Unliganded Progesterone Receptor Governs Estrogen Receptor Gene Expression by Regulating DNA Methylation in Breast Cancer Cells

 , , , , and
, , , , and 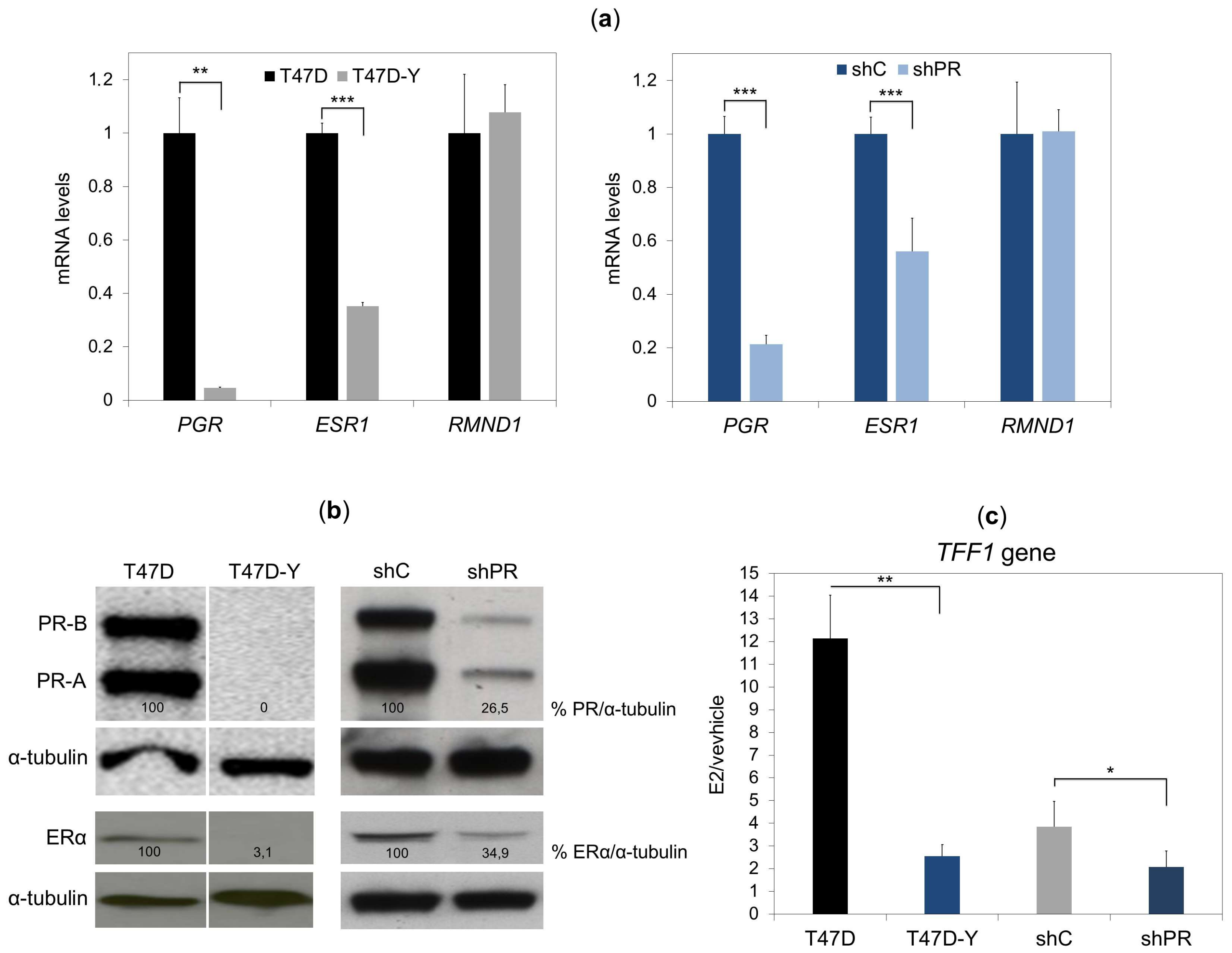{