Cell Origins of High-Grade Serous Ovarian Cancer



Abstract
1. Ovarian Cancer
1.1. Non-Epithelial Ovarian Cancer (NEOC)
1.2. Epithelial Ovarian Cancer (EOC)
High-Grade Serous Ovarian Cancer (High-Grade Serous Carcinoma: HGSC)
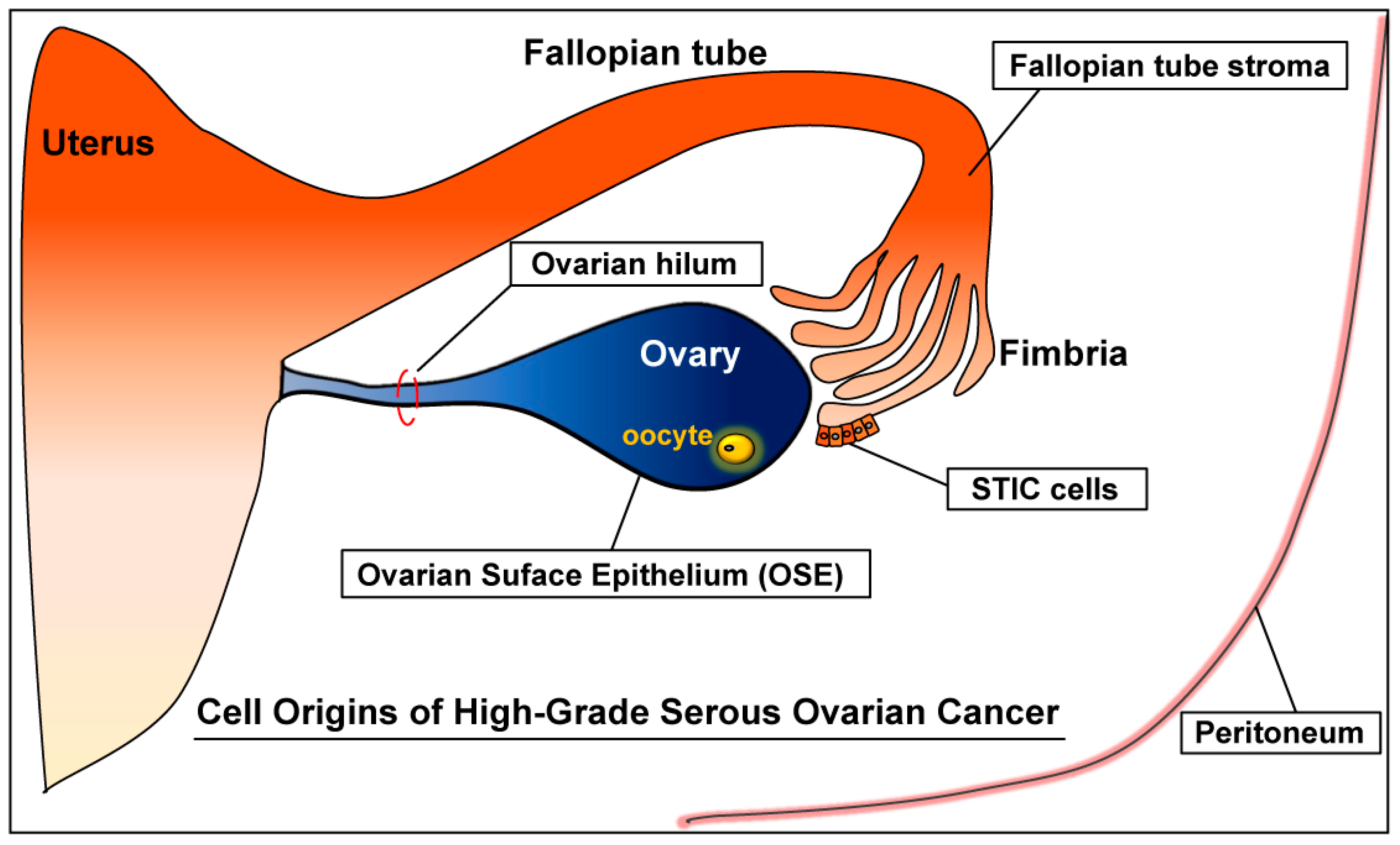
2. Origins of High-Grade Serous Ovarian Cancer (HGSC)
2.1. Ovary
Ovarian Surface Epithelium (OSE)
OSE: Developmental View
OSE: Mouse Models
2.2. Fallopian Tube
2.2.1. Serous Tubal Intraepithelial Carcinoma (STIC)
STIC: Clinical and Molecular Observations
STIC: Mouse Models
STIC: Clinical Significance
Insights from Ductal Carcinoma In Situ (DCIS) in Breast Cancer
2.2.2. Fallopian Tube Stroma
2.2.3. Ovarian Cancer Prevention: Salpingo-Oophorectomy vs. Salpingectomy
2.3. Other Potential Origins of HGSC
2.3.1. Secondary Müllerian System
2.3.2. Ovarian Hilum
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.R.; Shih Ie, M. Ovarian cancer. Annu. Rev. Pathol. 2009, 4, 287–313. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Zarkavelis, G.; Seraj, E.; Zerdes, I.; Tatsi, K.; Pentheroudakis, G. Non-epithelial Ovarian Cancer: Elucidating Uncommon Gynaecological Malignancies. AntiCancer Res. 2016, 36, 5031–5042. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Peiretti, M.; Garbi, A.; Carinelli, S.; Marini, C.; Sessa, C. Non-epithelial ovarian cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, VII20–VII26. [Google Scholar] [CrossRef] [PubMed]
- Koulouris, C.R.; Penson, R.T. Ovarian stromal and germ cell tumors. Semin. Oncol. 2009, 36, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanova, A.V.; Cosin, J.A.; Bidus, M.A.; Boice, C.R.; Seidman, J.D. Pathology of stage I versus stage III ovarian carcinoma with implications for pathogenesis and screening. Int. J. Gynecol. Cancer 2008, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.; Hung, T.; Morin, J.; Gilks, C.B. Molecular abnormalities in ovarian carcinoma: Clinical, morphological and therapeutic correlates. Histopathology 2013, 62, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Horkayne-Szakaly, I.; Haiba, M.; Boice, C.R.; Kurman, R.J.; Ronnett, B.M. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 2004, 23, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Peres, L.C.; Cushing-Haugen, K.L.; Kobel, M.; Harris, H.R.; Berchuck, A.; Rossing, M.A.; Schildkraut, J.M.; Doherty, J.A. Invasive Epithelial Ovarian Cancer Survival by Histotype and Disease Stage. J. Natl. Cancer Inst. 2018. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J. Whence epithelial ovarian carcinoma? Gynecol. Oncol. 2008, 109, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D. The genesis and evolution of high-grade serous ovarian cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A. Cancer of the ovary. N. Engl. J. Med. 2004, 351, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.A. Origins and molecular pathology of ovarian cancer. Mod. Pathol. 2005, 18, S19–S32. [Google Scholar] [CrossRef] [PubMed]
- Feeley, K.M.; Wells, M. Precursor lesions of ovarian epithelial malignancy. Histopathology 2001, 38, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Oktem, O.; Oktay, K. The ovary: Anatomy and function throughout human life. Ann. N. Y. Acad. Sci. 2008, 1127, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.S.; Pangas, S.A. The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, A.J.; Kawamura, K.; Cheng, Y.; Fauser, B.C. Intraovarian control of early folliculogenesis. Endocr. Rev. 2015, 36, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, A. Human ovarian follicular development: From activation of resting follicles to preovulatory maturation. Ann. Endocrinol. 2010, 71, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Baerwald, A.R.; Adams, G.P.; Pierson, R.A. Ovarian antral folliculogenesis during the human menstrual cycle: A review. Hum. Reprod. Update 2012, 18, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Edson, M.A.; Nagaraja, A.K.; Matzuk, M.M. The mammalian ovary from genesis to revelation. Endocr. Rev. 2009, 30, 624–712. [Google Scholar] [CrossRef] [PubMed]
- Matzuk, M.M.; Lamb, D.J. The biology of infertility: Research advances and clinical challenges. Nat. Med. 2008, 14, 1197–1213. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, S.M.; Matzuk, M.M. The menstrual cycle: Basic biology. Ann. N. Y. Acad. Sci. 2008, 1135, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Horta, M.; Cunha, T.M. Sex cord-stromal tumors of the ovary: A comprehensive review and update for radiologists. Diagn. Interv. Radiol. 2015, 21, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Leung, D.; Chu, S. Genetics and genomics of ovarian sex cord-stromal tumors. Clin. Genet. 2017, 91, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Oliva, E. Ovarian sex cord-stromal tumours: An update in recent molecular advances. Pathology 2018, 50, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Friedlander, M.; Backes, F.J.; Harter, P.; O’Connor, D.M.; de la Motte Rouge, T.; Lorusso, D.; Maenpaa, J.; Kim, J.W.; Tenney, M.E.; et al. Gynecologic Cancer Intergroup (GCIG) consensus review for ovarian germ cell tumors. Int. J. Gynecol. Cancer 2014, 24, S48–S54. [Google Scholar] [CrossRef] [PubMed]
- Low, J.J.; Ilancheran, A.; Ng, J.S. Malignant ovarian germ-cell tumours. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, D.; Pectasides, E.; Kassanos, D. Germ cell tumors of the ovary. Cancer Treat. Rev. 2008, 34, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, C.A.; Hatcher, H.M.; Ajithkumar, T.V. Management of malignant ovarian germ cell tumors. Obstet. Gynecol. Surv. 2011, 66, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.R.H.; Wang, X.; Hawkins, S.M. The Endometriotic Tumor Microenvironment in Ovarian Cancer. Cancers 2018, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Oliva, E. Precursors and pathogenesis of ovarian carcinoma. Pathology 2013, 45, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Nezhat, F.R.; Apostol, R.; Nezhat, C.; Pejovic, T. New insights in the pathophysiology of ovarian cancer and implications for screening and prevention. Am. J. Obstet. Gynecol. 2015, 213, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef] [PubMed]
