Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer

 ,
,  , , ,
, , , 
 and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
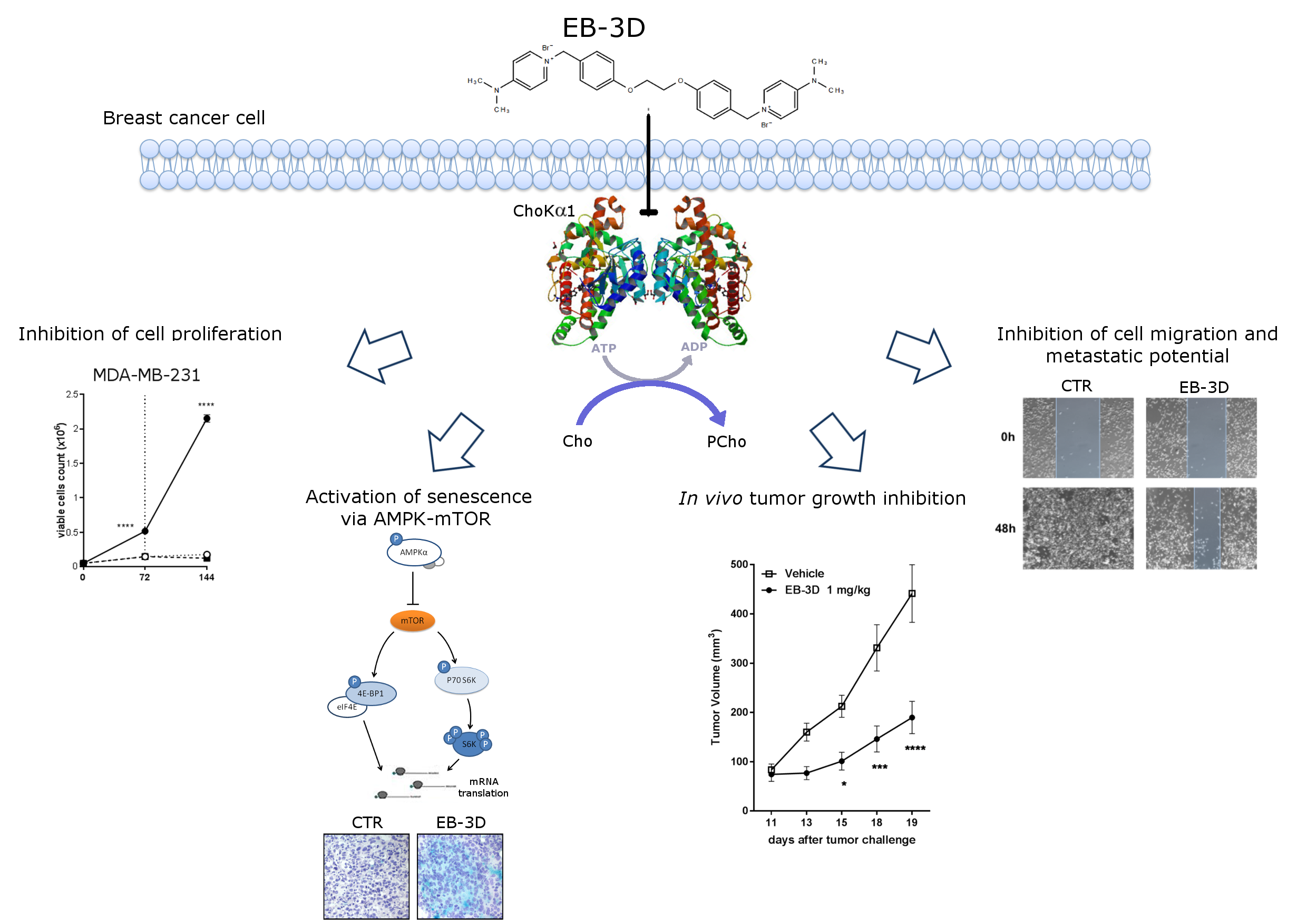
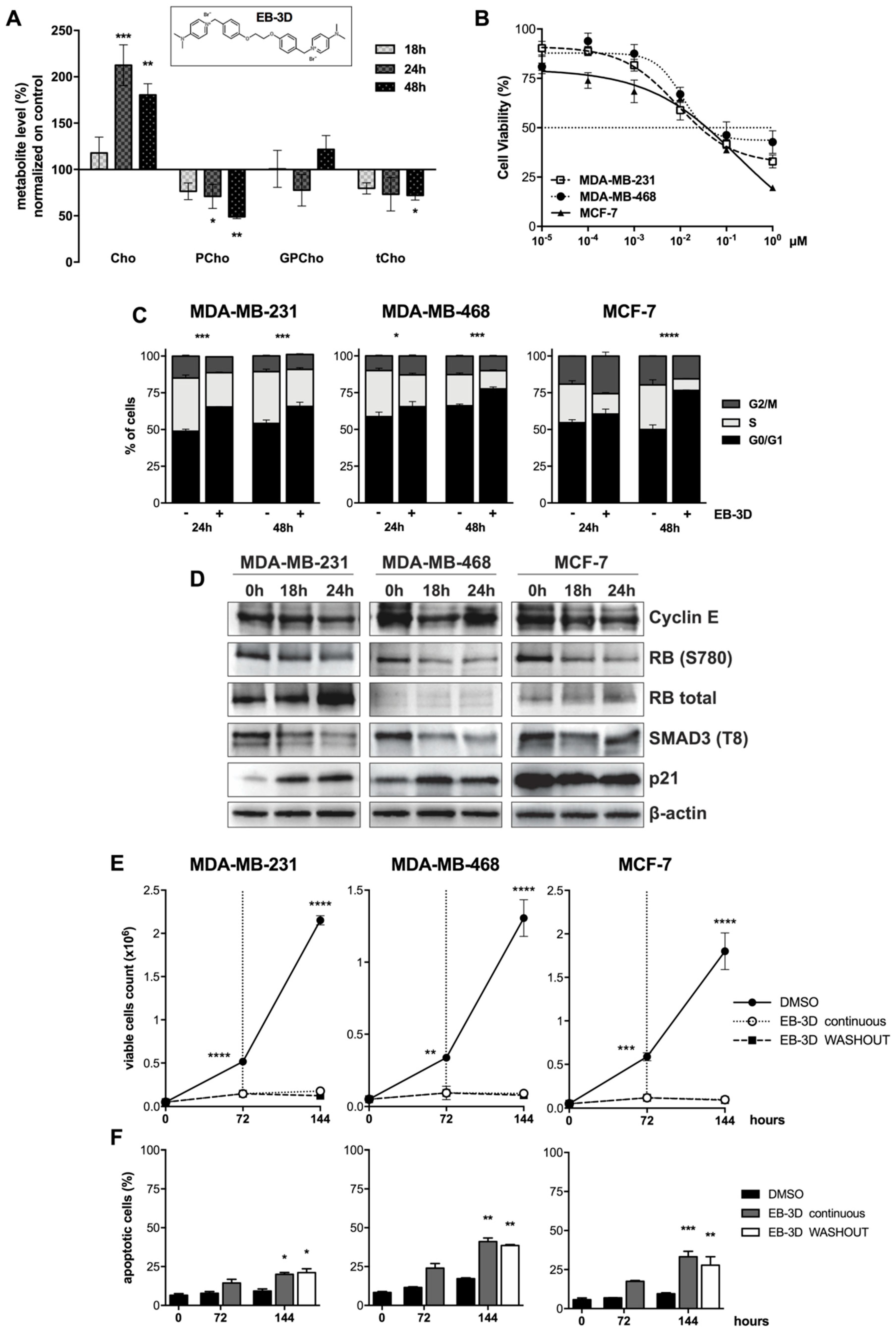
2.1. Inhibition of ChoKα by EB-3D Reduces Soluble Choline Metabolites
2.2. EB-3D Reduces Cell Proliferation in Breast Cancer Cell Lines
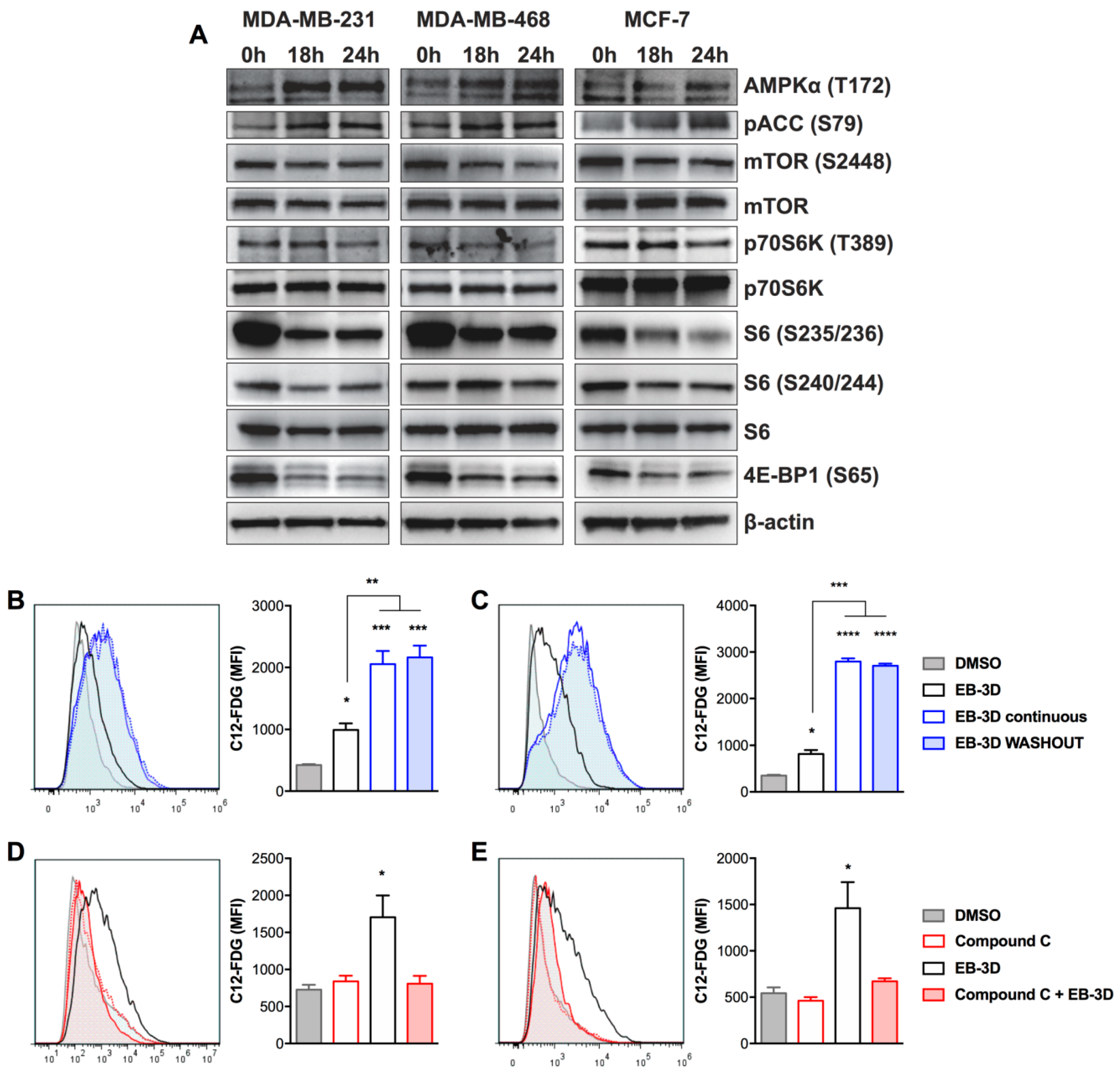
2.3. EB-3D Induces a Senescence-Like Phenotype via AMPK-mTOR Signaling
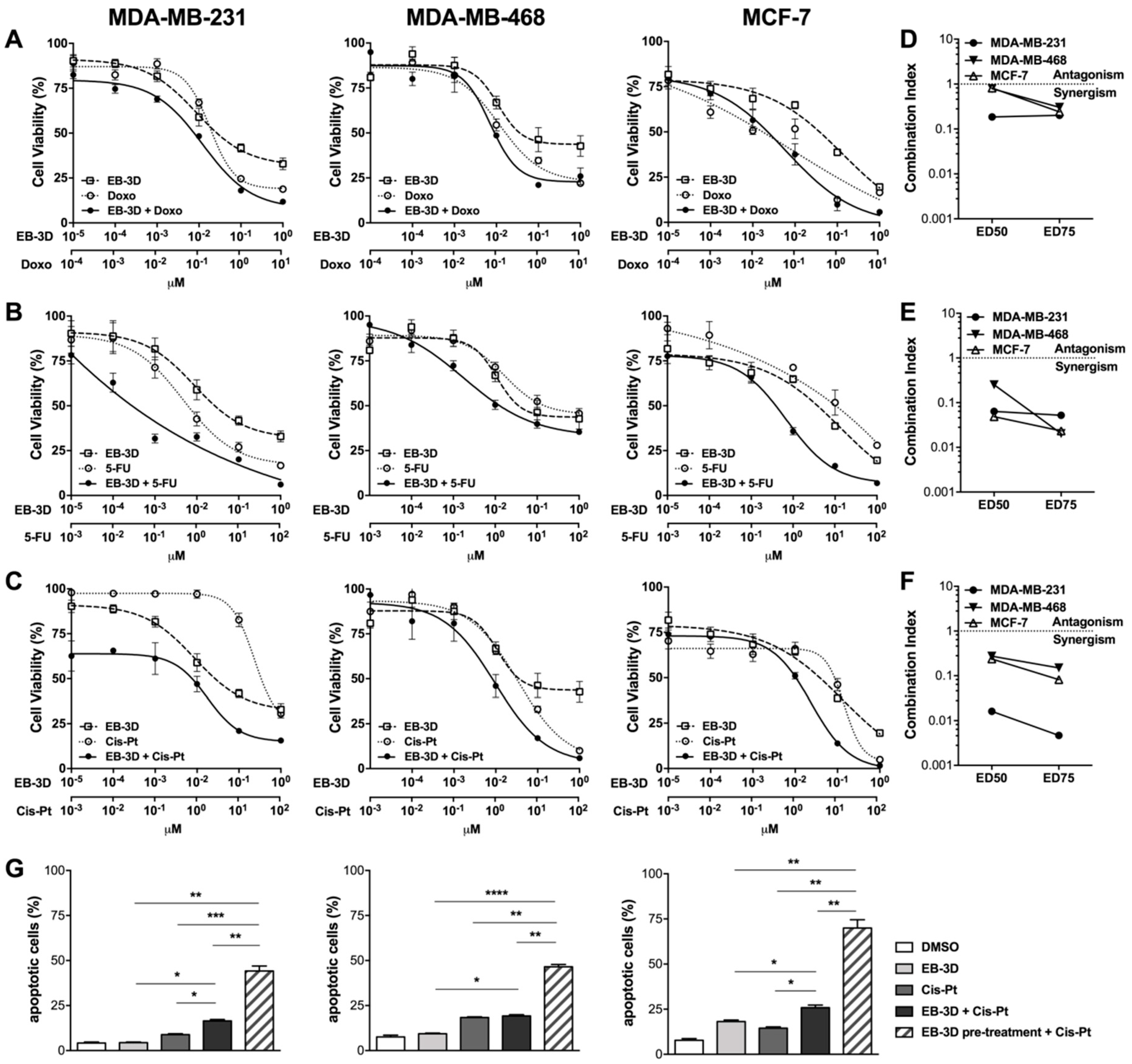
2.4. ChoKα Inhibition Synergistically Enhances Commonly Used Chemotherapeutics Effects and Sensitizes Breast Cancer Cell Lines to Cisplatin
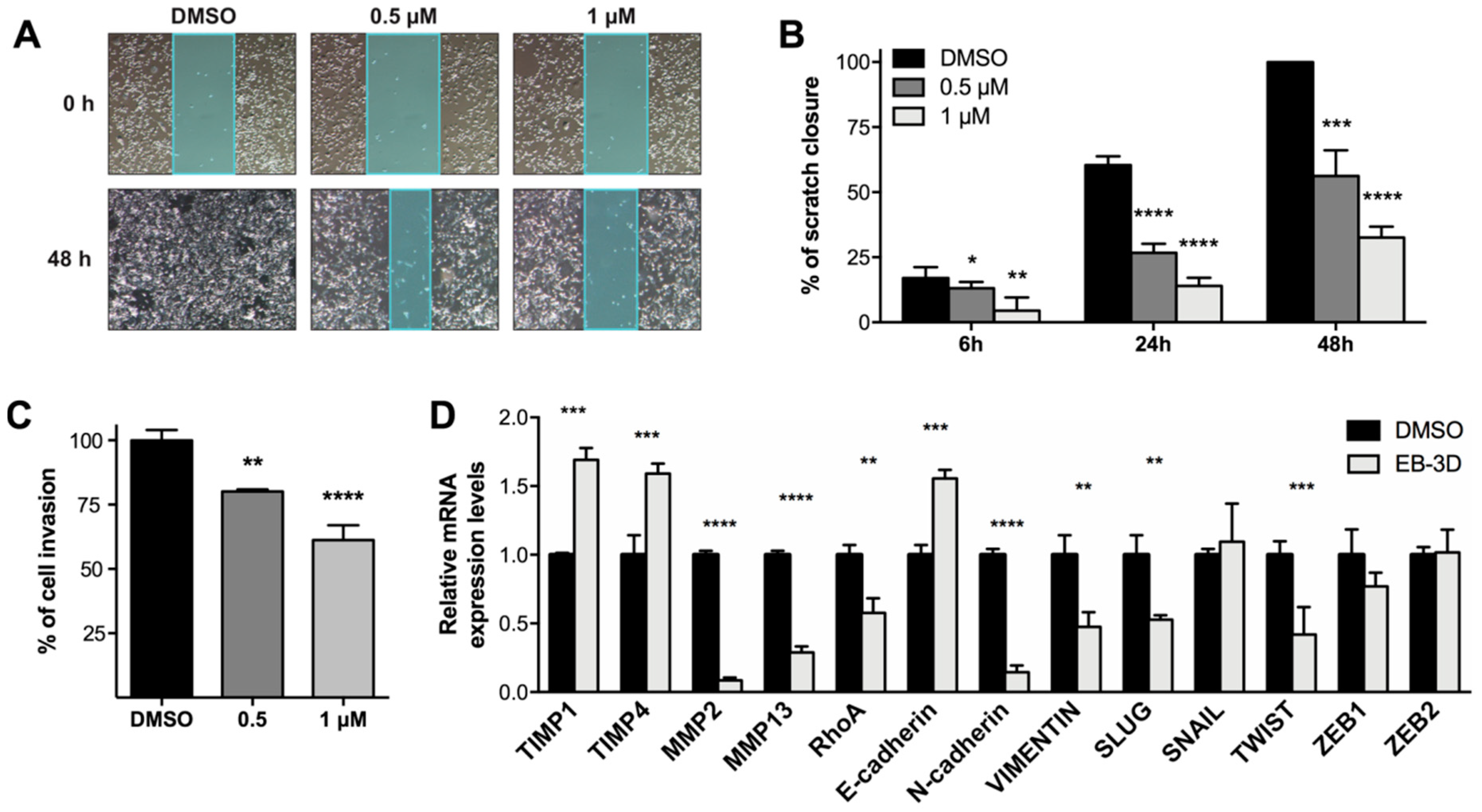
2.5. Targeting ChoKα Activity Impacts Cell Migration and Invasion in the Highly Aggressive MDA-MB-231 Cells
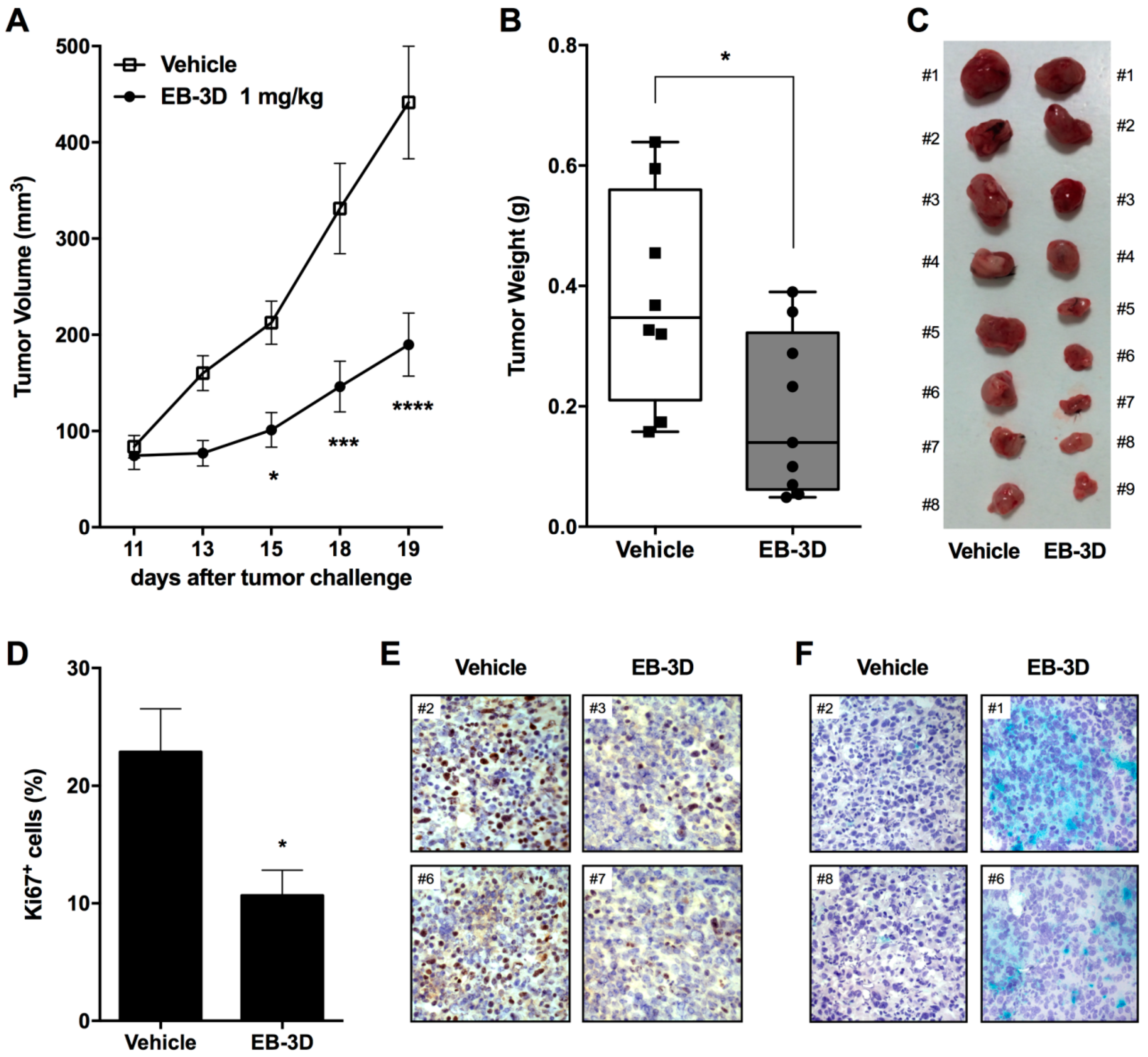
2.6. In Vivo Tumor Growth Inhibition and Senescence Induction
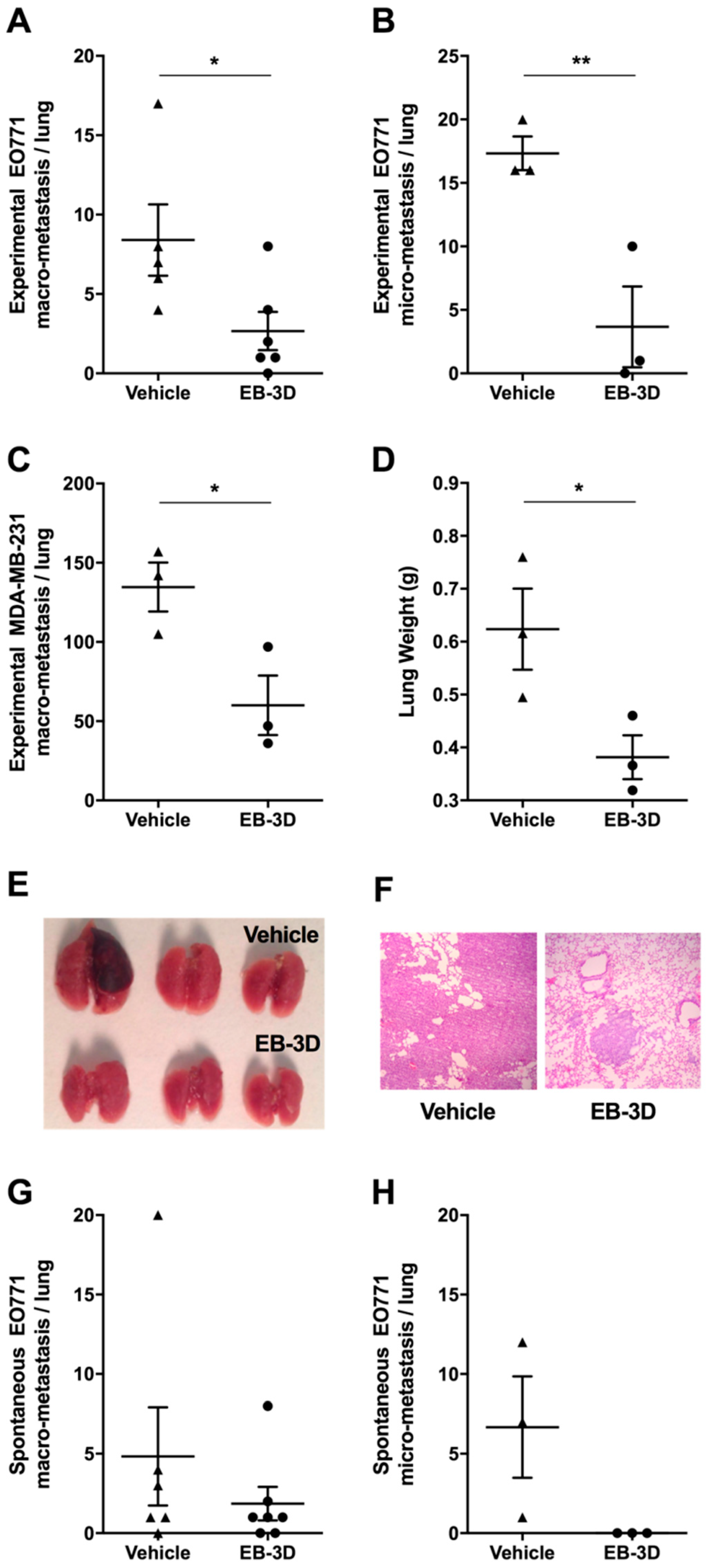
2.7. Metastasis Formation Is Reduced by EB-3D Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Cell Viability Assay and Drug Combination Sensitivity Assay
4.3. Cell Cycle Distribution Analysis
4.4. Magnetic Resonance Spectroscopy (1H-MRS)
4.5. Annexin-V/PI Assay
4.6. C12-FDG Senescence Assay
4.7. Western Blot Analysis
4.8. Scratch-Migration Assay
4.9. Cultrex BME Cell Invasion Assay
4.10. Quantitative Real-Time PCR (qRT-PCR)
4.11. In Vivo Tumor Growth
4.12. In Vivo Metastasis Models
4.13. Histological Analyses
4.14. Statistical Analyses