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
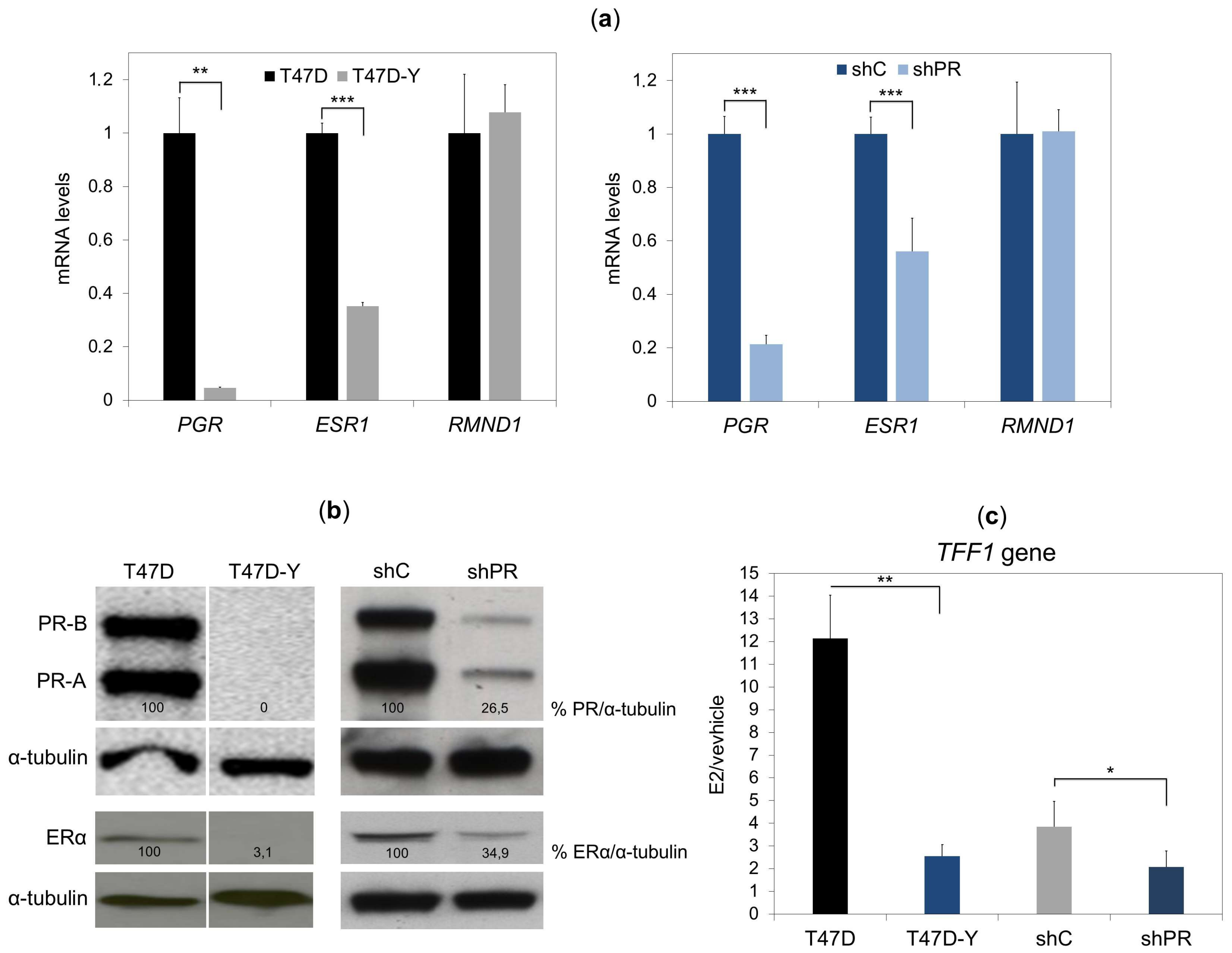