- Malpica, A.; Wong, K.K. The molecular pathology of ovarian serous borderline tumors. Ann. Oncol. 2016, 27, i16–i19. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- TCGA. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Abbas, A.K.; Fausto, N.; Aster, J.C. Robbins and Cotran Pathologic Basics of Disease, 9th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2014; p. 270. [Google Scholar]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Hogg, R.; Friedlander, M. Biology of epithelial ovarian cancer: Implications for screening women at high genetic risk. J. Clin. Oncol. 2004, 22, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Zhao, P.; Yemelyanova, A. “Primary peritoneal” high-grade serous carcinoma is very likely metastatic from serous tubal intraepithelial carcinoma: Assessing the new paradigm of ovarian and pelvic serous carcinogenesis and its implications for screening for ovarian cancer. Gynecol. Oncol. 2011, 120, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P.; Drapkin, R.; Miron, A.; Ince, T.A.; Muto, M.; Kindelberger, D.W.; Lee, Y. The distal fallopian tube: A new model for pelvic serous carcinogenesis. Curr. Opin. Obstet. Gynecol. 2007, 19, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jarboe, E.A.; Miron, A.; Carlson, J.W.; Hirsch, M.S.; Kindelberger, D.; Mutter, G.L.; Crum, C.P.; Nucci, M.R. Coexisting intraepithelial serous carcinomas of the endometrium and fallopian tube: Frequency and potential significance. Int. J. Gynecol. Pathol. 2009, 28, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Przybycin, C.G.; Kurman, R.J.; Ronnett, B.M.; Shih Ie, M.; Vang, R. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am. J. Surg. Pathol. 2010, 34, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Basso, O.; Sampalis, J.; Karp, I.; Martins, C.; Feng, J.; Piedimonte, S.; Quintal, L.; Ramanakumar, A.V.; Takefman, J.; et al. Assessment of symptomatic women for early diagnosis of ovarian cancer: Results from the prospective DOvE pilot project. Lancet Oncol. 2012, 13, 285–291. [Google Scholar] [CrossRef]
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2015; Based on November 2017 SEER Data Submission, Posted to the SEER Web Site; National Cancer Institute: Bethesda, MD, USA, 2018.
- Chan, A.; Gilks, B.; Kwon, J.; Tinker, A.V. New insights into the pathogenesis of ovarian carcinoma: Time to rethink ovarian cancer screening. Obstet. Gynecol. 2012, 120, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Skates, S.; Hernandez, M.A.; Bedi, D.; Bevers, T.; Leeds, L.; Moore, R.; Granai, C.; Harris, S.; Newland, W.; et al. A 2-stage ovarian cancer screening strategy using the Risk of Ovarian Cancer Algorithm (ROCA) identifies early-stage incident cancers and demonstrates high positive predictive value. Cancer 2013, 119, 3454–3461. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef]
- Buys, S.S.; Partridge, E.; Black, A.; Johnson, C.C.; Lamerato, L.; Isaacs, C.; Reding, D.J.; Greenlee, R.T.; Yokochi, L.A.; Kessel, B.; et al. Effect of screening on ovarian cancer mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011, 305, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.T.; Webber, E.M.; Sawaya, G.F. Screening for Ovarian Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2018, 319, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Yamada, Y.; Sado, T.; Sakata, M.; Yoshida, S.; Kawaguchi, R.; Kanayama, S.; Shigetomi, H.; Haruta, S.; Tsuji, Y.; et al. A randomized study of screening for ovarian cancer: A multicenter study in Japan. Int. J. Gynecol. Cancer 2008, 18, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.E. Pathology of ovarian cancer precursors. J. Cell. Biochem. Suppl. 1995, 23, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.; McCluggage, W.G. Current concepts in ovarian epithelial tumorigenesis: Correlation between morphological and molecular data. Histol. Histopathol. 2006, 21, 81–92. [Google Scholar] [PubMed]
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Burdette, J.E.; Oliver, R.M.; Ulyanov, V.; Kilen, S.M.; Mayo, K.E.; Woodruff, T.K. Ovarian epithelial inclusion cysts in chronically superovulated CD1 and Smad2 dominant-negative mice. Endocrinology 2007, 148, 3595–3604. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N. The origin of ovarian carcinomas: A unifying hypothesis. Int. J. Gynecol. Pathol. 2011, 30, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liu, J.; Yoshida, H.; Rosen, D.; Naora, H. Lineage infidelity of epithelial ovarian cancers is controlled by HOX genes that specify regional identity in the reproductive tract. Nat. Med. 2005, 11, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.C.; Bao, R.; Nikitin, A.Y.; Stephens, K.C.; Poole, T.W.; Hua, X.; Harris, S.S.; Vanderhyden, B.C.; Hamilton, T.C. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003, 63, 1389–1397. [Google Scholar] [PubMed]
- Orsulic, S.; Li, Y.; Soslow, R.A.; Vitale-Cross, L.A.; Gutkind, J.S.; Varmus, H.E. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell 2002, 1, 53–62. [Google Scholar] [CrossRef]
- Flesken-Nikitin, A.; Choi, K.C.; Eng, J.P.; Shmidt, E.N.; Nikitin, A.Y. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003, 63, 3459–3463. [Google Scholar] [PubMed]
- Dinulescu, D.M.; Ince, T.A.; Quade, B.J.; Shafer, S.A.; Crowley, D.; Jacks, T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med. 2005, 11, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.Y.; Liu, Z.; Paquet, M.; Wang, J.; Lydon, J.P.; DeMayo, F.J.; Richards, J.S. Cell type-specific targeted mutations of Kras and Pten document proliferation arrest in granulosa cells versus oncogenic insult to ovarian surface epithelial cells. Cancer Res. 2009, 69, 6463–6472. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Hendrix-Lucas, N.; Kuick, R.; Zhai, Y.; Schwartz, D.R.; Akyol, A.; Hanash, S.; Misek, D.E.; Katabuchi, H.; Williams, B.O.; et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell 2007, 11, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.A.; Mullany, L.K.; Liu, Z.; Herron, A.J.; Wong, K.K.; Richards, J.S. Mutant p53 Promotes Epithelial Ovarian Cancer by Regulating Tumor Differentiation, Metastasis, and Responsiveness to Steroid Hormones. Cancer Res. 2016, 76, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Clark-Knowles, K.V.; Senterman, M.K.; Collins, O.; Vanderhyden, B.C. Conditional inactivation of Brca1, p53 and Rb in mouse ovaries results in the development of leiomyosarcomas. PLoS ONE 2009, 4, e8534. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.A.; Brake, T.; Hua, X.; Baxter-Jones, K.; Litwin, S.; Ellenson, L.H.; Connolly, D.C. Induction of ovarian leiomyosarcomas in mice by conditional inactivation of Brca1 and p53. PLoS ONE 2009, 4, e8404. [Google Scholar] [CrossRef] [PubMed]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.J.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, p53, and BRCA1 or BRCA2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res. 2012, 72, 4141–4153. [Google Scholar] [CrossRef] [PubMed]