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Weinberg Cell—Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Iorio, E.; Ricci, A.; Bagnoli, M.; Pisanu, M.E.; Castellano, G.; Di Vito, M.; Venturini, E.; Glunde, K.; Bhujwalla, Z.M.; Mezzanzanica, D.; et al. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 2010, 70, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Massie, C.E.; Orafidiya, F.; Pértega-Gomes, N.; Warren, A.Y.; Esmaeili, M.; Selth, L.A.; Zecchini, H.I.; Luko, K.; Qureshi, A.; et al. Choline Kinase Alpha as an Androgen Receptor Chaperone and Prostate Cancer Therapeutic Target. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Ramírez de Molina, A.; Rodríguez-González, A.; Gutiérrez, R.; Martínez-Pieiro, L.; Sánchez, J.J.; Bonilla, F.; Rosell, R.; Lacal, J.C. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem. Biophys. Res. Commun. 2002, 296, 580–583. [Google Scholar] [CrossRef]
- De Molina, A.R.; Gutiérrez, R.; Ramos, M.A.; Silva, J.M.; Silva, J.; Bonilla, F.; Sá Nchez, J.J.; Lacal, J.C. Increased choline kinase activity in human breast carcinomas: Clinical evidence for a potential novel antitumor strategy. Oncogene 2002, 21, 4317–4322. [Google Scholar] [CrossRef] [PubMed]
- Ramírez de Molina, A.; Sarmentero-Estrada, J.; Belda-Iniesta, C.; Tarón, M.; Ramírez de Molina, V.; Cejas, P.; Skrzypski, M.; Gallego-Ortega, D.; de Castro, J.; Casado, E.; et al. Expression of choline kinase alpha to predict outcome in patients with early-stage non-small-cell lung cancer: A retrospective study. Lancet Oncol. 2007, 8, 889–897. [Google Scholar] [CrossRef]