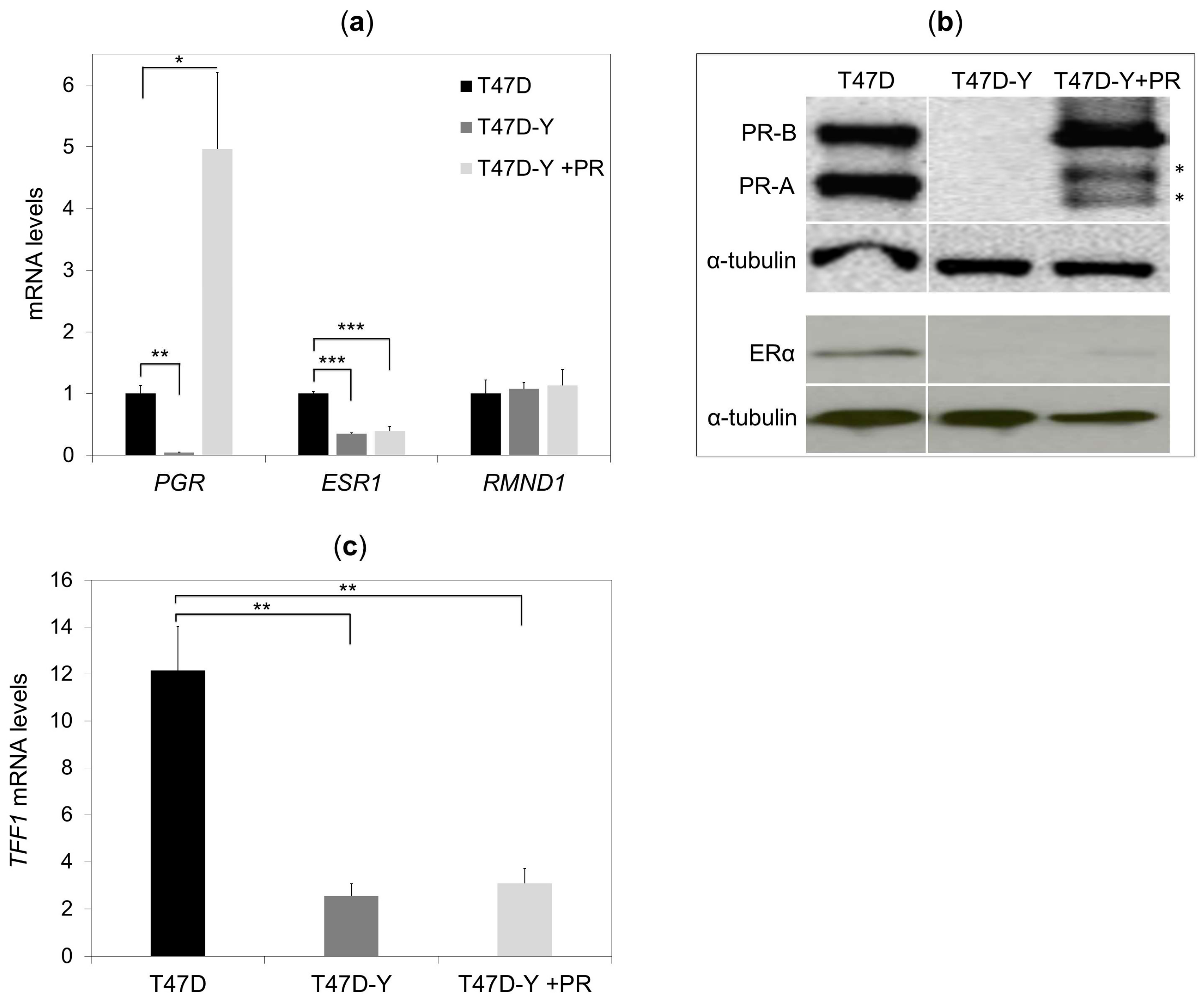
2.1. Unliganded PR Is Required to Maintain ESR1 Gene Basal Expression in Breast