- Flesken-Nikitin, A.; Hwang, C.I.; Cheng, C.Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, P.S.; Mohapatra, G.; Chiang, S.; Engler, D.A.; Zhang, L.; Kaneko-Tarui, T.; Ohguchi, Y.; Birrer, M.J.; Teixeira, J.M. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis 2014, 35, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Coffey, D.M.; Ma, L.; Matzuk, M.M. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 2015, 156, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P.; Drapkin, R.; Kindelberger, D.; Medeiros, F.; Miron, A.; Lee, Y. Lessons from BRCA: The tubal fimbria emerges as an origin for pelvic serous cancer. Clin. Med. Res. 2007, 5, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Coffey, D.M.; Creighton, C.J.; Yu, Z.; Hawkins, S.M.; Matzuk, M.M. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Piek, J.M.; van Diest, P.J.; Zweemer, R.P.; Jansen, J.W.; Poort-Keesom, R.J.; Menko, F.H.; Gille, J.J.; Jongsma, A.P.; Pals, G.; Kenemans, P.; et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 2001, 195, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P.; Herfs, M.; Ning, G.; Bijron, J.G.; Howitt, B.E.; Jimenez, C.A.; Hanamornroongruang, S.; McKeon, F.D.; Xian, W. Through the glass darkly: Intraepithelial neoplasia, top-down differentiation, and the road to ovarian cancer. J. Pathol. 2013, 231, 402–412. [Google Scholar] [CrossRef] [PubMed]
- George, S.H.; Shaw, P. BRCA and Early Events in the Development of Serous Ovarian Cancer. Front. Oncol. 2014, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.C.; Lindor, N.M. The Role of Risk-Reducing Surgery in Hereditary Breast and Ovarian Cancer. N. Engl. J. Med. 2016, 374, 454–468. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Press, J.Z.; De Luca, A.; Boyd, N.; Young, S.; Troussard, A.; Ridge, Y.; Kaurah, P.; Kalloger, S.E.; Blood, K.A.; Smith, M.; et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer 2008, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Lynch, H.T.; Neuhausen, S.L.; Narod, S.A.; Van’t Veer, L.; Garber, J.E.; Evans, G.; Isaacs, C.; Daly, M.B.; Matloff, E.; et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N. Engl. J. Med. 2002, 346, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.P.; Lubinski, J.; Moller, P.; Singer, C.F.; Karlan, B.; Senter, L.; Rosen, B.; Maehle, L.; Ghadirian, P.; Cybulski, C.; et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J. Clin. Oncol. 2014, 32, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Zweemer, R.P.; van Diest, P.J.; Verheijen, R.H.; Ryan, A.; Gille, J.J.; Sijmons, R.H.; Jacobs, I.J.; Menko, F.H.; Kenemans, P. Molecular evidence linking primary cancer of the fallopian tube to BRCA1 germline mutations. Gynecol. Oncol. 2000, 76, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Colgan, T.J.; Boerner, S.L.; Murphy, J.; Cole, D.E.; Narod, S.; Rosen, B. Peritoneal lavage cytology: An assessment of its value during prophylactic oophorectomy. Gynecol. Oncol. 2002, 85, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Leeper, K.; Garcia, R.; Swisher, E.; Goff, B.; Greer, B.; Paley, P. Pathologic findings in prophylactic oophorectomy specimens in high-risk women. Gynecol. Oncol. 2002, 87, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Paley, P.J.; Swisher, E.M.; Garcia, R.L.; Agoff, S.N.; Greer, B.E.; Peters, K.L.; Goff, B.A. Occult cancer of the fallopian tube in BRCA-1 germline mutation carriers at prophylactic oophorectomy: A case for recommending hysterectomy at surgical prophylaxis. Gynecol. Oncol. 2001, 80, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.L.; Powell, C.B.; Chen, L.M.; Carter, J.; Bae Jump, V.L.; Parker, L.P.; Borowsky, M.E.; Gibb, R.K. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer 2015, 121, 2108–2120. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, F.; Muto, M.G.; Lee, Y.; Elvin, J.A.; Callahan, M.J.; Feltmate, C.; Garber, J.E.; Cramer, D.W.; Crum, C.P. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am. J. Surg. Pathol. 2006, 30, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Barakat, R.R.; Federici, M.G.; Saigo, P.E.; Robson, M.E.; Offit, K.; Boyd, J. Absence of premalignant histologic, molecular, or cell biologic alterations in prophylactic oophorectomy specimens from BRCA1 heterozygotes. Cancer 2000, 89, 383–390. [Google Scholar] [CrossRef]
- Jarboe, E.; Folkins, A.; Nucci, M.R.; Kindelberger, D.; Drapkin, R.; Miron, A.; Lee, Y.; Crum, C.P. Serous carcinogenesis in the fallopian tube: A descriptive classification. Int. J. Gynecol. Pathol. 2008, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Vang, R.; Shaw, P.; Gross, A.; Soslow, R.; Parkash, V.; Shih Ie, M.; Kurman, R.J. Diagnosis of serous tubal intraepithelial carcinoma based on morphologic and immunohistochemical features: A reproducibility study. Am. J. Surg. Pathol. 2011, 35, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.; Shaw, P.; Rosen, B.; Murphy, J.; Narod, S.A.; Colgan, T.J. Clinical and pathologic findings of prophylactic salpingo-oophorectomies in 159 BRCA1 and BRCA2 carriers. Gynecol. Oncol. 2006, 100, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.A.; Rouzbahman, M.; Pizer, E.S.; Pintilie, M.; Begley, H. Candidate serous cancer precursors in fallopian tube epithelium of BRCA1/2 mutation carriers. Mod. Pathol. 2009, 22, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Wethington, S.L.; Park, K.J.; Soslow, R.A.; Kauff, N.D.; Brown, C.L.; Dao, F.; Otegbeye, E.; Sonoda, Y.; Abu-Rustum, N.R.; Barakat, R.R.; et al. Clinical outcome of isolated serous tubal intraepithelial carcinomas (STIC). Int. J. Gynecol. Cancer 2013, 23, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, S.W.; Kim, K.R.; Jung, K.H.; Lee, J.W.; Kim, Y.M. Pathologic findings at risk-reducing salpingo-oophorectomy (RRSO) in germline BRCA mutation carriers with breast cancer: Significance of bilateral RRSO at the optimal age in germline BRCA mutation carriers. J. Gynecol. Oncol. 