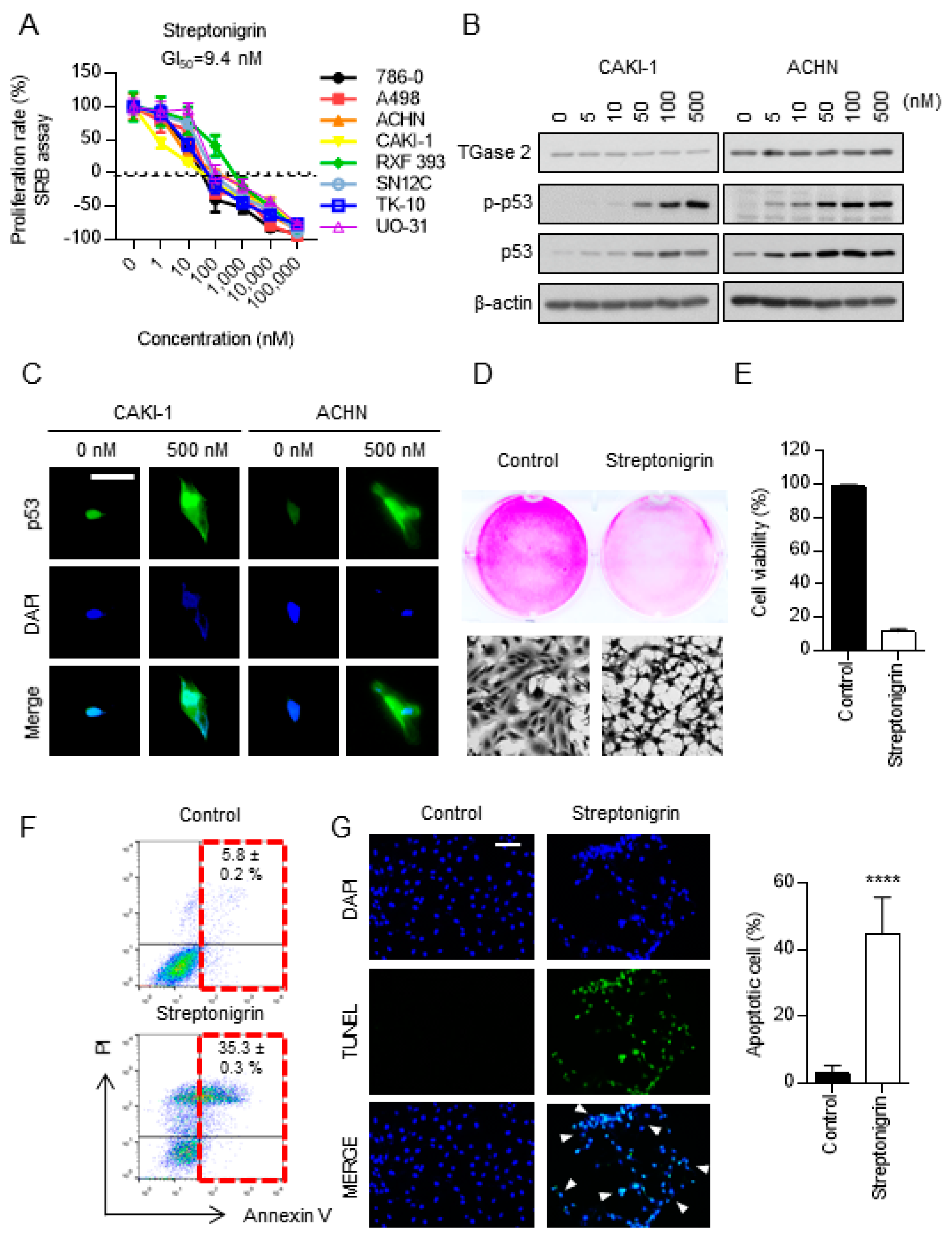
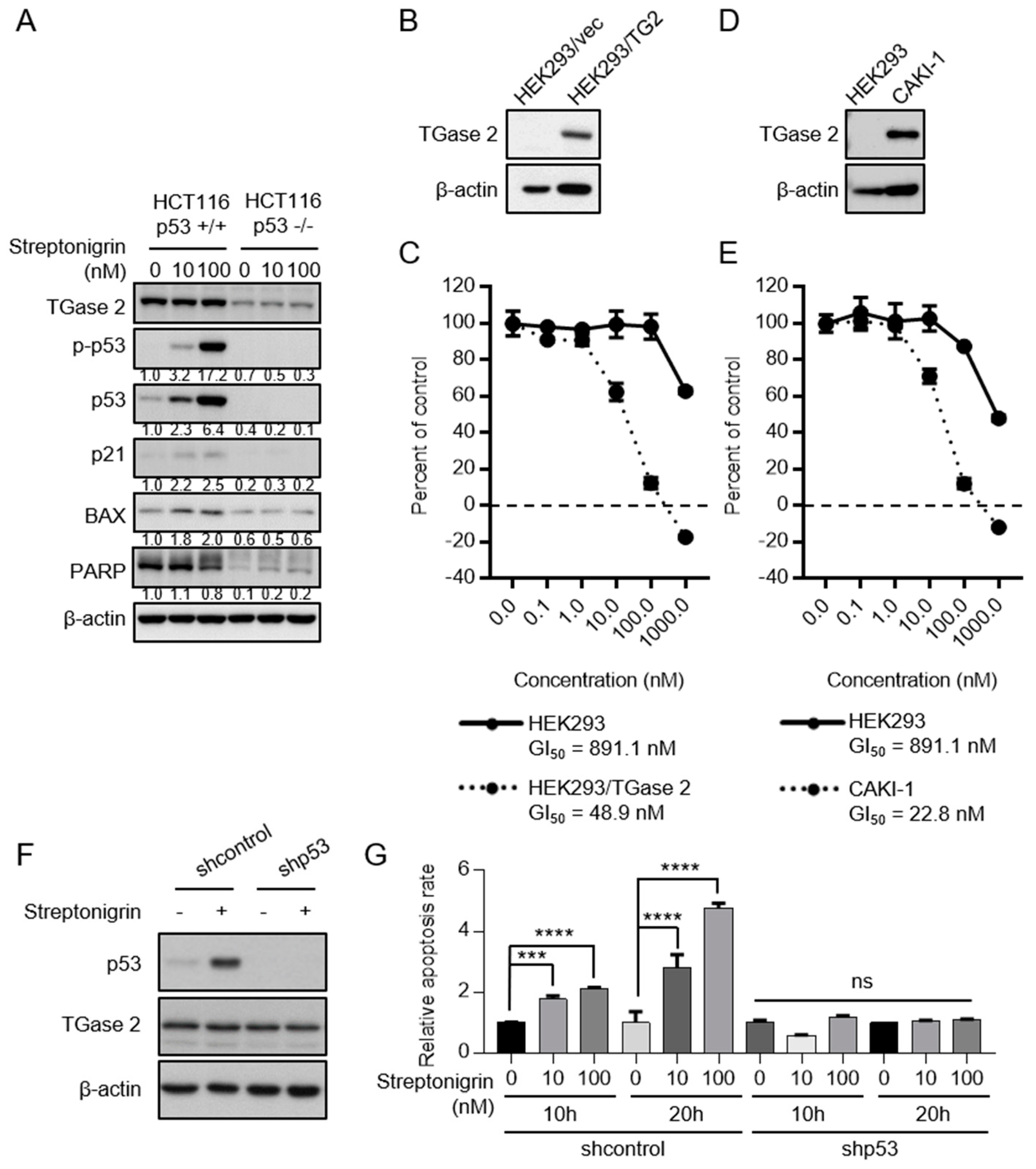
2.3. Streptonigrin Stabilizes p53 by Inhibiting TGase 2
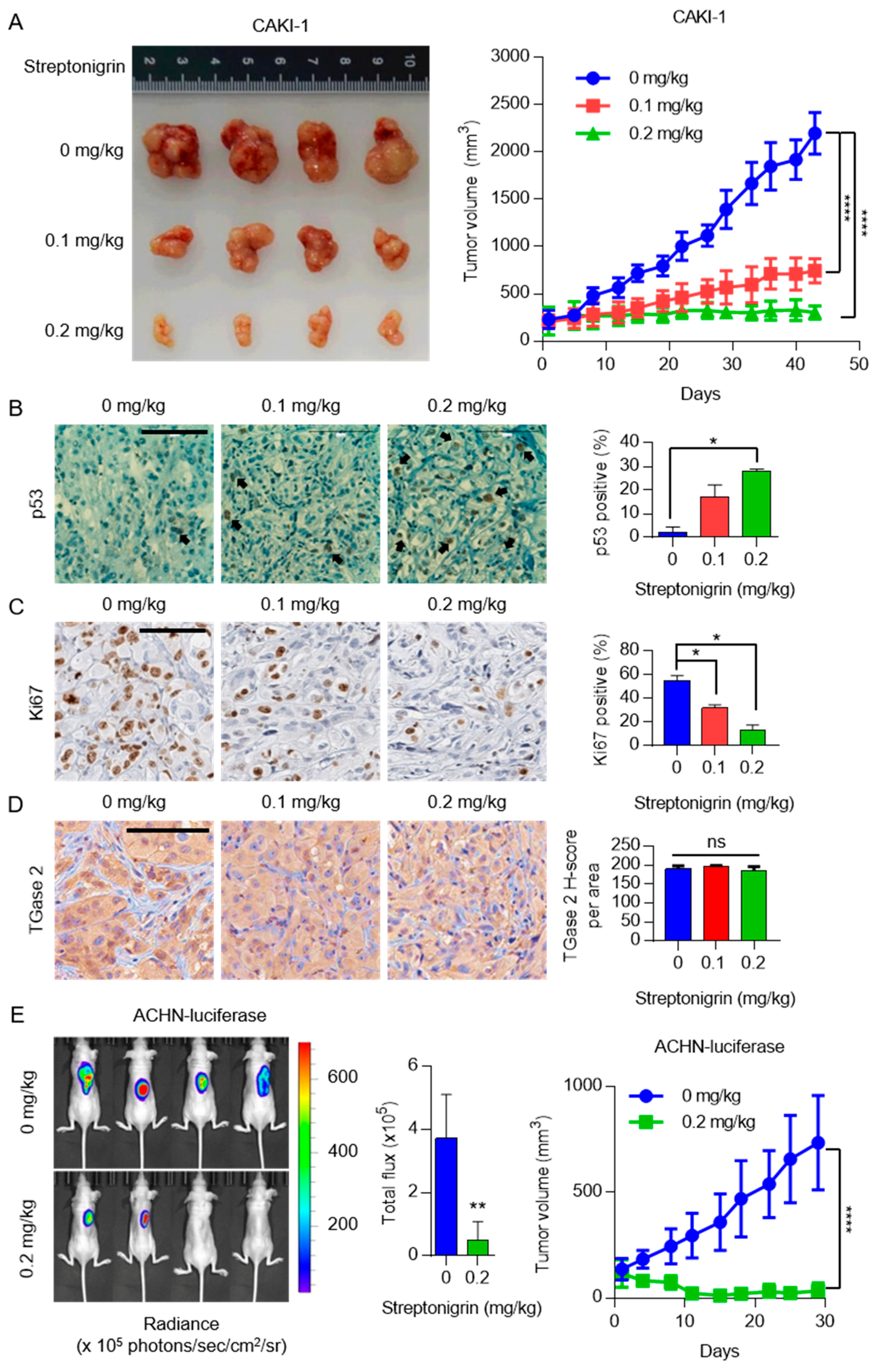
2.4. A Single Dose of Streptonigrin Inhibits the Growth of RCC Xenografts
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Immunofluorescence Analysis
4.4. TGase 2 Activity Assay
4.5. Construction of the TGase 2 Mutant
4.6. MS Analysis of the Streptonigrin Binding Site in TGase 2
4.7. Measurement of the Interaction between Streptonigrin and TGase 2
4.8. Molecular Docking Analysis
4.9. SRB Assay of Anti-Proliferative Activity
4.10. Viral Transduction
4.11. Western Blotting
4.12. Measurement of Cell Viability
4.13. Apoptosis Assay
4.14. Preclinical Xenograft Tumor Models
4.15. Automated Immunohistochemistry
4.16. In Vivo Bioluminescence Imaging
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lipworth, L.; Tarone, R.E.; McLaughlin, J.K. The epidemiology of renal cell carcinoma. J. Urol. 2006, 176, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, R.A.; Zelefsky, M.J.; Gaynor, J.J.; Fuks, Z. Patterns of failure following surgical resection of renal cell carcinoma: Implications for adjuvant local and systemic therapy. J. Clin. Oncol. 1994, 12, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Kim, D.S.; Kim, K.H.; Yoo, B.C.; Kim, S.H.; Gong, Y.D.; Kim, S.Y. Transglutaminase 2 inhibition found to induce p53 mediated apoptosis in renal cell carcinoma. FASEB J. 2013, 27, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, J.S.; Hong, D.; Lee, S.H.; Kim, N.; Lee, W.K.; Sung, T.W.; Gong, Y.D.; Kim, S.Y. Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy. Cell Death Dis. 2016, 7, e2163. [Google Scholar] [CrossRef] [PubMed]