Cancer Cells
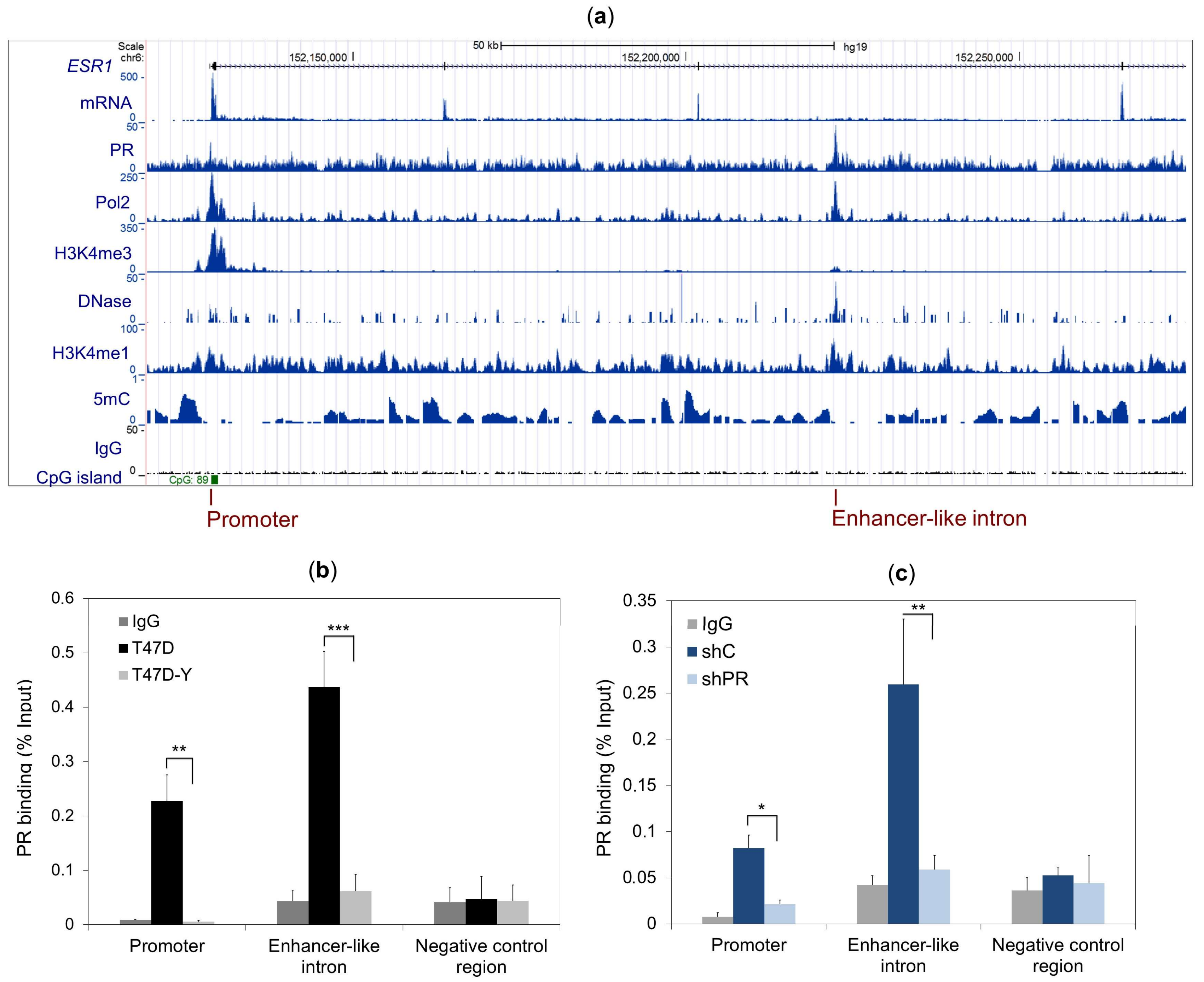
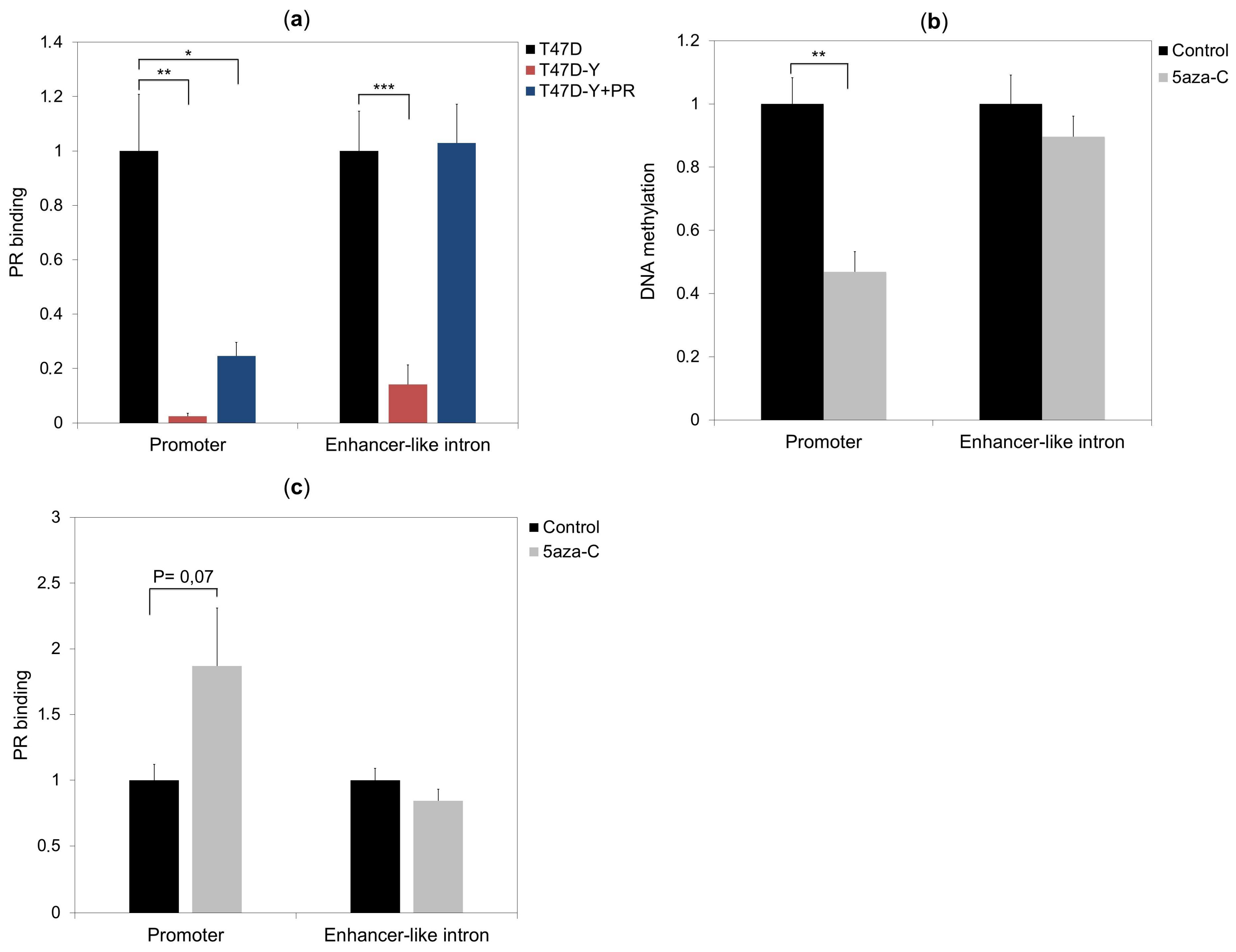