2017, 28, e3. [Google Scholar] [CrossRef] [PubMed]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Koc, N.; Ayas, S.; Uygur, L. The association of serous tubal intraepithelial carcinoma with gynecologic pathologies and its role in pelvic serous cancer. Gynecol. Oncol. 2014, 134, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Onuma, K.; Deb, P.; Wang, E.; Lytwyn, A.; Sur, M.; Daya, D. Frequency of serous tubal intraepithelial carcinoma in various gynecologic malignancies: A study of 300 consecutive cases. Int. J. Gynecol. Pathol. 2012, 31, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, K.; Klynning, C.; Floter-Radestad, A.; Carlson, J.W. Serous tubal intraepithelial carcinoma, chronic fallopian tube injury, and serous carcinoma development. Virchows Arch. Int. J. Pathol. 2016, 468, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Hanamornroongruang, S.; Lin, D.I.; Conner, J.E.; Schulte, S.; Horowitz, N.; Crum, C.P.; Meserve, E.E. Evidence for a dualistic model of high-grade serous carcinoma: BRCA mutation status, histology, and tubal intraepithelial carcinoma. Am. J. Surg. Pathol. 2015, 39, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007, 211, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Folkins, A.K.; Jarboe, E.A.; Saleemuddin, A.; Lee, Y.; Callahan, M.J.; Drapkin, R.; Garber, J.E.; Muto, M.G.; Tworoger, S.; Crum, C.P. A candidate precursor to pelvic serous cancer (p53 signature) and its prevalence in ovaries and fallopian tubes from women with BRCA mutations. Gynecol. Oncol. 2008, 109, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih Ie, M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Jarboe, E.A.; Folkins, A.K.; Drapkin, R.; Ince, T.A.; Agoston, E.S.; Crum, C.P. Tubal and ovarian pathways to pelvic epithelial cancer: A pathological perspective. Histopathology 2008, 53, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Meyer, L.A.; Deavers, M.T.; Daniels, M.S.; Keeler, E.R.; Mok, S.C.; Gershenson, D.M.; Lu, K.H. Microscopic and early-stage ovarian cancers in BRCA1/2 mutation carriers: Building a model for early BRCA-associated tumorigenesis. Cancer Prev. Res. 2011, 4, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Salvador, S.; Rempel, A.; Soslow, R.A.; Gilks, B.; Huntsman, D.; Miller, D. Chromosomal instability in fallopian tube precursor lesions of serous carcinoma and frequent monoclonality of synchronous ovarian and fallopian tube mucosal serous carcinoma. Gynecol. Oncol. 2008, 110, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Rong, G.; Zhao, J.; Wang, Z.; Shi, F. Identification of candidate genes associated with tubal origin of high-grade serous ovarian cancer. Oncol. Lett. 2018, 15, 7769–7775. [Google Scholar] [CrossRef] [PubMed]
- Piek, J.M.; Verheijen, R.H.; Kenemans, P.; Massuger, L.F.; Bulten, H.; van Diest, P.J. BRCA1/2-related ovarian cancers are of tubal origin: A hypothesis. Gynecol. Oncol. 2003, 90, 491. [Google Scholar] [CrossRef]
- Kroeger, P.T., Jr.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Bijron, J.G.; Seldenrijk, C.A.; Zweemer, R.P.; Lange, J.G.; Verheijen, R.H.; van Diest, P.J. Fallopian tube intraluminal tumor spread from noninvasive precursor lesions: A novel metastatic route in early pelvic carcinogenesis. Am. J. Surg. Pathol. 2013, 37, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Stuckelberger, S.; Drapkin, R. Precious GEMMs: Emergence of faithful models for ovarian Cancer Research. J. Pathol. 2018, 245, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, I.; Takahashi, K.; Kon, Y.; Okamura, T.; Mototani, Y.; Araki, Y.; Kasai, N. Mouse transgenic for murine oviduct-specific glycoprotein promoter-driven simian virus 40 large T-antigen: Tumor formation and its hormonal regulation. Mol. Reprod. Dev. 2002, 63, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Kuhn, E.; Valle, B.L.; Shih, I.M.; Kurman, R.J.; Wang, T.L.; Amano, T.; Ko, M.S.; Miyoshi, I.; Araki, Y.; et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J. Pathol. 2014, 2014, 4353. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, P.H.; van der Zee, M.; Heijmans-Antonissen, C.; Jia, Y.; DeMayo, F.J.; Lydon, J.P.; van Deurzen, C.H.; Ewing, P.C.; Burger, C.W.; Blok, L.J. A mouse model for endometrioid ovarian cancer arising from the distal oviduct. Int. J. Cancer 2014, 135, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zhai, Y.; Kuick, R.; Karnezis, A.N.; Garcia, P.; Naseem, A.; Hu, T.C.; Fearon, E.R.; Cho, K.R. Impact of oviductal versus ovarian epithelial cell of origin on ovarian endometrioid carcinoma phenotype in the mouse. J. Pathol. 2016, 240, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.W.; Miron, A.; Jarboe, E.A.; Parast, M.M.; Hirsch, M.S.; Lee, Y.; Muto, M.G.; Kindelberger, D.; Crum, C.P. Serous tubal intraepithelial carcinoma: Its potential role in primary peritoneal serous carcinoma and serous cancer prevention. J. Clin. Oncol. 2008, 26, 4160–4165. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gaitskell, K.; Garcia, M.J.; Albukhari, A.; Tsaltas, J.; Ahmed, A.A. Serous tubal intraepithelial carcinomas associated with high-grade serous ovarian carcinomas: A systematic review. BJOG 2017, 124, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.B.; Chen, L.M.; McLennan, J.; Crawford, B.; Zaloudek, C.; Rabban, J.T.; Moore, D.H.; Ziegler, J. Risk-reducing salpingo-oophorectomy (RRSO) in BRCA mutation carriers: Experience with a consecutive series of 111 patients using a standardized surgical-pathological protocol. Int. J. Gynecol. Cancer 2011, 21, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.B.; Swisher, E.M.; Cass, I.; McLennan, J.; Norquist, B.; Garcia, R.L.; Lester, J.; Karlan, B.Y.; Chen, L. Long term follow up of BRCA1 and BRCA2 mutation carriers with unsuspected neoplasia identified at risk reducing salpingo-oophorectomy. Gynecol. Oncol. 2013, 129, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.J.; Crum, C.P.; Medeiros, F.; Kindelberger, D.W.; Elvin, J.A.; Garber, J.E.; Feltmate, C.M.; Berkowitz, R.S.; Muto, M.G. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J. Clin. Oncol. 2007, 25, 3985–3990. [Google Scholar] [CrossRef] [PubMed]