- Balklava, Z.; Verderio, E.; Collighan, R.; Gross, S.; Adams, J.; Griffin, M. Analysis of tissue transglutaminase function in the migration of Swiss 3T3 fibroblasts: The active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J. Biol. Chem. 2002, 277, 16567–16575. [Google Scholar] [CrossRef] [PubMed]
- Verderio, E.A.; Johnson, T.; Griffin, M. Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids 2004, 26, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Sarang, Z.; Molnar, P.; Nemeth, T.; Piacentini, M.; Mastroberardino, P.G.; Falasca, L.; Aeschlimann, D.; Kovacs, J.; Kiss, I.; et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7812–7817. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Kerr, C. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol. Carcinog. 2015, 54, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cerione, R.A.; Antonyak, M. Tissue transglutaminase and its role in human cancer progression. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 78, 247–293. [Google Scholar] [PubMed]
- Mehta, K. Biological and therapeutic significance of tissue transglutaminase in pancreatic cancer. Amino Acids 2009, 36, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jeitner, T.M.; Steinert, P.M. Transglutaminases in disease. Neurochem. Int. 2002, 40, 85–103. [Google Scholar] [CrossRef]
- Muma, N.A. Transglutaminase is linked to neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Ronchetti, R.D.; Quezado, M.M.; Kim, S.Y.; Mouradian, M.M. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y. Transglutaminase 2: A new paradigm for NF-κB involvement in disease. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 78, 161–195. [Google Scholar] [PubMed]
- Lee, J.; Kim, Y.S.; Choi, D.H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.Y. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Choi, Y.B.; Han, B.G.; Park, S.Y.; Jeon, Y.; Kim, D.H.; Ahn, E.R.; Shin, J.E.; Lee, B.I.; Lee, H.; et al. Cancer cells promote survival through depletion of the von Hippel-Lindau tumor suppressor by protein crosslinking. Oncogene 2011, 30, 4780–4790. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, H.; Seki, N.; Yoshino, H.; Yamasaki, T.; Yamada, Y.; Nohata, N.; Fuse, M.; Nakagawa, M.; Enokida, H. Tumor suppressive microRNA-1285 regulates novel molecular targets: Aberrant expression and functional significance in renal cell carcinoma. Oncotarget 2012, 3, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Erdem, S.; Yegen, G.; Telci, D.; Yildiz, I.; Tefik, T.; Issever, H.; Kilicaslan, I.; Sanli, O. The increased transglutaminase 2 expression levels during initial tumorigenesis predict increased risk of metastasis and decreased disease-free and cancer-specific survivals in renal cell carcinoma. World J. Urol. 2015, 33, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Baek, H.W.; Rhee, Y.Y.; Lee, C.; Park, J.W.; Kim, H.W.; Moon, K.C. Transglutaminase 2 expression and its prognostic significance in clear cell renal cell carcinoma. J. Pathol. Transl. Med. 2015, 49, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Kim, S.J.; Kim, N.; Hong, D.; Choi, Y.B.; Lee, S.H.; Gong, Y.D.; Kim, S.Y. Transglutaminase 2 inhibitor abrogates renal cell carcinoma in xenograft models. J. Cancer Res. Clin. Oncol. 2014, 140, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Folk, J.E.; Chung, S.I. Molecular and catalytic properties of transglutaminases. Adv. Enzymol. Relat. Areas Mol. Biol. 1973, 38, 109–191. [Google Scholar] [PubMed]
- Mandrusiak, L.M.; Beitel, L.K.; Wang, X.; Scanlon, T.C.; Chevalier-Larsen, E.; Merry, D.E.; Trifiro, M.A. Transglutaminase potentiates ligand-dependent proteasome dysfunction induced by polyglutamine-expanded androgen receptor. Hum. Mol. Genet. 2003, 12, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Pinto, J.T.; Cooper, A.J.L. Cystamine and cysteamine as inhibitors of transglutaminase activity in vivo. Biosci. Rep. 2018, 38, BSR20180691. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [Google Scholar] [CrossRef] [PubMed]