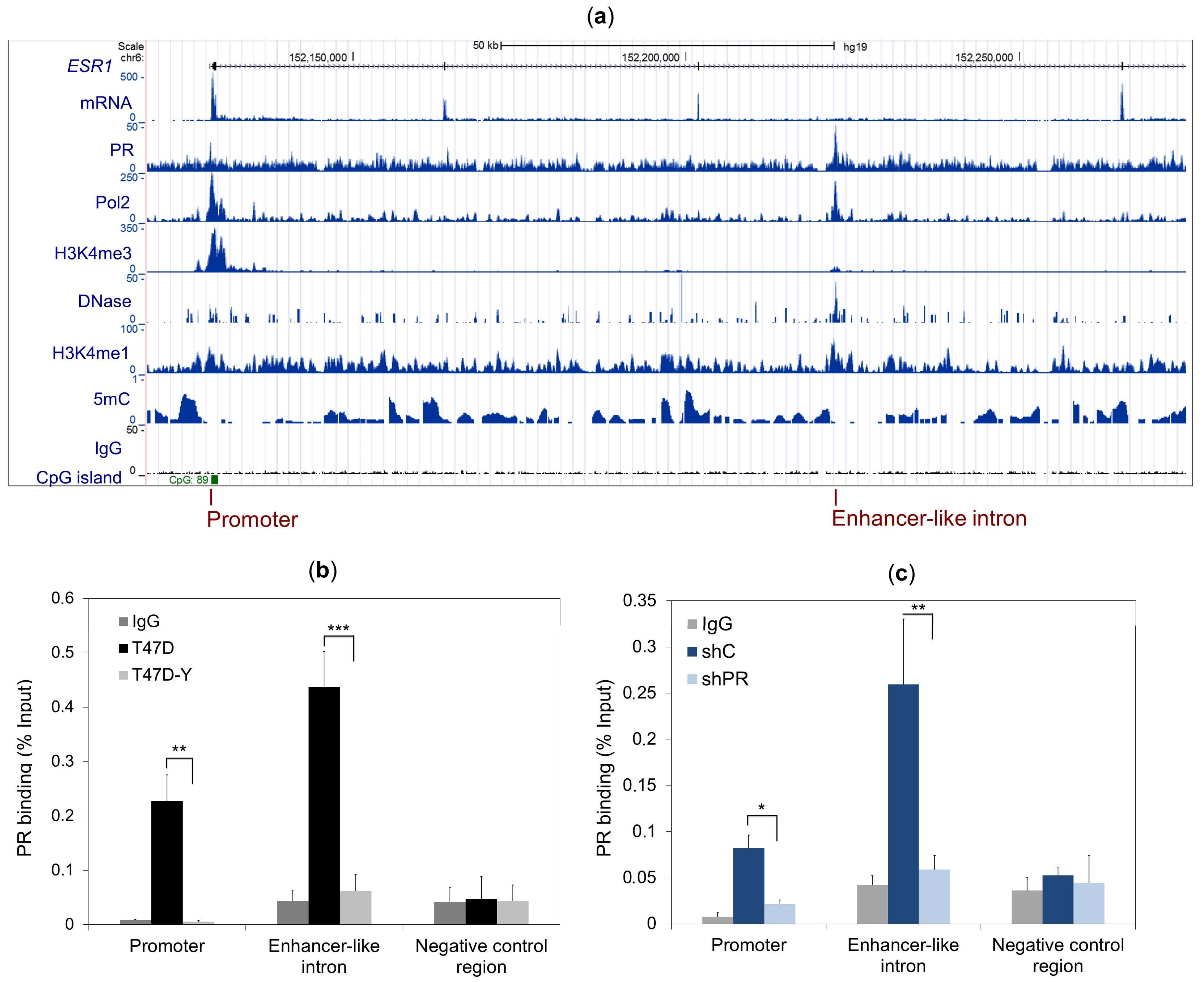
2.2. PR Binds to the ESR1 Locus in Hormone-Free Breast Cancer Cells