- Carcangiu, M.L.; Peissel, B.; Pasini, B.; Spatti, G.; Radice, P.; Manoukian, S. Incidental carcinomas in prophylactic specimens in BRCA1 and BRCA2 germ-line mutation carriers, with emphasis on fallopian tube lesions: Report of 6 cases and review of the literature. Am. J. Surg. Pathol. 2006, 30, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Cass, I.; Walts, A.E.; Barbuto, D.; Lester, J.; Karlan, B. A cautious view of putative precursors of serous carcinomas in the fallopian tubes of BRCA mutation carriers. Gynecol. Oncol. 2014, 134, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Colgan, T.J.; Murphy, J.; Cole, D.E.; Narod, S.; Rosen, B. Occult carcinoma in prophylactic oophorectomy specimens: Prevalence and association with BRCA germline mutation status. Am. J. Surg. Pathol. 2001, 25, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.E.; Gard, G.B.; McIllroy, K.; Nevell, D.; Field, M. High rates of occult fallopian tube cancer diagnosed at prophylactic bilateral salpingo-oophorectomy. Int. J. Gynecol. Cancer 2009, 19, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Manchanda, R.; Abdelraheim, A.; Johnson, M.; Rosenthal, A.N.; Benjamin, E.; Brunell, C.; Burnell, M.; Side, L.; Gessler, S.; Saridogan, E.; et al. Outcome of risk-reducing salpingo-oophorectomy in BRCA carriers and women of unknown mutation status. BJOG 2011, 118, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Mingels, M.J.; Roelofsen, T.; van der Laak, J.A.; de Hullu, J.A.; van Ham, M.A.; Massuger, L.F.; Bulten, J.; Bol, M. Tubal epithelial lesions in salpingo-oophorectomy specimens of BRCA-mutation carriers and controls. Gynecol. Oncol. 2012, 127, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.B.; Kenley, E.; Chen, L.M.; Crawford, B.; McLennan, J.; Zaloudek, C.; Komaromy, M.; Beattie, M.; Ziegler, J. Risk-reducing salpingo-oophorectomy in BRCA mutation carriers: Role of serial sectioning in the detection of occult malignancy. J. Clin. Oncol. 2005, 23, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, W.; de Bock, G.H.; Oosterwijk, J.C.; Bart, J.; Hollema, H.; Mourits, M.J. Support of the ‘fallopian tube hypothesis’ in a prospective series of risk-reducing salpingo-oophorectomy specimens. Eur. J. Cancer 2013, 49, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.E.; Piedmonte, M.; Mai, P.L.; Ioffe, O.B.; Ronnett, B.M.; Van Le, L.; Ivanov, I.; Bell, M.C.; Blank, S.V.; DiSilvestro, P.; et al. Pathologic findings at risk-reducing salpingo-oophorectomy: Primary results from Gynecologic Oncology Group Trial GOG-0199. J. Clin. Oncol. 2014, 32, 3275–3283. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Krishnan, J.; Yemelyanova, A.; Vang, R. Incidental Serous Tubal Intraepithelial Carcinoma and Non-Neoplastic Conditions of the Fallopian Tubes in Grossly Normal Adnexa: A Clinicopathologic Study of 388 Completely Embedded Cases. Int. J. Gynecol. Pathol. 2016, 35, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Mingels, M.J.; van Ham, M.A.; de Kievit, I.M.; Snijders, M.P.; van Tilborg, A.A.; Bulten, J.; Massuger, L.F. Mullerian precursor lesions in serous ovarian cancer patients: Using the SEE-Fim and SEE-End protocol. Mod. Pathol. 2014, 27, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Rabban, J.T.; Garg, K.; Crawford, B.; Chen, L.M.; Zaloudek, C.J. Early detection of high-grade tubal serous carcinoma in women at low risk for hereditary breast and ovarian cancer syndrome by systematic examination of fallopian tubes incidentally removed during benign surgery. Am. J. Surg. Pathol. 2014, 38, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Armstrong, M.; Brennan, B.A.; Hammond, I.G.; Havlat, M.; Rene Kee, A.; Koay, E.; Leung, Y.; Netreba, A.N.; Ruba, S. Coexisting serous carcinoma of the endometrium and the fallopian tube. Int. J. Gynecol. Pathol. 2010, 29, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, M.C.; Swisher, E.M.; Medeiros, F.; Lima, J.F.; Hilderbrand, J.L.; Donovan, J.L.; Garcia, R.L.; Cliby, W.A.; Dowdy, S.C. Characterization of precursor lesions in the endometrium and fallopian tube epithelium of early-stage uterine serous carcinoma. Int. J. Gynecol. Pathol. 2015, 34, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Klotz, D.M.; Wimberger, P. Cells of origin of ovarian cancer: Ovarian surface epithelium or fallopian tube? Arch. Gynecol. Obstet. 2017, 296, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G.; Hirschowitz, L.; Gilks, C.B.; Wilkinson, N.; Singh, N. The Fallopian Tube Origin and Primary Site Assignment in Extrauterine High-grade Serous Carcinoma: Findings of a Survey of Pathologists and Clinicians. Int. J. Gynecol. Pathol. 2017, 36, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Esserman, L.J.; Thompson, I.M., Jr.; Reid, B. Overdiagnosis and overtreatment in cancer: An opportunity for improvement. JAMA 2013, 310, 797–798. [Google Scholar] [CrossRef] [PubMed]
- Esserman, L.J.; Thompson, I.M.; Reid, B.; Nelson, P.; Ransohoff, D.F.; Welch, H.G.; Hwang, S.; Berry, D.A.; Kinzler, K.W.; Black, W.C.; et al. Addressing overdiagnosis and overtreatment in cancer: A prescription for change. Lancet Oncol. 2014, 15, e234–242. [Google Scholar] [CrossRef]
- Brawley, O.W. Accepting the Existence of Breast Cancer Overdiagnosis. Ann. Intern. Med. 2017, 166, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.; Barletta, J.A.; Wenig, B.M.; Al Ghuzlan, A.; Kakudo, K.; et al. Nomenclature Revision for Encapsulated Follicular Variant of Papillary Thyroid Carcinoma: A Paradigm Shift to Reduce Overtreatment of Indolent Tumors. JAMA Oncol. 2016, 2, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Polyak, K.; Wong, J.S.; Lester, S.C.; Kaelin, C.M. Ductal carcinoma in situ of the breast. N. Engl. J. Med. 2004, 350, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bleyer, A.; Welch, H.G. Effect of three decades of screening mammography on breast-cancer incidence. N. Engl. J. Med. 2012, 367, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Shieh, Y.; Eklund, M.; Sawaya, G.F.; Black, W.C.; Kramer, B.S.; Esserman, L.J. Population-based screening for cancer: Hope and hype. Nat. Rev. Clin. Oncol. 2016, 13, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Thomsen, J.L.; Primdahl, S.; Dyreborg, U.; Andersen, J.A. Breast cancer and atypia among young and middle-aged women: A study of 110 medicolegal autopsies. Br. J. Cancer 1987, 56, 814–819. [Google Scholar] [CrossRef] [PubMed]
- SEER Cancer Statistics Review, 1975–2013; Howlader, N., Noone, A.M., Krapcho, M., Miller, D., Bishop, K., Altekruse, S.F., Kosary, C.L., Yu, M., Ruhl, J., Tatalovich, Z., et al., Eds.; National Cancer Institute: Bethesda, MD, USA, 2016.
