Prognostic Values of EPDR1 Hypermethylation and Its Inhibitory Function on Tumor Invasion in Colorectal Cancer

 ,
,
Abstract
:1. Introduction
2. Results
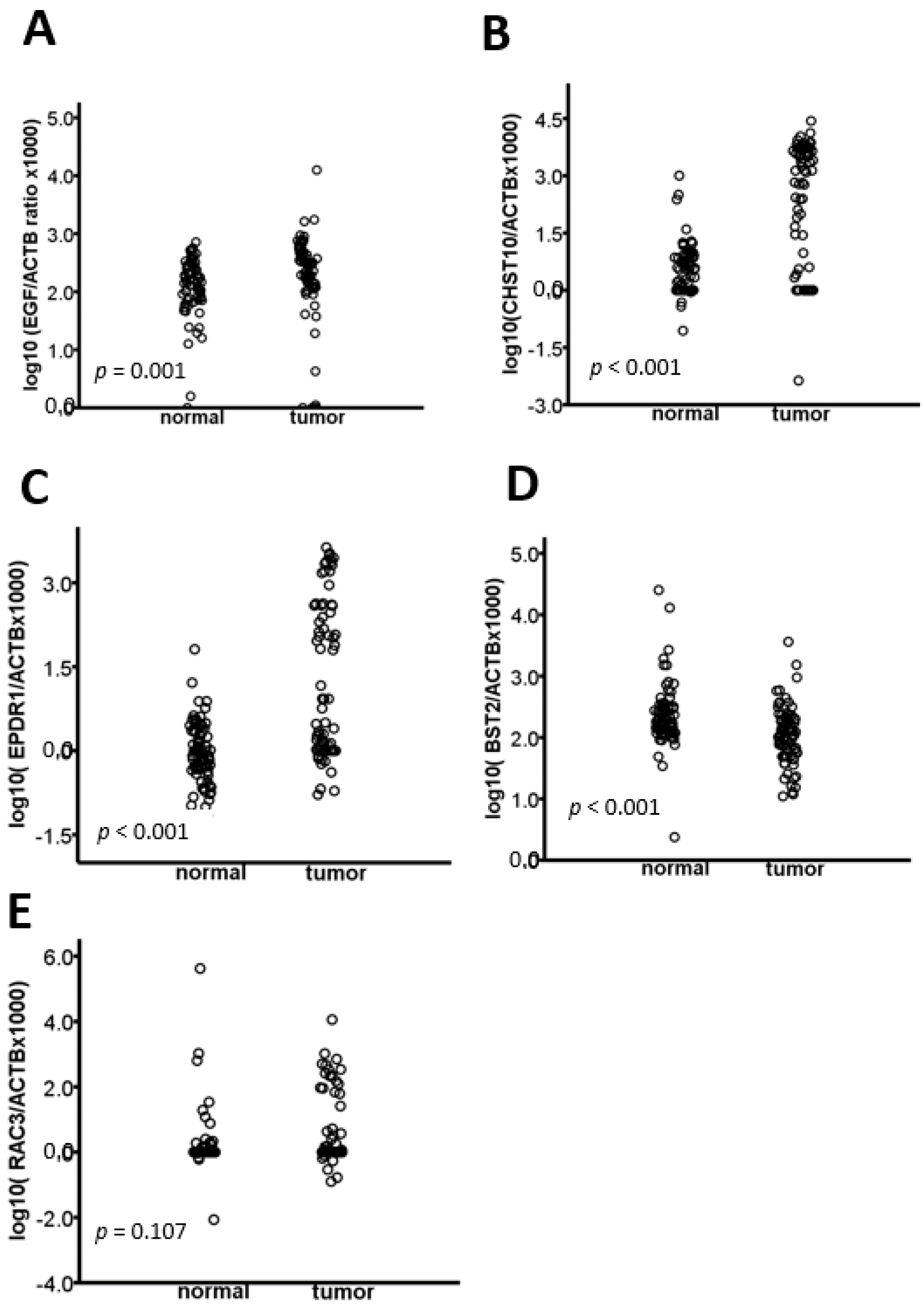
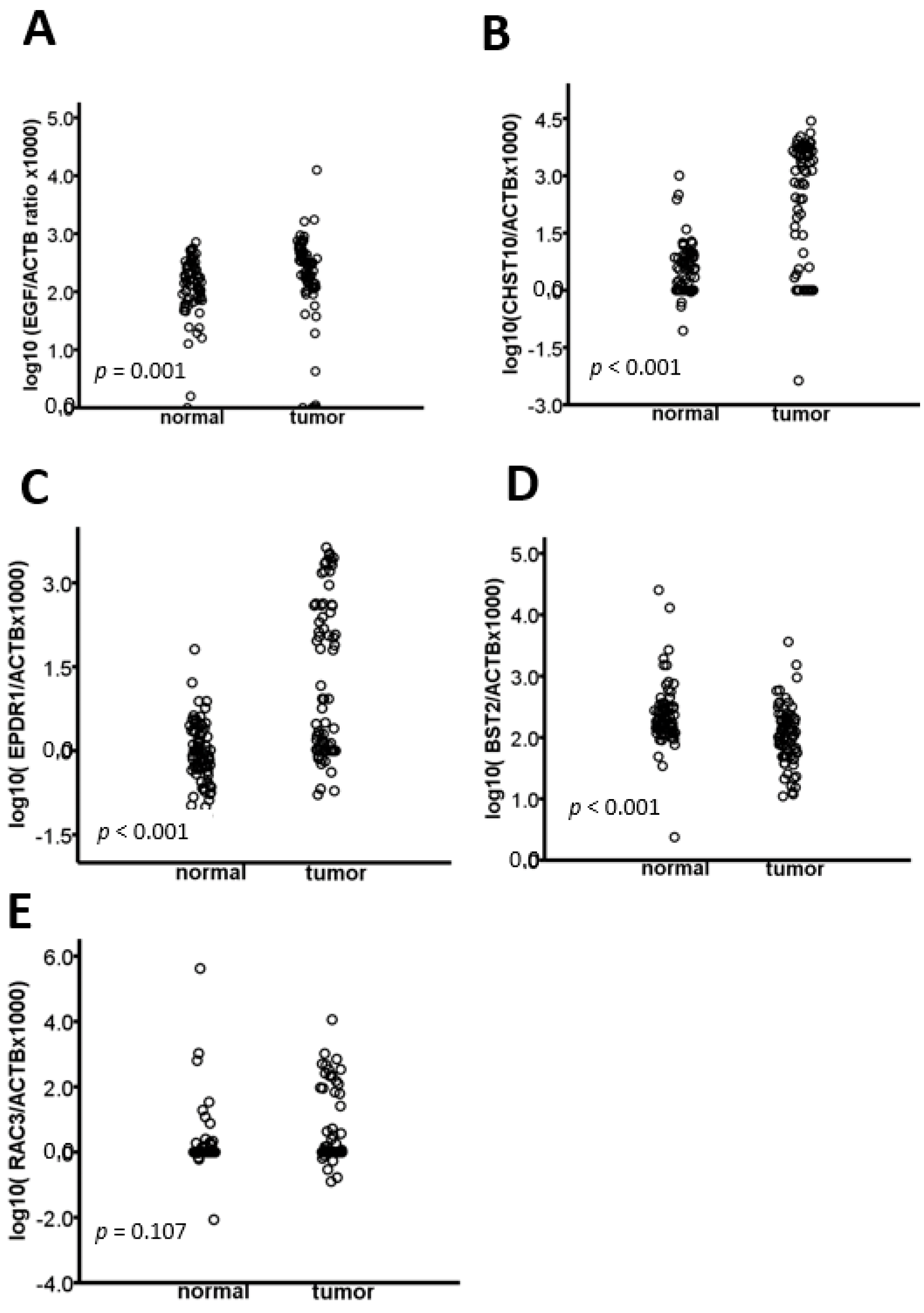
2.1. The EGF, CHST10, EPDR1, BST2, and RAC3 Methylation Levels Are Validated in CRC
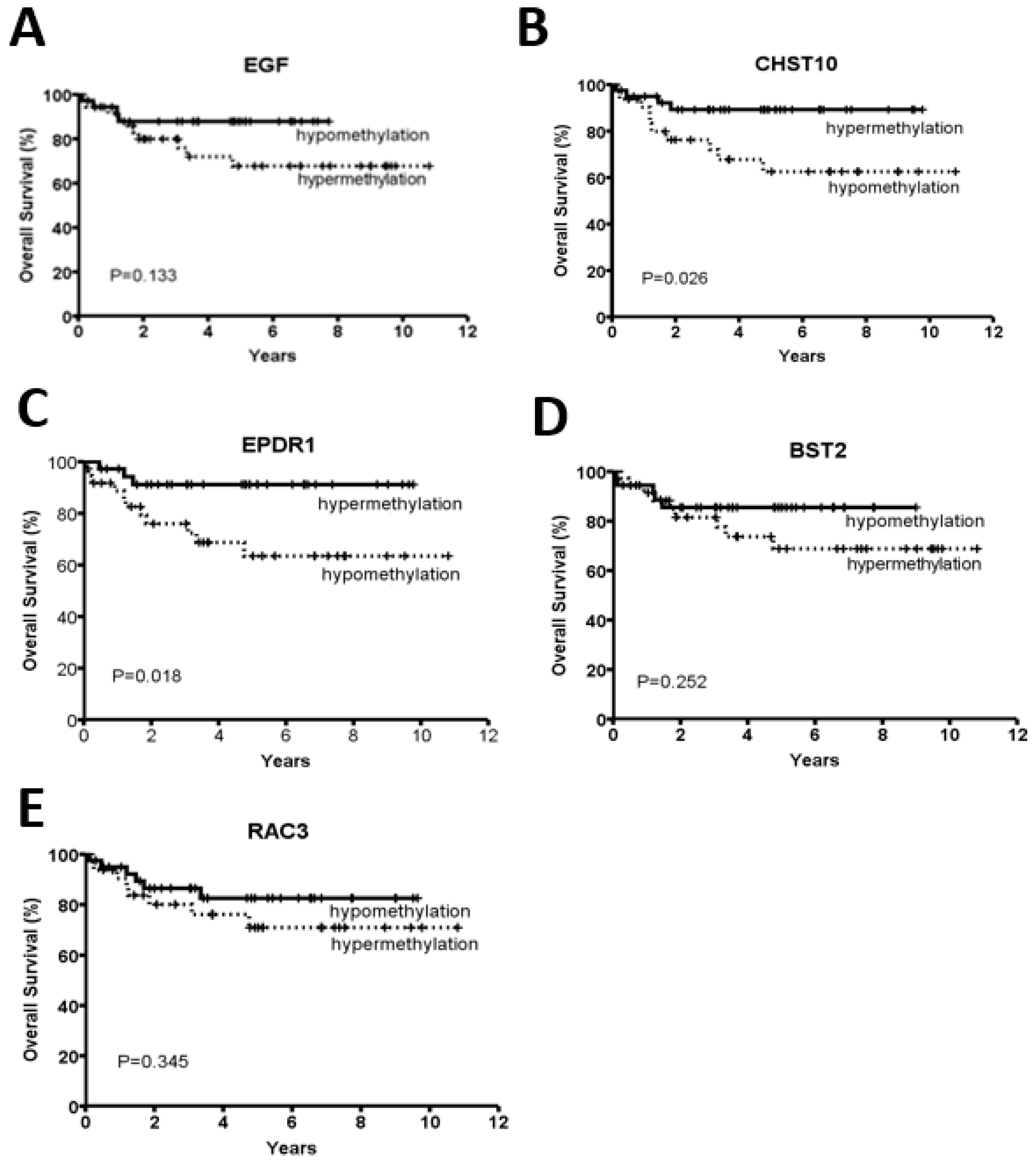
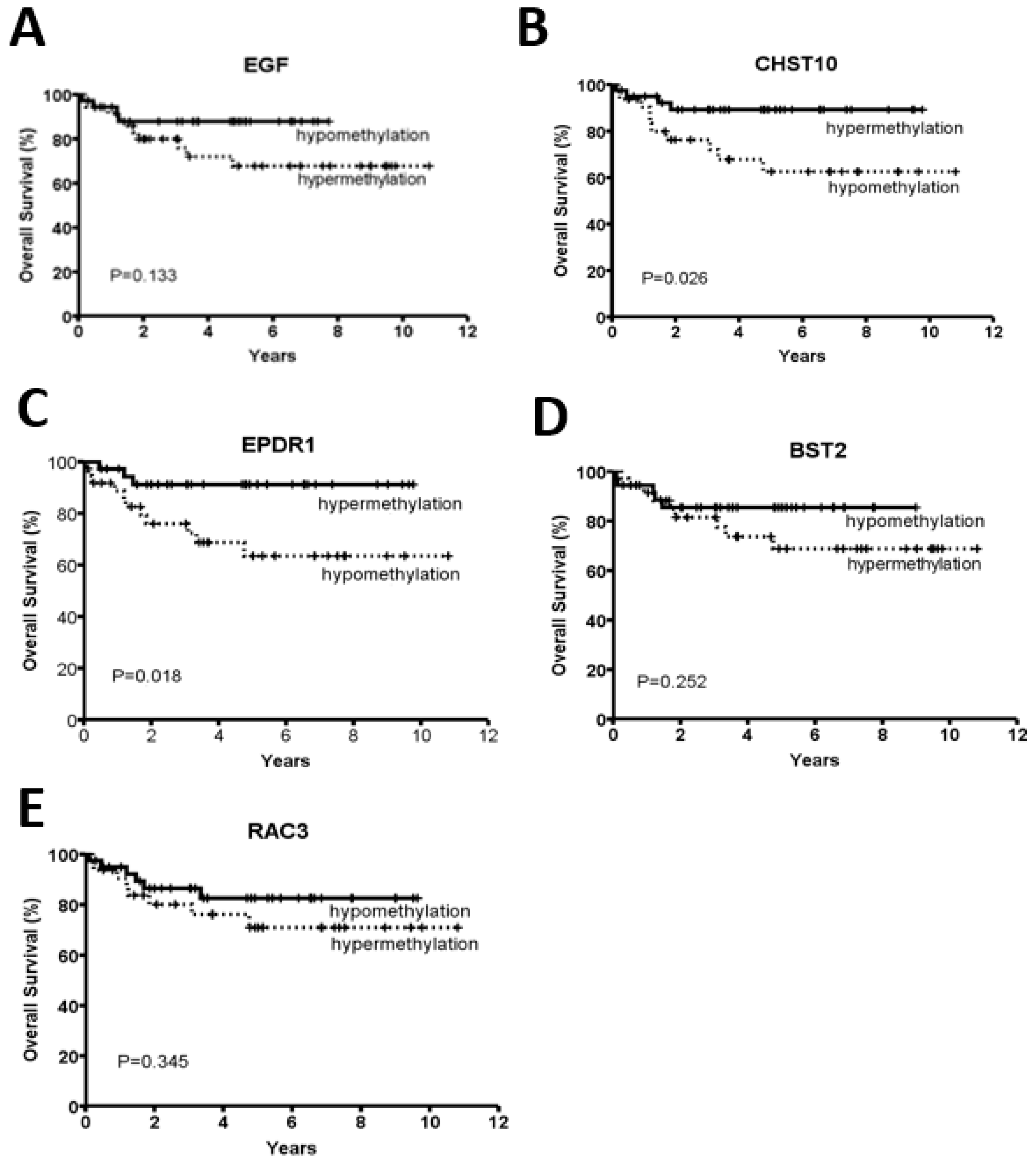
2.2. CHST10 and EPDR1 Hypermethylation Is Significantly Correlated with a Better Prognosis
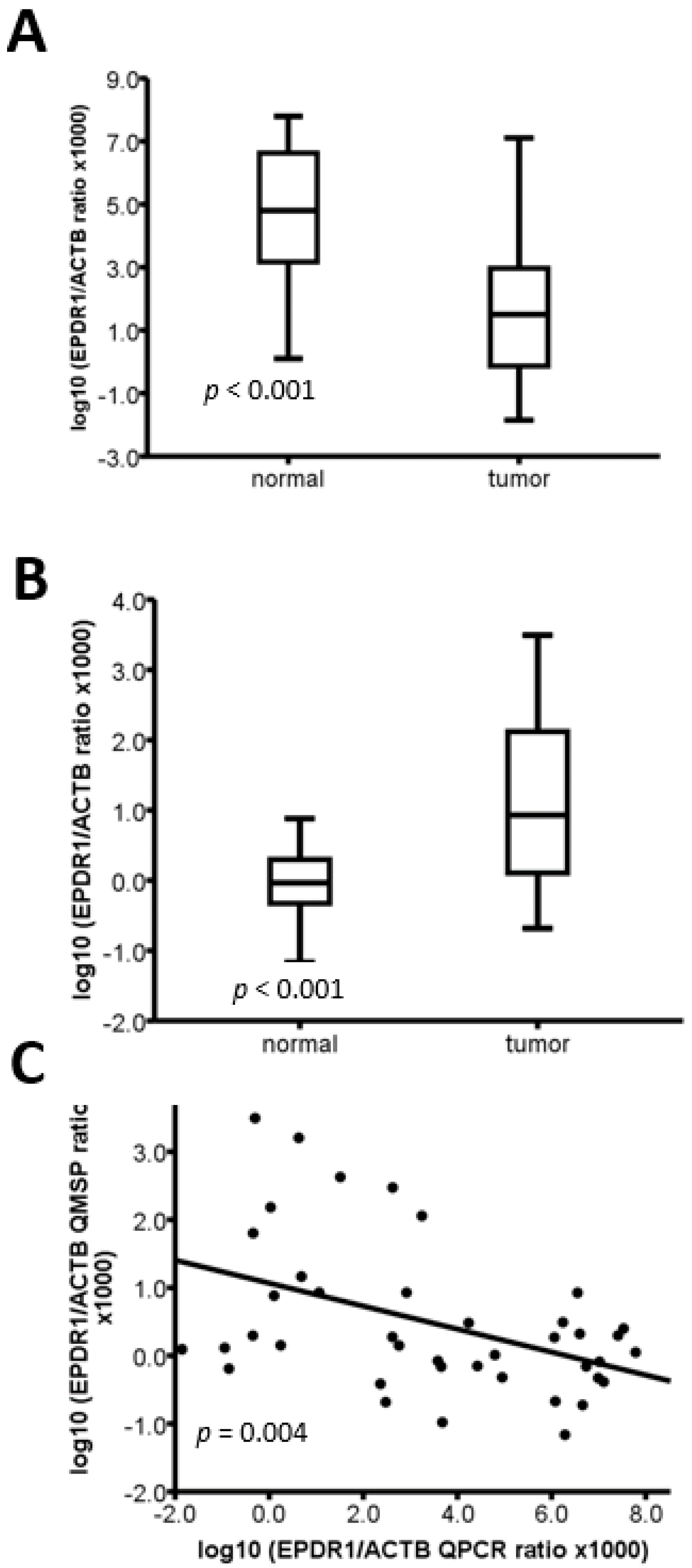
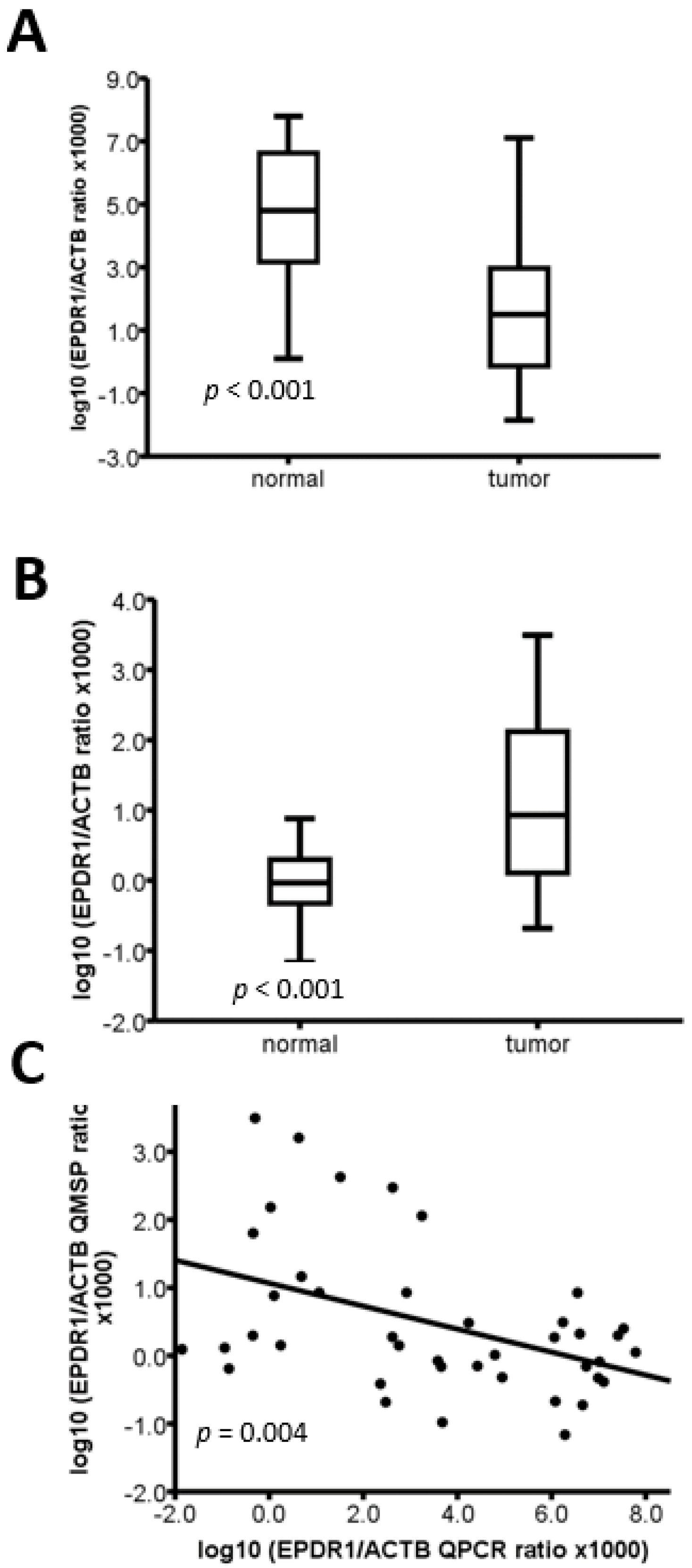
2.3. The Methylation Level of EPDR1 Is Correlated with Its mRNA Expression in CRC Tumor Tissues
2.4. EPDR1 Methylation Is Associated with BRAF and TGFβR2 Mutations in CRC Tumor Tissues
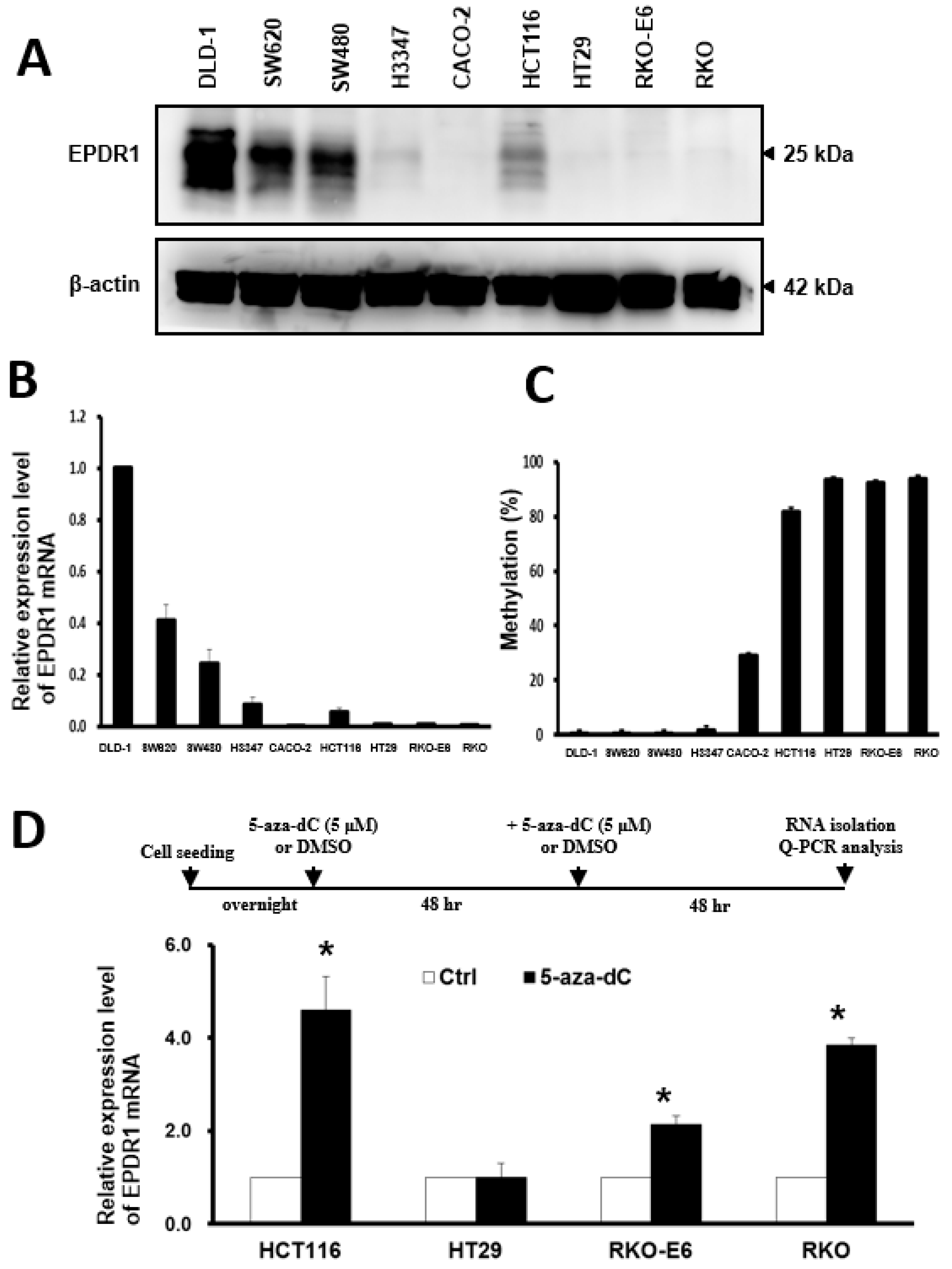
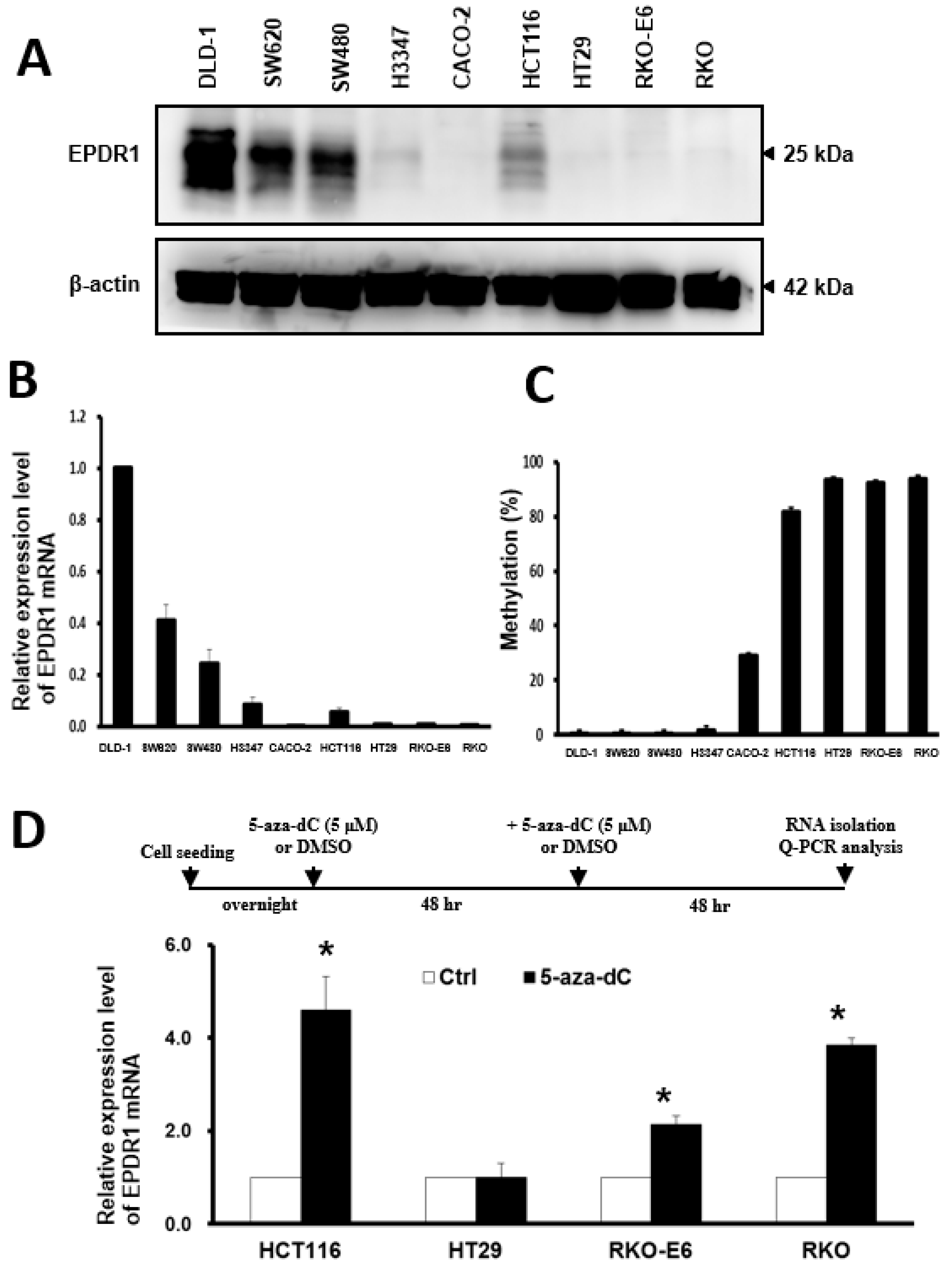
2.5. DNA Methylation Is Involved in the Regulation of EPDR1 Expression in CRC Cell Lines
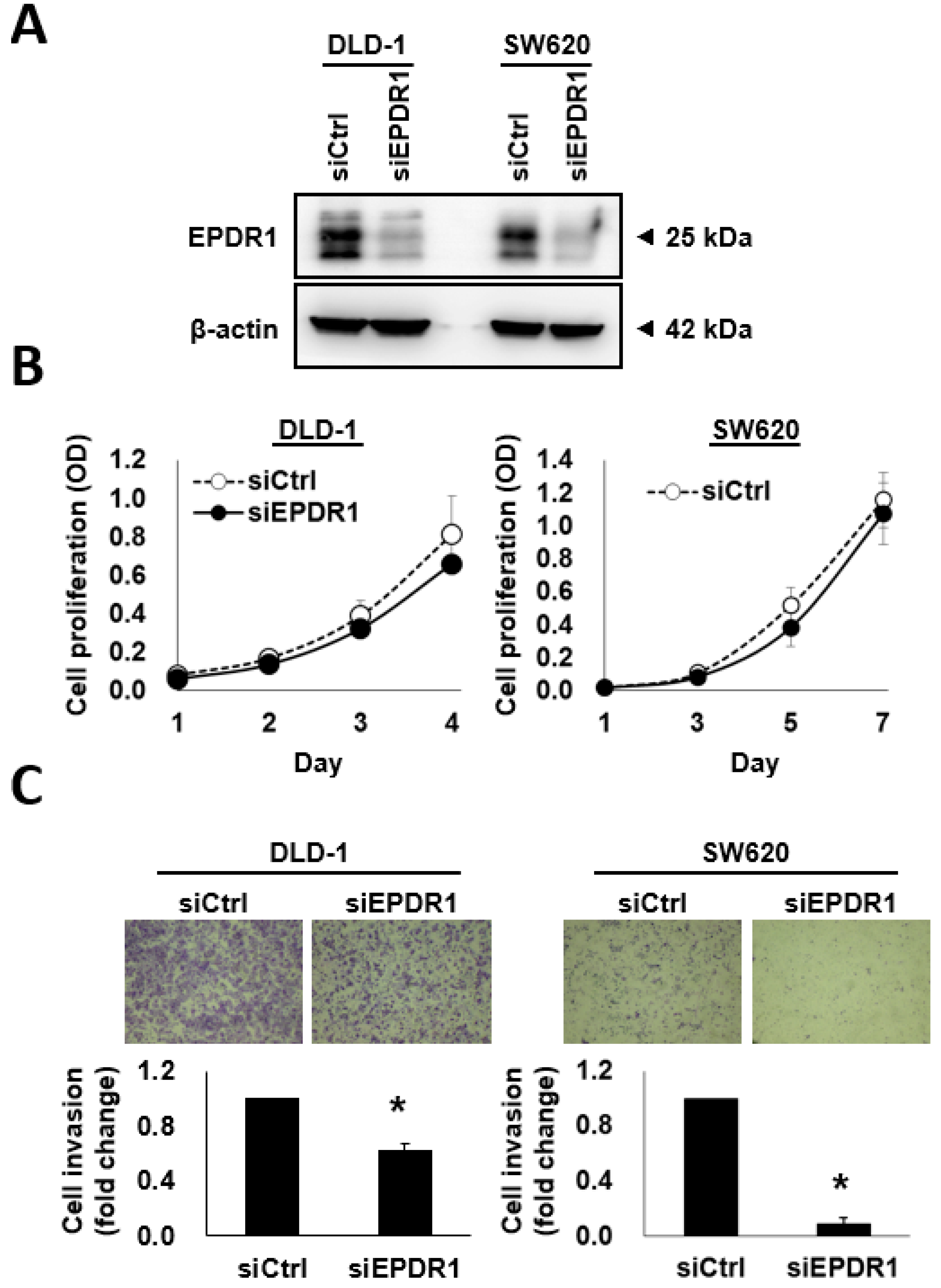
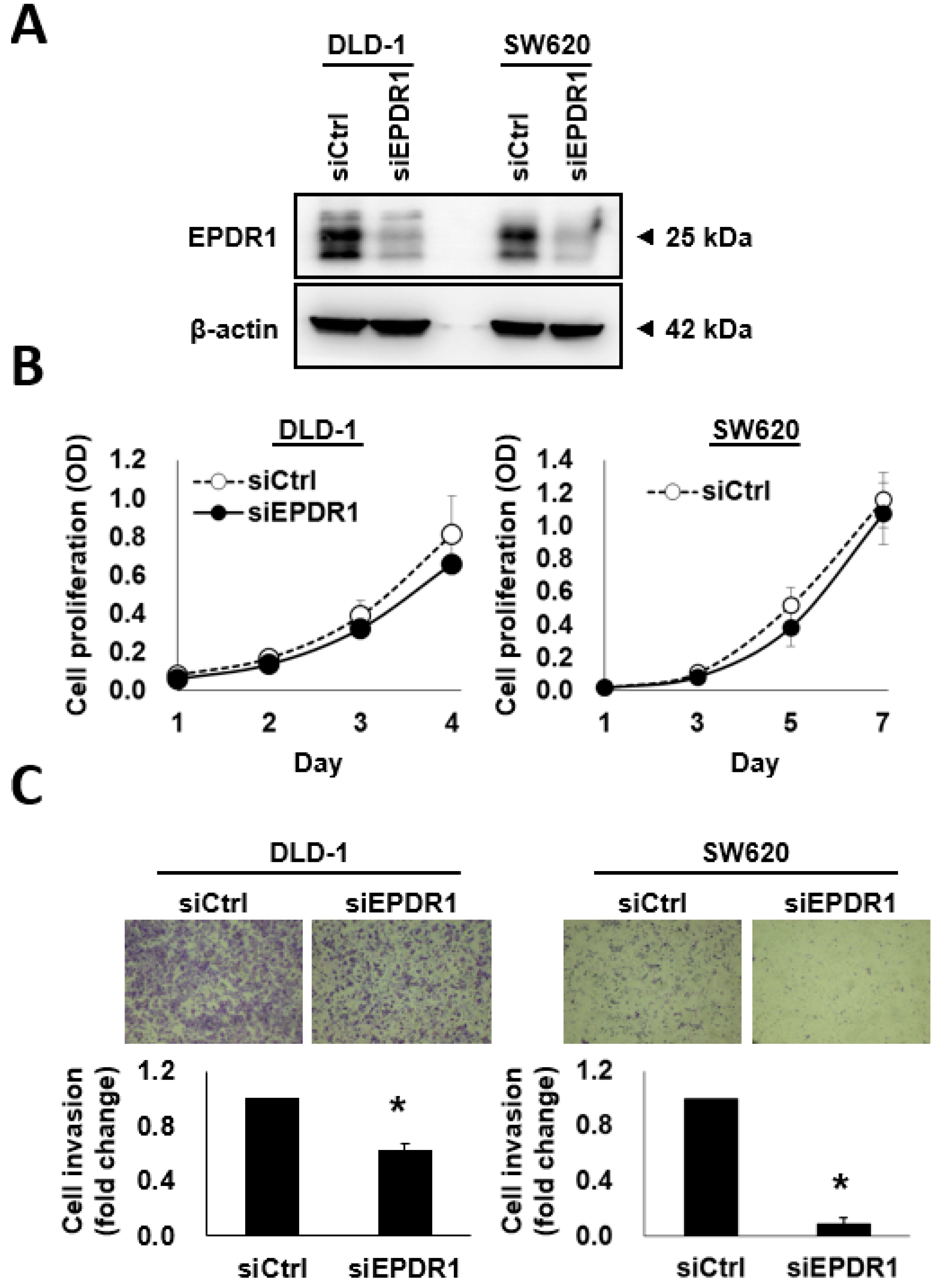
2.6. EPDR1 Knockdown Suppresses CRC Cell Invasion
3. Discussion
4. Materials and Methods
4.1. Patient and Tumor Samples
4.2. DNA Extraction from Tumor Samples
4.3. MSI Analysis
4.4. Mutation Analysis of MLH1
4.5. Methylation Analysis of MLH1
4.6. Genome-Wide DNA Methylation Profiling
4.7. Quantitative Methylation-Specific PCR (Q-MSP)
4.8. Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
4.9. MassArray-Based Mutation Characterization
4.10. Cell Lines and Cell Culture
4.11. Bisulfite Sequencing PCR (BSP)
4.12. Western Blotting
4.13. 5-Aza-2’-Deoxycytidine (5-aza-dC) Treatment
4.14. siRNA Transfection
4.15. Invasion Assay
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2016. [Google Scholar]
- Marmol, I.; Sanchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Biasco, G.; Derenzini, E.; Grazi, G.; Ercolani, G.; Ravaioli, M.; Pantaleo, M.A.; Brandi, G. Treatment of hepatic metastases from colorectal cancer: Many doubts, some certainties. Cancer Treat. Rev. 2006, 32, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.; Tekkis, P.P.; Welsh, F.K.; O’Rourke, T.; John, T.G. Evaluation of long-term survival after hepatic resection for metastatic colorectal cancer: A multifactorial model of 929 patients. Ann. Surg. 2008, 247, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T. Molecular pathology of colorectal cancer. Cytogenet. Cell Genet. 