- Nakagami, K.; Uchida, T.; Ohwada, S.; Koibuchi, Y.; Suda, Y.; Sekine, T.; Morishita, Y. Increased choline kinase activity and elevated phosphocholine levels in human colon cancer. Jpn. J. Cancer Res. 1999, 90, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Mariotto, E.; Bortolozzi, R.; Volpin, I.; Carta, D.; Serafin, V.; Accordi, B.; Basso, G.; Navarro, P.L.; López-Cara, L.C.; Viola, G. EB-3D a novel choline kinase inhibitor induces deregulation of the AMPK-mTOR Pathway and apoptosis in leukemia T-cells. Biochem. Pharmacol. 2018, 155, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Granata, A.; Nicoletti, R.; Perego, P.; Iorio, E.; Krishnamachary, B.; Benigni, F.; Ricci, A.; Podo, F.; Bhujwalla, Z.M.; Canevari, S.; et al. Global metabolic profile identifies choline kinase alpha as a key regulator of glutathione-dependent antioxidant cell defense in ovarian carcinoma. Oncotarget 2015, 6, 11216–11230. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Raman, V.; Mori, N.; Bhujwalla, Z.M. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res. 2005, 65, 11034–11043. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Glunde, K.; Wildes, F.; Mori, N.; Takagi, T.; Raman, V.; Bhujwalla, Z.M. Noninvasive detection of lentiviral-mediated choline kinase targeting in a human breast cancer xenograft. Cancer Res. 2009, 69, 3464–3471. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Glunde, K.; Takagi, T.; Raman, V.; Bhujwalla, Z.M. Choline kinase down-regulation increases the effect of 5-fluorouracil in breast cancer cells. Cancer Res. 2007, 67, 11284–11290. [Google Scholar] [CrossRef] [PubMed]
- Lacal, J.C. Choline kinase: A novel target for antitumor drugs. IDrugs 2001, 4, 419–426. [Google Scholar] [PubMed]