- Han, B.G.; Cho, J.W.; Cho, Y.D.; Jeong, K.C.; Kim, S.Y.; Lee, B.I. Crystal structure of human transglutaminase 2 in complex with adenosine triphosphate. Int. J. Biol. Macromol. 2010, 47, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Lee, W.K.; Lee, S.H.; Jin, K.S.; Kim, K.H.; Lee, Y.; Song, M.; Kim, S.Y. Inter-molecular crosslinking activity is engendered by the dimeric form of transglutaminase 2. Amino Acids 2017, 49, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y. New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds. Med. Sci. 2018, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kang, J.H.; Lee, W.K.; Kim, S.G.; Lee, J.S.; Lee, S.H.; Park, J.B.; Kim, K.H.; Gong, Y.D.; Hwang, K.Y.; et al. Allosteric inhibition site of transglutaminase 2 is unveiled in the N terminus. Amino Acids 2018, 50, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.Y.; Streets, A.J.; Watson, P.F.; Huang, L.; Verderio, E.A.; Johnson, T.S. A crucial sequence for transglutaminase type 2 extracellular trafficking in renal tubular epithelial cells lies in its N-terminal beta-sandwich domain. J. Biol. Chem. 2011, 286, 27825–27835. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cerione, R.A.; Clardy, J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. USA 2016, 99, 2743–2747. [Google Scholar] [CrossRef] [PubMed]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef] [PubMed]
- Marsh, W.S.; Garretson, A.L.; Wesel, E.M. Streptonigrin, an antitumor agent produced by strains of Streptomyces flocculus. I. Microbiological studies. Antibiot. Chemother. 1961, 11, 151–157. [Google Scholar]