1999, 86, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Gonzalez, S.; Risques, R.A.; Marcuello, E.; Mangues, R.; Germa, J.R.; Herman, J.G.; Capella, G.; Peinado, M.A. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J. Clin. Oncol. 2001, 19, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004, 23, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Breivik, J.; Lothe, R.A.; Meling, G.I.; Rognum, T.O.; Borresen-Dale, A.L.; Gaudernack, G. Different genetic pathways to proximal and distal colorectal cancer influenced by sex-related factors. Int. J. Cancer 1997, 74, 664–669. [Google Scholar] [CrossRef] [Green Version]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 268. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bae, J.M.; Cho, N.Y.; Kang, G.H. Distinct features between MLH1-methylated and unmethylated colorectal carcinomas with the CpG island methylator phenotype: Implications in the serrated neoplasia pathway. Oncotarget 2016, 7, 14095–14111. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.; Meagher, A.; Tomlinson, I.; O’Connor, T.; Norrie, M.; Wu, R.; Hawkins, N. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut 2001, 48, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jass, J.R.; Do, K.A.; Simms, L.A.; Iino, H.; Wynter, C.; Pillay, S.P.; Searle, J.; Radford-Smith, G.; Young, J.; Leggett, B. Morphology of sporadic colorectal cancer with DNA replication errors. Gut 1998, 42, 673–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.M.; Banerjea, A.; Feakins, R.; Li, S.R.; Bustin, S.A.; Dorudi, S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br. J. Surg. 2004, 91, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Storojeva, I.; Boulay, J.L.; Heinimann, K.; Ballabeni, P.; Terracciano, L.; Laffer, U.; Mild, G.; Herrmann, R.; Rochlitz, C. Prognostic and predictive relevance of microsatellite instability in colorectal cancer. Oncol. Rep. 2005, 14, 241–249. [Google Scholar] [PubMed]