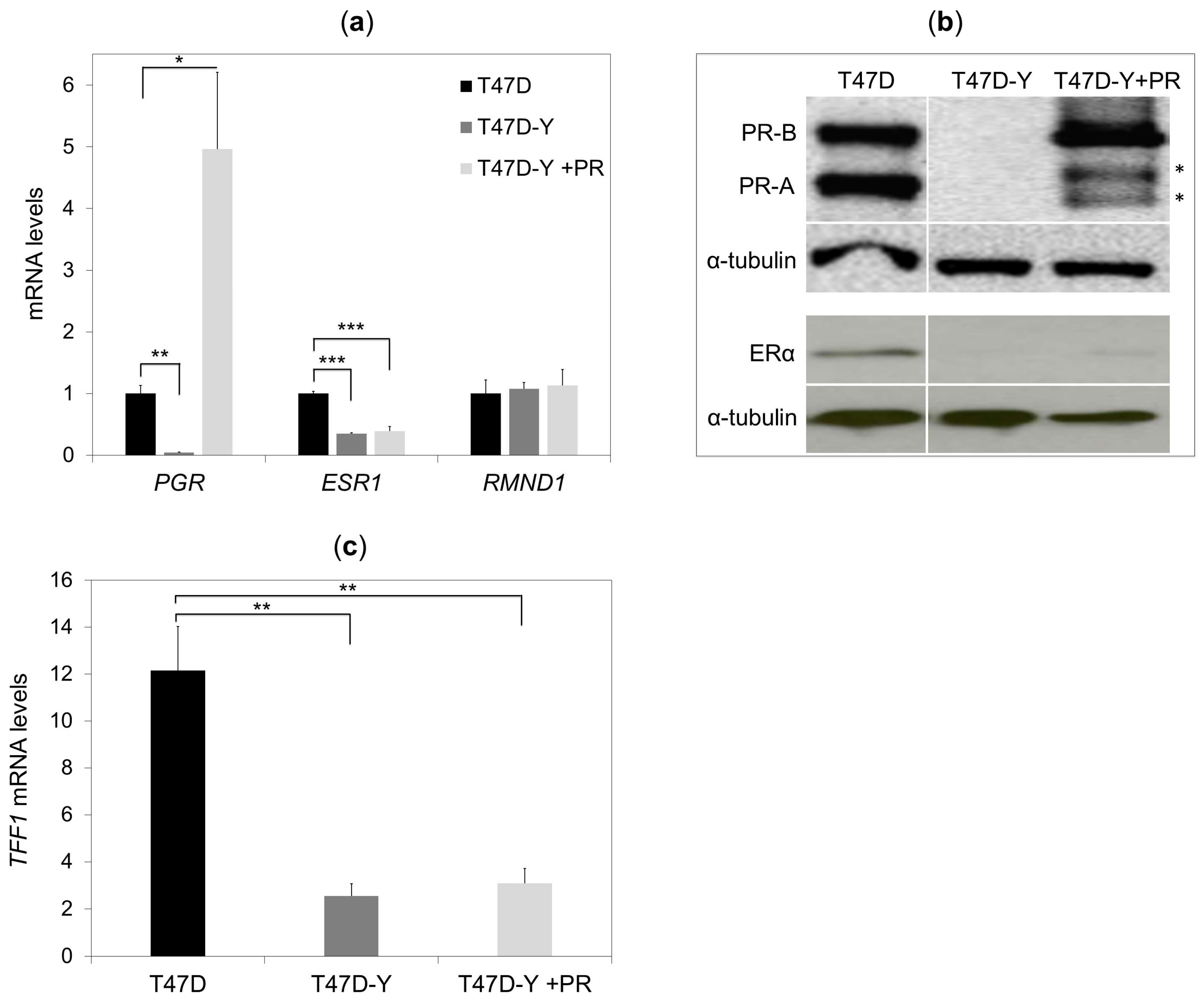
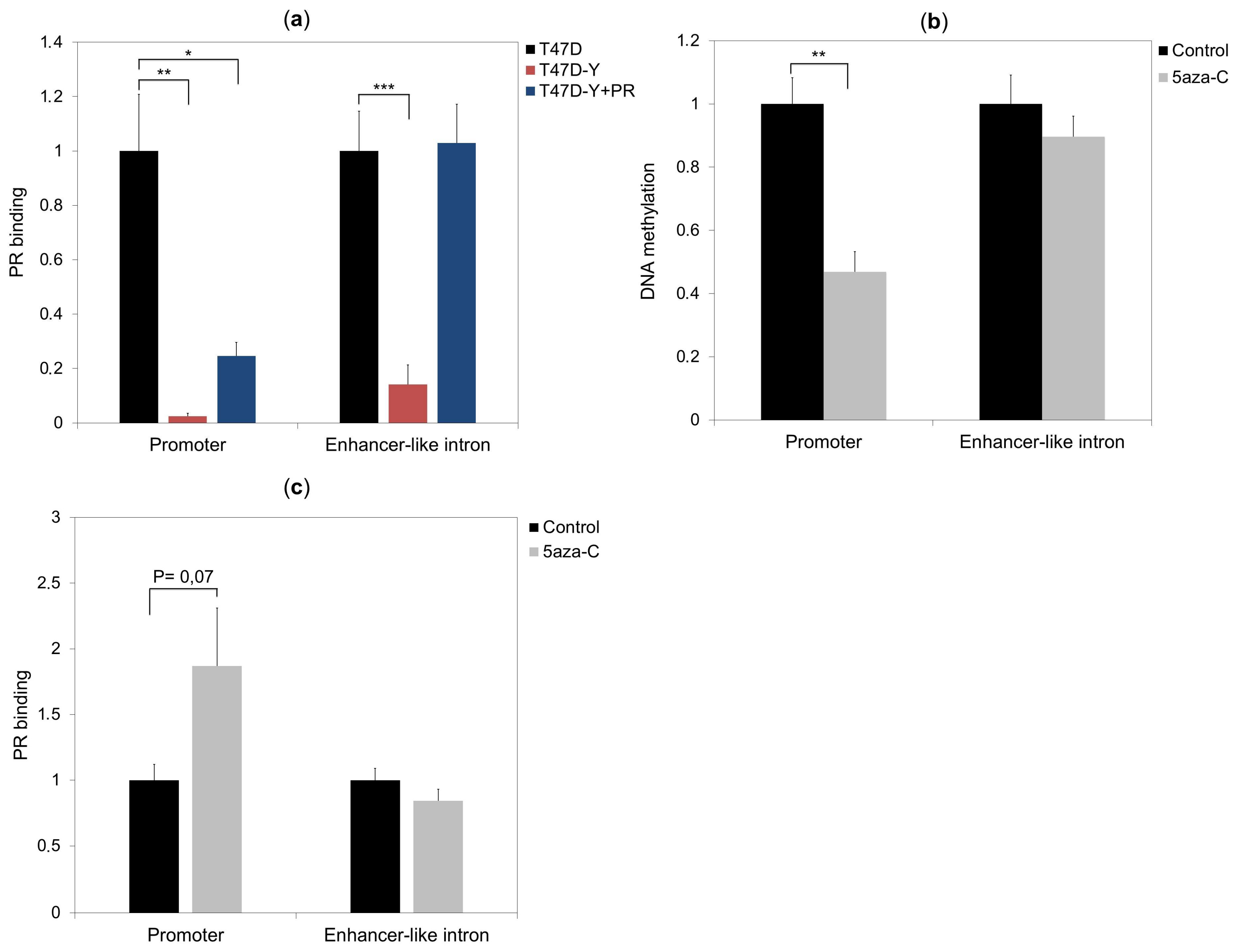