- Lacal, J.C.; Campos, J.M. Preclinical Characterization of RSM-932A, a Novel Anticancer Drug Targeting the Human Choline Kinase Alpha, an Enzyme Involved in Increased Lipid Metabolism of Cancer Cells. Mol. Cancer Ther. 2015, 14, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Alcoceba, R.; Fernández, F.; Lacal, J.C. In vivo antitumor activity of choline kinase inhibitors: A novel target for anticancer drug discovery. Cancer Res. 1999, 59, 3112–3118. [Google Scholar] [PubMed]
- Rodríguez-González, A.; Ramírez De Molina, A.; Fernández, F.; Ramos, M.A.; Núñez, M.D.C.; Campos, J.; Lacal, J.C. Inhibition of choline kinase as a specific cytotoxic strategy in oncogene-transformed cells. Oncogene 2003, 22, 8803–8812. [Google Scholar] [CrossRef] [PubMed]
- Falcon, S.C.; Hudson, C.S.; Huang, Y.; Mortimore, M.; Golec, J.M.; Charlton, P.A.; Weber, P.; Sundaram, H. A non-catalytic role of choline kinase alpha is important in promoting cancer cell survival. Oncogenesis 2013, 2. [Google Scholar] [CrossRef]
- Castro-Navas, F.F.; Schiaffino-Ortega, S.; Carrasco-Jimenez, M.P.; Ríos-Marco, P.; Marco, C.; Espinosa, A.; Gallo, M.A.; Mariotto, E.; Basso, G.; Viola, G.; et al. New more polar symmetrical bipyridinic compounds: New strategy for the inhibition of choline kinase α1. Future Med. Chem. 2015, 7, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Clem, B.F.; Clem, A.L.; Yalcin, A.; Goswami, U.; Arumugam, S.; Telang, S.; Trent, J.O.; Chesney, J. A novel small molecule antagonist of choline kinase-α that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene 2011, 30, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Clem, B.; Makoni, S.; Clem, A.; Nelson, K.; Thornburg, J.; Siow, D.; Lane, A.N.; Brock, S.E.; Goswami, U.; et al. Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene 2010, 29, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Kumar, M.; Popov, A.V.; Poptani, H.; Delikatny, E.J. Near infrared fluorescent imaging of choline kinase alpha expression and inhibition in breast tumors. Oncotarget 2017, 8, 16518–16530. [Google Scholar] [CrossRef] [PubMed]
- Trousil, S.; Kaliszczak, M.; Schug, Z.; Nguyen, Q.-D.; Tomasi, G.; Favicchio, R.; Brickute, D.; Fortt, R.; Twyman, F.J.; Carroll, L.; et al. The novel choline kinase inhibitor ICL-CCIC-0019 reprograms cellular metabolism and inhibits cancer cell growth. Oncotarget 2016, 7, 37103–37120. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino-Ortega, S.; Baglioni, E.; Mariotto, E.; Bortolozzi, R.; Serrán-Aguilera, L.; Riós-Marco, P.; Carrasco-Jimenez, M.P.; Gallo, M.A.; Hurtado-Guerrero, R.; Marco, C.; et al. Design, synthesis, crystallization and biological evaluation of new symmetrical biscationic compounds as selective inhibitors of human Choline Kinase α1 (ChoKα1). Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- De Molina, A.R.; Báñez-Coronel, M.; Gutiérrez, R.; Rodríguez-González, A.; Olmeda, D.; Megías, D.; Lacal, J.C. Choline kinase activation is a critical requirement for the proliferation of primary human mammary epithelial cells and breast tumor progression. Cancer Res. 2004, 64, 6732–6739. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.R.; Kim, E.J.; Choi, H.J.; Park, J.-J.; Kim, H.-S.; Lee, Y.-J.; Park, M.-J.; Lee, M. Aspirin Targets SIRT1 and AMPK to Induce Senescence of Colorectal Carcinoma Cells. Mol. Pharmacol. 2015, 88, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Granata, A.; Nicoletti, R.; Tinaglia, V.; De Cecco, L.; Pisanu, M.E.; Ricci, A.; Podo, F.; Canevari, S.; Iorio, E.; Bagnoli, M.; et al. Choline kinase-alpha by regulating cell aggressiveness and drug sensitivity is a potential druggable target for ovarian cancer. Br. J. Cancer 2014, 110, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Ramírez De Molina, A.; Gallego-Ortega, D.; Sarmentero, J.; Bañez-Coronel, M.; Martín-Cantalejo, Y.; Lacal, J.C. Choline kinase is a novel oncogene that potentiates RhoA-induced carcinogenesis. Cancer Res. 2005, 65, 5647–5653. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Zimmerman, T.; Gomez del Pulgar, T.; Moyer, M.P.; Lacal Sanjuan, J.C.; Cebrian, A. Choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis via CHOP in cancer cells. Cell Death Dis. 2013, 4, e933–e944. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Wildes, F.; Kakkad, S.; Jacob, D.; Solaiyappan, M.; Glunde, K.; Bhujwalla, Z.M. Choline kinase-α protein and phosphatidylcholine but not phosphocholine are required for breast cancer cell survival. NMR Biomed. 2015, 28, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Tercé, F.; Brun, H.; Vance, D.E. Requirement of phosphatidylcholine for normal progression through the cell cycle in C3H/10T1/2 fibroblasts. J. Lipid Res. 1994, 35, 2130–2142. [Google Scholar] [PubMed]