- Lanza, G.; Gafa, R.; Santini, A.; Maestri, I.; Guerzoni, L.; Cavazzini, L. Immunohistochemical test for MLH1 and MSH2 expression predicts clinical outcome in stage II and III colorectal cancer patients. J. Clin. Oncol. 2006, 24, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolli, M.M.; Niedzwiecki, D.; Compton, C.C.; Hahn, H.P.; Hall, M.; Damas, B.; Jewell, S.D.; Mayer, R.J.; Goldberg, R.M.; Saltz, L.B.; et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and leukemia group b protocol 89803. J. Clin. Oncol. 2009, 27, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with braf mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Esteller, M.; Risques, R.A.; Toyota, M.; Capella, G.; Moreno, V.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Promoter hypermethylation of the DNA repair gene o(6)-methylguanine-DNA methyltransferase is associated with the presence of g:C to a:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 2001, 61, 4689–4692. [Google Scholar] [PubMed]
- Kim, Y.H.; Lee, H.C.; Kim, S.Y.; Yeom, Y.I.; Ryu, K.J.; Min, B.H.; Kim, D.H.; Son, H.J.; Rhee, P.L.; Kim, J.J.; et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann. Surg. Oncol. 2011, 18, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Brim, H. DNA methylation and colorectal cancer. Curr. Colorectal Cancer Rep. 2014, 10, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Dhir, M.; Van Neste, L.; Downing, S.R.; Jeschke, J.; Glockner, S.C.; de Freitas Calmon, M.; Hooker, C.M.; Funes, J.M.; Boshoff, C.; et al. Genomic and epigenomic integration identifies a prognostic signature in colon cancer. Clin. Cancer Res. 2011, 17, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Melotte, V.; Lentjes, M.H.; van den Bosch, S.M.; Hellebrekers, D.M.; de Hoon, J.P.; Wouters, K.A.; Daenen, K.L.; Partouns-Hendriks, I.E.; Stessels, F.; Louwagie, J.; et al. N-myc downstream-regulated gene 4 (NDRG4): A candidate tumor suppressor gene and potential biomarker for colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Lentjes, M.H.; van den Bosch, S.M.; Melotte, V.; Wouters, K.A.; Daenen, K.L.; Smits, K.M.; Akiyama, Y.; Yuasa, Y.; Sanduleanu, S.; et al. GATA4 and GATA5 are potential tumor suppressors and biomarkers in colorectal cancer. Clin. Cancer Res. 2009, 15, 3990–3997. [Google Scholar] [CrossRef] [PubMed]
- Nimmrich, I.; Erdmann, S.; Melchers, U.; Chtarbova, S.; Finke, U.; Hentsch, S.; Hoffmann, I.; Oertel, M.; Hoffmann, W.; Muller, O. The novel ependymin related gene UCC1 is highly expressed in colorectal tumor cells. Cancer Lett. 2001, 165, 71–79. [Google Scholar] [CrossRef]
- Feng, Y.; Dai, X.; Li, X.; Wang, H.; Liu, J.; Zhang, J.; Du, Y.; Xia, L. EGF signalling pathway regulates colon cancer stem cell proliferation and apoptosis. Cell Prolif. 2012, 45, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.W.; Lee, S.K.; Lee, J.O.; Kim, N.; Lee, Y.W.; Kim, S.J.; Kang, H.J.; Kim, J.; Kim, H.S.; Park, S.H. Identification of novel hypermethylated genes and demethylating effect of vincristine in colorectal cancer. J. Exp. Clin. Cancer Res. 2014, 33, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, S.; Oue, N.; Oshima, T.; Mukai, R.; Tatsumoto, Y.; Sakamoto, N.; Sentani, K.; Tanabe, K.; Egi, H.; Hinoi, T.; et al. Overexpression of transmembrane protein BST2 is associated with poor survival of patients with esophageal, gastric, or colorectal cancer. Ann. Surg. Oncol. 2017, 24, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Q.; Wang, G.B.; Li, Z.J.; Tong, X.D.; Liu, H.X. Silencing of rac3 inhibits proliferation and induces apoptosis of human lung cancer cells. Asian Pac. J. Cancer Prev. 2015, 16, 3061–3065. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Simmer, F.; Brinkman, A.B.; Assenov, Y.; Matarese, F.; Kaan, A.; Sabatino, L.; Villanueva, A.; Huertas, D.; Esteller, M.; Lengauer, T.; et al. Comparative genome-wide DNA methylation analysis of colorectal tumor and matched normal tissues. Epigenetics 2012, 7, 1355–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morkel, M.; Riemer, P.; Blaker, H.; Sers, C. Similar but different: Distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance. Oncotarget 2015, 6, 20785–20800. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.I.; Limburg, P.J.; Baron, J.A.; Burnett-Hartman, A.N.; Weisenberger, D.J.; Laird, P.W.; Sinicrope, F.A.; Rosty, C.; Buchanan, D.D.; Potter, J.D.; et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology 2015, 148, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Ou, J.; Hutchinson, L.; Green, M.R. The braf oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG island methylator phenotype. Mol. Cell 2014, 55, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hagan, H.M.; Wang, W.; Sen, S.; Destefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Riviere, G.; Lienhard, D.; Andrieu, T.; Vieau, D.; Frey, B.M.; Frey, F.J. Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics 2011, 6, 478–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashoua, V.E.; Hesse, G.W.; Milinazzo, B. Evidence for the in vivo polymerization of ependymin: A brain extracellular glycoprotein. Brain Res. 1990, 522, 181–190. [Google Scholar] [CrossRef]
- Miranda, E.; Destro, A.; Malesci, A.; Balladore, E.; Bianchi, P.; Baryshnikova, E.; Franchi, G.; Morenghi, E.; Laghi, L.; Gennari, L.; et al. Genetic and epigenetic changes in primary metastatic and nonmetastatic colorectal cancer. Br. J. Cancer 2006, 95, 1101–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirahata, A.; Sakata, M.; Sakuraba, K.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y.; et al. Vimentin methylation as a marker for advanced colorectal carcinoma. Anticancer Res. 2009, 29, 279–281. [Google Scholar] [PubMed]
- Mizukami, H.; Shirahata, A.; Goto, T.; Sakata, M.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y.; Hibi, K. PGP9.5 methylation as a marker for metastatic colorectal cancer. Anticancer Res. 2008, 28, 2697–2700. [Google Scholar] [PubMed]