- Dreyton, C.J.; Anderson, E.D.; Subramanian, V.; Boger, D.L.; Thompson, P.R. Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg. Med. Chem. 2014, 22, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.; Hasan, S.K.; Lown, J.W.; Morgan, A.R. The mechanism of the degradation of DNA by streptonigrin. Can. J. Biochem. 1976, 54, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Tabor, E.; Becker, Y. Abnormal response of ataxia-telangiectasia cells to agents that break the deoxyribose moiety of DNA via a targeted free radical mechanism. Carcinogenesis 1983, 4, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.M.; Gordon, J.A.; Sewell, I.A.; Ellis, H. A trial of streptonigrin in the treatment of advanced malignant disease. Br. J. Cancer 1967, 21, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Farber, N.J.; Kim, C.J.; Modi, P.K.; Hon, J.D.; Sadimin, E.T.; Singer, E.A. Renal cell carcinoma: The search for a reliable biomarker. Transl. Cancer Res. 2017, 6, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kakutani, S.; Sato, Y.; Hanashi, A.; Kinoshita, Y.; Ishikawa, A. Drug review: Pazopanib. Jpn. J. Clin. Oncol. 2018, 48, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.L.; Ding, R.; Sharma, V.K.; Chon, W.J.; Lagman, M.; Suthanthiran, M. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney Int. 2003, 63, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, S.H.; Kim, S.Y. Discovery of a novel target for renal cell carcinoma: Transglutaminase 2. Cell Death Dis. 2016, 7, e2200. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Park, S.S.; Nam, B.H.; Kim, I.H.; Kim, S.Y. Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-κB inactivation. Cancer Res. 2006, 66, 10936–10943. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Kim, J.M.; Kim, D.S.; Kim, I.H.; Kim, S.Y. Transglutaminase 2 mediates polymer formation of I-kappaBalpha through C-terminal glutamine cluster. J. Biol. Chem. 2006, 281, 34965–34972. [Google Scholar] [CrossRef] [PubMed]