- Rodríguez-González, A.; De Molina, A.R.; Fernández, F.; Lacal, J.C. Choline kinase inhibition induces the increase in ceramides resulting in a highly specific and selective cytotoxic antitumoral strategy as a potential mechanism of action. Oncogene 2004, 23, 8247–8259. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A.; Yellen, P.; Xu, L.; Saqcena, M. Regulation of G1 cell cycle progression: Distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 2011, 1, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Houweling, M. Phosphatidylcholine and cell death. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2002, 1585, 87–96. [Google Scholar] [CrossRef]
- Gey, C.; Seeger, K. Metabolic changes during cellular senescence investigated by proton NMR-spectroscopy. Mech. Ageing Dev. 2013, 134, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, X.; De Silanes, I.L.; Carling, D.; Gorospe, M. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J. Biol. Chem. 2003, 278, 27016–27023. [Google Scholar] [CrossRef] [PubMed]
- Liao, E.-C.; Hsu, Y.-T.; Chuah, Q.-Y.; Lee, Y.-J.; Hu, J.-Y.; Huang, T.-C.; Yang, P.-M.; Chiu, S.-J. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis. 2014, 5, e1255. [Google Scholar] [CrossRef] [PubMed]
- Ido, Y.; Duranton, A.; Lan, F.; Cacicedo, J.M.; Chen, T.C.; Breton, L.; Ruderman, N.B. Acute Activation of AMP-Activated Protein Kinase Prevents H2O2-Induced Premature Senescence in Primary Human Keratinocytes. PLoS ONE 2012, 7, e35092. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD + elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xie, B.; Mao, Z. Autophagy in premature senescent cells is activated via AMPK pathway. Int. J. Mol. Sci. 2012, 13, 3563–3582. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.Z.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Hartmann, R.; Schroeter, F.; Suwala, A.K.; Maciaczyk, D.; Krueger, A.C.; Willbold, D.; Kahlert, U.D.; Maciaczyk, J. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 2016, 7, 73414–73431. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mariotto, E.; Viola, G.; Ronca, R.; Persano, L.; Aveic, S.; Bhujwalla, Z.M.; Mori, N.; Accordi, B.; Serafin, V.; López-Cara, L.C.; et al. Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers 2018, 10, 391. https://doi.org/10.3390/cancers10100391
Mariotto E, Viola G, Ronca R, Persano L, Aveic S, Bhujwalla ZM, Mori N, Accordi B, Serafin V, López-Cara LC, et al. Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers. 2018; 10(10):391. https://doi.org/10.3390/cancers10100391
Chicago/Turabian StyleMariotto, Elena, Giampietro Viola, Roberto Ronca, Luca Persano, Sanja Aveic, Zaver M. Bhujwalla, Noriko Mori, Benedetta Accordi, Valentina Serafin, Luisa Carlota López-Cara, and et al. 2018. "Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer" Cancers 10, no. 10: 391. https://doi.org/10.3390/cancers10100391
APA StyleMariotto, E., Viola, G., Ronca, R., Persano, L., Aveic, S., Bhujwalla, Z. M., Mori, N., Accordi, B., Serafin, V., López-Cara, L. C., & Bortolozzi, R. (2018). Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers, 10(10), 391. https://doi.org/10.3390/cancers10100391