- Yamazaki, T.; Hibi, K.; Takase, T.; Tezel, E.; Nakayama, H.; Kasai, Y.; Ito, K.; Akiyama, S.; Nagasaka, T.; Nakao, A. PGP9.5 as a marker for invasive colorectal cancer. Clin. Cancer Res. 2002, 8, 192–195. [Google Scholar] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Chadwick, R.B.; Pyatt, R.E.; Niemann, T.H.; Richards, S.K.; Johnson, C.K.; Stevens, M.W.; Meek, J.E.; Hampel, H.; Prior, T.W.; de la Chapelle, A. Hereditary and somatic DNA mismatch repair gene mutations in sporadic endometrial carcinoma. J. Med. Genet. 2001, 38, 461–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grady, W.M.; Rajput, A.; Lutterbaugh, J.D.; Markowitz, S.D. Detection of aberrantly methylated hMLH1 promoter DNA in the serum of patients with microsatellite unstable colon cancer. Cancer Res. 2001, 61, 900–902. [Google Scholar] [PubMed]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. Methylight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Li, C.F.; Lin, L.F.; Wang, A.S.; Pu, Y.S.; Wang, H.H.; Mar, A.C.; Chen, C.J.; Lee, T.C. Promoter hypermethylation of LGALS4 correlates with poor prognosis in patients with urothelial carcinoma. Oncotarget 2017, 8, 23787–23802. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.K.; Lin, P.C.; Lin, C.H.; Jiang, J.K.; Yang, S.H.; Liang, W.Y.; Chen, W.S.; Chang, S.C. Clinical relevance of alterations in quantity and quality of plasma DNA in colorectal cancer patients: Based on the mutation spectra detected in primary tumors. Ann. Surg. Oncol. 2014, 2, S680–S686. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Wu, M.M.; Chan, M.W.; Pu, Y.S.; Chen, C.J.; Lee, T.C. Long-term low-dose exposure of human urothelial cells to sodium arsenite activates lipocalin-2 via promoter hypomethylation. Arch. Toxicol. 2014, 88, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.C.; Liu, C.J.; Chang, K.W.; Lee, T.C. Depleting IFIT2 mediates atypical PKC signaling to enhance the migration and metastatic activity of oral squamous cell carcinoma cells. Oncogene 2013, 32, 3686–3697. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | MSS * | MSI # | p Value | |
|---|---|---|---|---|
| Gender | ||||