- Duffy, S.W.; Dibden, A.; Michalopoulos, D.; Offman, J.; Parmar, D.; Jenkins, J.; Collins, B.; Robson, T.; Scorfield, S.; Green, K.; et al. Screen detection of ductal carcinoma in situ and subsequent incidence of invasive interval breast cancers: A retrospective population-based study. Lancet Oncol. 2016, 17, 109–114. [Google Scholar] [CrossRef]
- Sopik, V.; Nofech-Mozes, S.; Sun, P.; Narod, S.A. The relationship between local recurrence and death in early-stage breast cancer. Breast Cancer Res. Treat. 2016, 155, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Welch, H.G.; Prorok, P.C.; O’Malley, A.J.; Kramer, B.S. Breast-Cancer Tumor Size, Overdiagnosis, and Mammography Screening Effectiveness. N. Engl. J. Med. 2016, 375, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Autier, P.; Boniol, M.; Koechlin, A.; Pizot, C.; Boniol, M. Effectiveness of and overdiagnosis from mammography screening in the Netherlands: Population based study. BMJ 2017, 359, j5224. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, K.J.; Gotzsche, P.C.; Kalager, M.; Zahl, P.H. Breast Cancer Screening in Denmark: A Cohort Study of Tumor Size and Overdiagnosis. Ann. Intern. Med. 2017, 166, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Orvis, G.D.; Wang, Y.; Behringer, R.R. Stromal-to-Epithelial Transition during Postpartum Endometrial Regeneration. PLoS ONE 2012, 7, e44285. [Google Scholar] [CrossRef] [PubMed]
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.F.; Nick, A.M.; et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- NIH Consensus Conference. Ovarian cancer. Screening, treatment, and follow-up. NIH Consensus Development Panel on Ovarian Cancer. JAMA 1995, 273, 491–497. [Google Scholar] [CrossRef]
- Lancaster, J.M.; Powell, C.B.; Chen, L.M.; Richardson, D.L. Society of Gynecologic Oncology statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol. Oncol. 2015, 136, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Kauff, N.D.; Domchek, S.M.; Friebel, T.M.; Robson, M.E.; Lee, J.; Garber, J.E.; Isaacs, C.; Evans, D.G.; Lynch, H.; Eeles, R.A.; et al. Risk-reducing salpingo-oophorectomy for the prevention of BRCA1- and BRCA2-associated breast and gynecologic cancer: A multicenter, prospective study. J. Clin. Oncol. 2008, 26, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Friebel, T.M.; Neuhausen, S.L.; Wagner, T.; Evans, G.; Isaacs, C.; Garber, J.E.; Daly, M.B.; Eeles, R.; Matloff, E.; et al. Mortality after bilateral salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers: A prospective cohort study. Lancet Oncol. 2006, 7, 223–229. [Google Scholar] [CrossRef]
- Eisen, A.; Lubinski, J.; Klijn, J.; Moller, P.; Lynch, H.T.; Offit, K.; Weber, B.; Rebbeck, T.; Neuhausen, S.L.; Ghadirian, P.; et al. Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: An international case-control study. J. Clin. Oncol. 2005, 23, 7491–7496. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Kauff, N.D.; Domchek, S.M. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J. Natl. Cancer Inst. 2009, 101, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.L.; Velazquez, I.A.; Chen, B.E.; Rosenberg, P.S.; Struewing, J.P.; Greene, M.H. Prophylactic oophorectomy reduces breast cancer penetrance during prospective, long-term follow-up of BRCA1 mutation carriers. J. Clin. Oncol. 2005, 23, 8629–8635. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk-Gerritsen, B.A.; Seynaeve, C.; van Asperen, C.J.; Ausems, M.G.; Collee, J.M.; van Doorn, H.C.; Gomez Garcia, E.B.; Kets, C.M.; van Leeuwen, F.E.; Meijers-Heijboer, H.E.; et al. Breast cancer risk after salpingo-oophorectomy in healthy BRCA1/2 mutation carriers: Revisiting the evidence for risk reduction. J. Natl. Cancer Inst. 2015, 107, djv033. [Google Scholar] [CrossRef] [PubMed]
- Kotsopoulos, J.; Huzarski, T.; Gronwald, J.; Singer, C.F.; Moller, P.; Lynch, H.T.; Armel, S.; Karlan, B.; Foulkes, W.D.; Neuhausen, S.L.; et al. Bilateral Oophorectomy and Breast Cancer Risk in BRCA1 and BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 2017, 109, djw177. [Google Scholar] [CrossRef] [PubMed]
- Practice Bulletin No 182: Hereditary Breast and Ovarian Cancer Syndrome. Obstet. Gynecol. 2017, 130, e110–e126. [CrossRef] [PubMed]
- Russo, A.; Calo, V.; Bruno, L.; Rizzo, S.; Bazan, V.; Di Fede, G. Hereditary ovarian cancer. Crit. Rev. Oncol. Hematol. 2009, 69, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Madalinska, J.B.; van Beurden, M.; Bleiker, E.M.; Valdimarsdottir, H.B.; Hollenstein, J.; Massuger, L.F.; Gaarenstroom, K.N.; Mourits, M.J.; Verheijen, R.H.; van Dorst, E.B.; et al. The impact of hormone replacement therapy on menopausal symptoms in younger high-risk women after prophylactic salpingo-oophorectomy. J. Clin. Oncol. 2006, 24, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Stan, D.L.; Shuster, L.T.; Wick, M.J.; Swanson, C.L.; Pruthi, S.; Bakkum-Gamez, J.N. Challenging and complex decisions in the management of the BRCA mutation carrier. J. Women’s Health 2013, 22, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.H.; Mai, P.L.; Schwartz, P.E. Does bilateral salpingectomy with ovarian retention warrant consideration as a temporary bridge to risk-reducing bilateral oophorectomy in BRCA1/2 mutation carriers? Am. J. Obstet. Gynecol. 2011, 204, 19.e1–19.e6. [Google Scholar] [CrossRef] [PubMed]