- De Laurenzi, V.; Melino, G. Gene disruption of tissue transglutaminase. Mol. Cell. Biol. 2001, 21, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Kim, B.; Tahk, H.; Kim, D.H.; Ahn, E.R.; Choi, C.; Jeon, Y.; Park, S.Y.; Lee, H.; Oh, S.H.; et al. Transglutaminase 2 gene ablation protects against renal ischemic injury by blocking constant NF-kappaB activation. Biochem. Biophys. Res. Commun. 2010, 403, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, N.; Kim, S.J.; Song, J.; Gong, Y.D.; Kim, S.Y. Anti-cancer effect of a quinoxaline derivative GK13 as a transglutaminase 2 inhibitor. J. Cancer Res. Clin. Oncol. 2013, 139, 1279–1294. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-H.; Lee, W.-K.; Kim, N.; Kang, J.H.; Kim, K.-H.; Kim, S.-G.; Lee, J.-S.; Lee, S.; Lee, J.; Joo, J.; et al. Renal Cell Carcinoma Is Abrogated by p53 Stabilization through Transglutaminase 2 Inhibition. Cancers 2018, 10, 455. https://doi.org/10.3390/cancers10110455
Lee S-H, Lee W-K, Kim N, Kang JH, Kim K-H, Kim S-G, Lee J-S, Lee S, Lee J, Joo J, et al. Renal Cell Carcinoma Is Abrogated by p53 Stabilization through Transglutaminase 2 Inhibition. Cancers. 2018; 10(11):455. https://doi.org/10.3390/cancers10110455
Chicago/Turabian StyleLee, Seon-Hyeong, Won-Kyu Lee, Nayeon Kim, Joon Hee Kang, Kyung-Hee Kim, Seul-Gi Kim, Jae-Seon Lee, Soohyun Lee, Jongkook Lee, Jungnam Joo, and et al. 2018. "Renal Cell Carcinoma Is Abrogated by p53 Stabilization through Transglutaminase 2 Inhibition" Cancers 10, no. 11: 455. https://doi.org/10.3390/cancers10110455
APA StyleLee, S.-H., Lee, W.-K., Kim, N., Kang, J. H., Kim, K.-H., Kim, S.-G., Lee, J.-S., Lee, S., Lee, J., Joo, J., Kwon, W. S., Rha, S. Y., & Kim, S.-Y. (2018). Renal Cell Carcinoma Is Abrogated by p53 Stabilization through Transglutaminase 2 Inhibition. Cancers, 10(11), 455. https://doi.org/10.3390/cancers10110455