| Male | 42 | 28 (66.7) | 14 (33.3) | 0.004 |
| Female | 33 | 11 (33.3) | 22 (66.7) | |
| Age | 76.9 ± 3.6 | 62.9 ± 14.3 | <0.001 | |
| Stage | ||||
| I–II | 44 | 19 (43.2) | 25 (56.8) | 0.069 |
| III–IV | 31 | 20 (64.5) | 11 (35.5) | |
| Differentiation | ||||
| Well-moderate | 63 | 38 (60.3) | 25 (39.7) | 0.001 |
| Poor | 12 | 1 (8.3) | 11 (91.7) | |
| Location | ||||
| Proximal colon | 27 | 5 (18.5) | 22 (81.5) | <0.001 |
| Distal colon | 48 | 34 (70.8) | 14 (29.2) | |
| Histology | ||||
| Adenocarcinoma | 68 | 36 (52.9) | 32 (47.1) | 0.704 |
| Mucinous | 7 | 3 (42.9) | 4 (57.1) |
| EGF | CHST10 | EPDR1 | BST2 | RAC3 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypo-M | Hyper-M | p | Hypo-M | Hyper-M | p | Hypo-M | Hyper-M | p | Hypo-M | Hyper-M | p | Hypo-M | Hyper-M | p | |
| Differentiation | |||||||||||||||
| Well-moderate | 33 (52.4) | 30 (47.6) | 29 (46.0) | 34 (56.0) | 33 (52.4) | 30 (47.6) | 29 (46.0) | 34 (54.0) | 32 (52.5) | 29 (47.5) | |||||
| Poor | 5 (41.7) | 7 (58.3) | 0.496 | 3 (25.0) | 9 (75.0) | 0.177 | 5 (41.7) | 7 (58.3) | 0.496 | 8 (66.7) | 4 (33.3) | 0.190 | 9 (75.0) | 3 (25.0) | 0.150 |
| Invasion depth | |||||||||||||||
| T1/T2 | 5 (38.5) | 8 (61.5) | 5 (38.5) | 8 (61.5) | 6 (46.2) | 7 (53.8) | 6 (46.2) | 7 (53.8) | 6 (54.5) | 5 (45.5) | |||||
| T3/T4 | 33 (53.2) | 29 (46.8) | 0.333 | 27 (43.5) | 35 (56.5) | 0.736 | 32 (51.6) | 30 (48.4) | 0.720 | 31 (50.0) | 31 (50.0) | 0.801 | 35 (56.5) | 27 (43.5) | 1.00 |
| Node stage | |||||||||||||||
| Negative | 23 (52.3) | 21 (47.7) | 16 (36.4) | 28 (63.6) | 18 (40.9) | 26 (59.1) | 22 (50.0) | 22 (50.0) | 26 (61.9) | 16 (38.1) | |||||
| Positive | 15 (48.4) | 16 (51.6) | 0.740 | 16 (51.6) | 15 (48.4) | 0.189 | 20 (64.5) | 11 (35.5) | 0.044 | 15 (48.4) | 16 (51.6) | 0.891 | 15 (48.4) | 16 (51.6) | 0.250 |
| AJCC stage | |||||||||||||||
| I–II | 23 (52.3) | 21 (47.7) | 16 (36.4) | 28 (63.6) | 18 (40.9) | 26 (59.1) | 22 (50.0) | 22 (50.0) | 26 (61.9) | 16 (38.1) | |||||
| III–IV | 15 (48.4) | 16 (51.6) | 0.740 | 16 (51.6) | 15 (48.4) | 0.189 | 20 (64.5) | 11 (35.5) | 0.044 | 15 (48.4) | 16 (51.6) | 0.891 | 15 (48.4) | 16 (51.6) | 0.250 |