- Beattie, M.S.; Crawford, B.; Lin, F.; Vittinghoff, E.; Ziegler, J. Uptake, time course, and predictors of risk-reducing surgeries in BRCA carriers. Genet. Test. Mol. Biomark. 2009, 13, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.H.; Piedmonte, M.; Alberts, D.; Gail, M.; Hensley, M.; Miner, Z.; Mai, P.L.; Loud, J.; Rodriguez, G.; Basil, J.; et al. A prospective study of risk-reducing salpingo-oophorectomy and longitudinal CA-125 screening among women at increased genetic risk of ovarian cancer: Design and baseline characteristics: A Gynecologic Oncology Group study. Cancer Epidemiol. Biomark. Prev. 2008, 17, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Kram, V.; Peretz, T.; Sagi, M. Acceptance of preventive surgeries by Israeli women who had undergone BRCA testing. Fam. Cancer 2006, 5, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.A.; Birenbaum-Carmeli, D.; Lubinski, J.; Gronwald, J.; Lynch, H.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Klijn, J.; Friedman, E.; et al. International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int. J. Cancer 2008, 122, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Schmeler, K.M.; Sun, C.C.; Bodurka, D.C.; White, K.G.; Soliman, P.T.; Uyei, A.R.; Erlichman, J.L.; Arun, B.K.; Daniels, M.S.; Rimes, S.A.; et al. Prophylactic bilateral salpingo-oophorectomy compared with surveillance in women with BRCA mutations. Obstet. Gynecol. 2006, 108, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.G.; Arts-de Jong, M.; Hoogerbrugge, N.; Maas, A.H.; Prins, J.B.; Bulten, J.; Teerenstra, S.; Adang, E.M.; Piek, J.M.; van Doorn, H.C.; et al. Early salpingectomy (TUbectomy) with delayed oophorectomy to improve quality of life as alternative for risk-reducing salpingo-oophorectomy in BRCA1/2 mutation carriers (TUBA study): A prospective non-randomised multicentre study. BMC Cancer 2015, 15, 593. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.S.; Tinker, A.; Pansegrau, G.; McAlpine, J.; Housty, M.; McCullum, M.; Gilks, C.B. Prophylactic salpingectomy and delayed oophorectomy as an alternative for BRCA mutation carriers. Obstet. Gynecol. 2013, 121, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A. Salpingectomy to prevent ovarian cancer: A Countercurrents Series. Curr. Oncol. 2013, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Falconer, H.; Yin, L.; Gronberg, H.; Altman, D. Ovarian cancer risk after salpingectomy: A nationwide population-based study. J. Natl. Cancer Inst. 2015, 107, dju410. [Google Scholar] [CrossRef] [PubMed]
- Lessard-Anderson, C.R.; Handlogten, K.S.; Molitor, R.J.; Dowdy, S.C.; Cliby, W.A.; Weaver, A.L.; Sauver, J.S.; Bakkum-Gamez, J.N. Effect of tubal sterilization technique on risk of serous epithelial ovarian and primary peritoneal carcinoma. Gynecol. Oncol. 2014, 135, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Dietl, J.; Wischhusen, J.; Hausler, S.F. The post-reproductive Fallopian tube: Better removed? Hum. Reprod. 2011, 26, 2918–2924. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; McAlpine, J.N.; Gilks, C.B.; Huntsman, D.G. Opportunistic salpingectomy: The way forward-response to Steven Narod. Curr. Oncol. 2013, 20, 143–144. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists. Salpingectomy for Ovarian Cancer Prevention; Number 620; American Congress of Obstetricians and Gynecologists: Washington, DC, USA, 2015. [Google Scholar]
- Whiteman, M.K.; Hillis, S.D.; Jamieson, D.J.; Morrow, B.; Podgornik, M.N.; Brett, K.M.; Marchbanks, P.A. Inpatient hysterectomy surveillance in the United States, 2000–2004. Am. J. Obstet. Gynecol. 2008, 198, 34.e1–34.e7. [Google Scholar]
- Parker, W.H. Bilateral oophorectomy versus ovarian conservation: Effects on long-term women’s health. J. Minim. Invasive Gynecol. 2010, 17, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.H.; Broder, M.S.; Chang, E.; Feskanich, D.; Farquhar, C.; Liu, Z.; Shoupe, D.; Berek, J.S.; Hankinson, S.; Manson, J.E. Ovarian conservation at the time of hysterectomy and long-term health outcomes in the nurses’ health study. Obstet. Gynecol. 2009, 113, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Dubeau, L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: Does the emperor have no clothes? Gynecol. Oncol. 1999, 72, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Dubeau, L. The cell of origin of ovarian epithelial tumours. Lancet Oncol. 2008, 9, 1191–1197. [Google Scholar] [CrossRef]
- Kauff, N.D.; Satagopan, J.M.; Robson, M.E.; Scheuer, L.; Hensley, M.; Hudis, C.A.; Ellis, N.A.; Boyd, J.; Borgen, P.I.; Barakat, R.R.; et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N. Engl. J. Med. 2002, 346, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Tone, A.A.; Salvador, S.; Finlayson, S.J.; Tinker, A.V.; Kwon, J.S.; Lee, C.H.; Cohen, T.; Ehlen, T.; Lee, M.; Carey, M.S.; et al. The role of the fallopian tube in ovarian cancer. Clin. Adv. Hematol. Oncol. 2012, 10, 296–306. [Google Scholar] [PubMed]


{kind=link}
| Targeted Genes | Promoter | Ovarian Tumor | Metastasis | Ascites | Ref. |
|---|---|---|---|---|---|
| p53, Myc, Kras G12D; p53, Kras G12D, Akt; p53, Akt, Myc | RCAS viral vector | Poorly differentiated or serous carcinoma (in nude mice) | Peritoneal lining, Omentum, Diaphragm, Liver, Pancreas, Intestines, Kidneys | Yes | [64] |
| p53, Rb1 | Adenovirus cre | Serous carcinoma: 97% (33/34 mice) | Peritoneal: 27% (9/33) Lungs: 18% (6/33) Liver: 6% (1/33) | 24% (8/33) | [65] |
| p53, Rb1; p53; Brca1, Rb1; p53, Rb1, Brca1 | Adenovirus cre | Leiomyosarcoma: 100% (44/44) | No | 27% (12/44) | [70] |
| p53, Brca1 | Adenovirus cre | Leiomyosarcoma or high-grade sarcoma: 56% (23/41) | No | No | [71] |
| SV40 TAg | Amhr2 (MISIIR) | Serous carcinoma: 50% (18/36) | Peritoneal metastasis including omentum: ?% | Yes (?%) | [63] |
| Hox9; Hox10; Hox11 | pCMV-Tag | HGSC (Hox9) Endometrioid (Hox10) Mucinous (Hox11) (in nude mice) | No description (ND) | ND | [62] |
| Pten, Apc | Adenovirus cre | Endometrioid carcinoma: 100% (29/29) | Peritoneal: 21% (6/29) | 76% (22/29) | [68] |
| Pten, Kras G12D | Adenovirus cre | Endometrioid carcinoma: ?% (?/9) | Peritoneal: ?% Lungs: 43% | Yes (?%) | [66] |
| Pten, Kras G12D | Amhr2 cre/+ | Low-grade serous carcinoma:100% (8/8) | Omentum: 100% (8/8) | No | [67,69] |
| Rb1, p53, Brca1; Rb1, p53 R172H, Brca1/2 | Adenovirus cre | HGSC: Stage I and II: 29% (46/158; 21–32%) | Peritoneal: 16% (25/158; 0–26%) Liver or lung or pleural: 17% (28/158; 0–25%) | Yes (?%) | [72] |
| p53, Rb1 in the OSE hilum | Adenovirus cre | HGSC: 88% (7/8) (in NOD scid mice) | Lungs: 71% (5/7) | No | [73] |
| Pten, Kras G12D, p53 R172H | Amhr2 cre/+ | Mucinous carcinoma: 80% (8/10) Mucinous & serous: 100% (10/10) | Omentum: 100% (36/36) | No | [69] |
| Lkb1, Pten | Amhr2 cre/+ | HGSC: 100% (12/12) | No description | 25% (3/12) | [74] |
| p53 R172H, Pten | Amhr2 cre/+ | HGSC: 30% (15/50) mixed with granulosa cell tumor | Peritoneal HGSC: 100% (15/15); omentum, diaphragm, mesentery, peritoneal lining | 80% (12/15) | [75] |
| p53 R172H, Pten | Amhr2 cre/+ | Granulosa cell tumor: 70% (35/50) | Lungs: 53.3% (19/35) | No | [75] |
| Targeted Genes | Promoter | STIC | Fallopian Tube HGSC | Ovarian HGSC Metastasis | Peritoneal HGSC Metastasis | Ascites | Ref. |
|---|---|---|---|---|---|---|---|
| SV40 TAg | Ovgp1 | – | Oviductal tumors (?%) | No ovarian tumor | No; Uterine tumor: 100% (26/26) Vaginal tumor: 62% (16/26) | No | [122] |
| - Monitoring of tumor development: 6–13 weeks of age | |||||||
| Brca1, p53 R172H, Pten | Pax8 | 100% (4/4) | No | 25% (1/4) | 25% (1/4): peritoneal mass | No | [78] |
| - Monitoring of tumor development: 5–7 weeks of age | |||||||
| Brca2, p53 R172H, Pten | Pax8 | 75% (9/12) | No | 75% (9/12) | 67% (8/12): peritoneal mass | No | [78] |
| - Monitoring of tumor development: 7–15 weeks of age | |||||||
| p53 R172H, Pten | Pax8 | 67% (4/6) | No | 0% (0/6) | 0% (0/6) | No | [78] |
| - Monitoring of tumor development: 19–38 weeks of age | |||||||
| SV40 TAg | Ovgp1 | Yes (?%) | No | Adeno-carcinoma (56%) | No | No | [123] |
| - Monitoring of tumor development: 8–10 weeks of age | |||||||
| Brca1, p53, Rb1, Nf1 | Ovgp1-iCreER | 37.5% (18/48) | HGSC: 60% (29/48) MMMT: 25% (12/48) | HGSC or MMMT: 40% (19/48) | HGSC or MMMT: 13% (6/48) | 13% (6/48) | [125] |
| - Monitoring of tumor development: 3.5–26 months of age | |||||||
| Brca1, p53, Rb1 | Ovgp1-iCreER | 34.5% (10/29) | HGSC: 17% (5/29) MMMT: 7% (2/29) | 0% | 0% | 0% | [125] |
| - Monitoring of tumor development: 3.5–26 months of age | |||||||
| Brca1, p53, Nf1 | Ovgp1-iCreER | 0% (0/3) | HGSC: 67% (2/3) MMMT: 67% (2/2) | HGSC or MMMT: 100% (3/3) | HGSC or MMMT: 33% (1/3) | 0% | [125] |
| - Monitoring of tumor development: 3.5–26 months of age | |||||||
| Brca1, p53, Pten | Ovgp1-iCreER | 40% (4/10) | HGSC: 80% (8/10) MMMT: 10% (1/10) | MMMT: 10% (1/10) | 0% | 10% (1/10) | [125] |
| - Monitoring of tumor development: 3–8 months of age | |||||||
| Dicer1, Pten | Amhr2 cre/+ | No | 100% (24/24) | 100% (24/24) | 100% (24/24): omentum, diaphragm, mesentery, peritoneal lining | 100% (24/24) | [77] |
| - Survival range: 6.2–13 months of age (mean survival = 9.4 months; n = 24) | |||||||
| Sample Tissue | Population | Incidence of STIC or Occult Tubal Carcinoma | Number of Cases | Reference |
|---|---|---|---|---|
| Fallopian tubes from prophylactic salpingo-oophorectomy | High risk | 50% (6) | 12 | Piek et al., 2001 [80] |
| 37% (16?) | 44 | Piek et al., 2003 [118] | ||
| 6.7% (4) | 60 | Colgan et al., 2001 [133] | ||
| 10% (3) | 30 | Leeper et al., 2002 [93] | ||
| 6% (4) | 67 | Powell et al., 2005 [137] | ||
| 8% (4) | 50 | Carcangiu et al., 2006 [131] | ||
| 3.8% (6) | 159 | Finch et al., 2006 [100] | ||
| 5.7% (7) | 122 | Callahan et al., 2007 [130] | ||
| 8.5% (15) | 176 | Shaw et al., 2009 [101] | ||
| 8.9% (4) | 45 | Hirst et al., 2009 [134] | ||
| 8.1% (9) | 111 | Powell et al., 2011 [128] | ||
| 8.5% (10) | 117 | Manchanda et al., 2011 [135] | ||
| 7.1% (16) | 226 | Mingels et al., 2012 [136] | ||
| 1.7% (5) | 303 | Reitsma et al., 2013 [138] | ||
| 4.2% (17) | 405 | Powell et al., 2013 [129] | ||
| 2.0% (12) | 593 | Wethington et al., 2013 [102] | ||
| 11.5% (9) | 78 | Cass et al., 2014 [132] | ||
| 2.6% (25) | 966 | Sherman et al., 2014 [139] | ||
| 0% (0) | 111 | Seidman et al., 2016 [140] | ||
| 5.6% (2) | 36 | Lee et al., 2017 [103] | ||
| Fallopian tubes from HGSC cases | High risk | 30.8% (8) | 26 | Howitt et al., 2015 [108] |
| 3.3% (2) | 60 | Malmberg et al., 2016 [107] | ||
| Fallopian tubes from HGSC cases | General | 47.6% (20) | 42 | Kindelberger et al., 2007 [104] |
| 58.5% (24) | 41 | Przybycin et al., 2010 [48] | ||
| 37.3% (19) | 51 | Seidman et al., 2011 [45] | ||
| 20.5% (8) | 39 | Tang et al., 2012 [106] | ||
| 38.3% (23) | 60 | Mingels et al., 2014 [141] | ||
| 38.2% (13) | 34 | Koc et al., 2014 [105] | ||
| 33.3% (6) | 18 | Malmberg et al., 2016 [107] | ||
| Fallopian tubes from non-ovarian-cancer or benign cases | General | 3.1% (2) | 64 | Shaw et al., 2009 [101] |
| 0.8% (4) | 522 | Rabban et al., 2014 [142] | ||
| 1.1% (3) | 277 | Seidman et al., 2016 [140] | ||
| Fallopian tubes from endometrial serous carcinoma cases | General | 22.7% (5) | 22 | Jarboe et al., 2009 [47] |
| 21.8% (12) | 55 | Stewart et al., 2010 [143] | ||
| 14.3% (4) | 28 | Tang et al., 2012 [106] | ||
| 7.9% (3) | 38 | Tolcher et al., 2015 [144] | ||
| Fallopian tubes from endometrial carcinoma or hyperplasia cases | General | 1.7% (3) | 175 | Seidman et al., 2016 [140] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.-H.; Bast, R.C. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. https://doi.org/10.3390/cancers10110433
Kim J, Park EY, Kim O, Schilder JM, Coffey DM, Cho C-H, Bast RC. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers. 2018; 10(11):433. https://doi.org/10.3390/cancers10110433
Chicago/Turabian StyleKim, Jaeyeon, Eun Young Park, Olga Kim, Jeanne M. Schilder, Donna M. Coffey, Chi-Heum Cho, and Robert C. Bast. 2018. "Cell Origins of High-Grade Serous Ovarian Cancer" Cancers 10, no. 11: 433. https://doi.org/10.3390/cancers10110433
APA StyleKim, J., Park, E. Y., Kim, O., Schilder, J. M., Coffey, D. M., Cho, C.-H., & Bast, R. C. (2018). Cell Origins of High-Grade Serous Ovarian Cancer. Cancers, 10(11), 433. https://doi.org/10.3390/cancers10110433