| Microsatellite status | |||||||||||||||
| MSS | 18 (46.2) | 21 (53.8) | 23 (59.0) | 16 (41.0) | 28 (71.8) | 11 (28.2) | 14 (35.9) | 25 (64.1) | 14 (35.9) | 25 (64.1) | |||||
| MSI | 20 (55.6) | 16 (44.4) | 0.416 | 9 (25.0) | 27 (75.0) | 0.003 | 10 (27.8) | 26 (72.2) | <0.001 | 23 (63.9) | 13 (36.1) | 0.015 | 28 (77.8) | 8 (22.2) | <0.001 |
| n | EPDR1 HYPER-M # (n = 27) | EPDR1 HYPO-M (n = 32) | p Value | |
|---|---|---|---|---|
| APC | ||||
| Wild type | 41 | 20 (74.1) | 21 (65.6) | 0.483 |
| Mutation | 18 | 7 (25.9) | 11 (34.4) | |
| TP53 | ||||
| Wild type | 46 | 22 (81.5) | 24 (75.0) | 0.550 |
| Mutation | 13 | 5 (18.5) | 8 (25.0) | |
| KRAS | ||||
| Wild type | 45 | 21 (77.8) | 24 (75.0) | 0.803 |
| Mutation | 14 | 6 (22.2) | 8 (25.0) | |
| BRAF | ||||
| Wild type | 49 | 17 (63.0) | 32 (100.0) | <0.001 |
| Mutation | 10 | 10 (37.0) | 0 (0.0) | |
| TGFBR2 | ||||
| Wild type | 52 | 21 (77.8) | 31 (96.9) | 0.040 |
| Mutation | 7 | 6 (22.2) | 1 (3.1) | |
| PIK3CA | ||||
| Wild type | 46 | 19 (70.4) | 27 (84.4) | 0.196 |
| Mutation | 13 | 8 (29.6) | 5 (15.6) | |
| SMAD4 | ||||
| Wild type | 57 | 25 (92.6) | 32(100.0) | 0.205 |
| Mutation | 2 | 2 (7.4) | 0 (0.0) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, C.-H.; Chang, S.-C.; Wang, H.-H.; Yang, S.-H.; Lai, K.-C.; Lee, T.-C. Prognostic Values of EPDR1 Hypermethylation and Its Inhibitory Function on Tumor Invasion in Colorectal Cancer. Cancers 2018, 10, 393. https://doi.org/10.3390/cancers10100393
Chu C-H, Chang S-C, Wang H-H, Yang S-H, Lai K-C, Lee T-C. Prognostic Values of EPDR1 Hypermethylation and Its Inhibitory Function on Tumor Invasion in Colorectal Cancer. Cancers. 2018; 10(10):393. https://doi.org/10.3390/cancers10100393
Chicago/Turabian StyleChu, Chun-Ho, Shih-Ching Chang, Hsiu-Hua Wang, Shung-Haur Yang, Kuo-Chu Lai, and Te-Chang Lee. 2018. "Prognostic Values of EPDR1 Hypermethylation and Its Inhibitory Function on Tumor Invasion in Colorectal Cancer" Cancers 10, no. 10: 393. https://doi.org/10.3390/cancers10100393