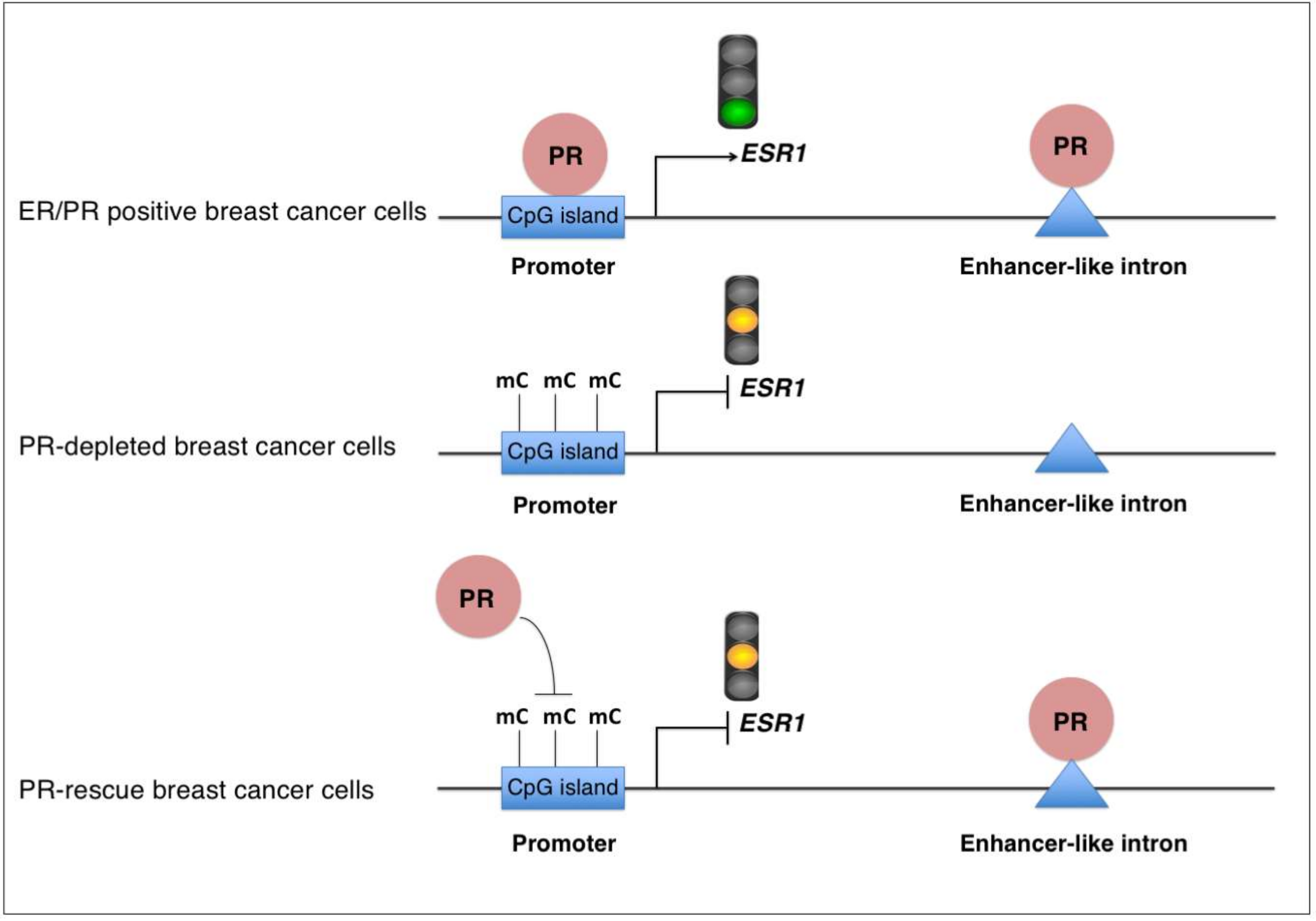
2.3. Rescue of PR Does Not Restore ESR1 Gene Expression in PR-Deficient Cells
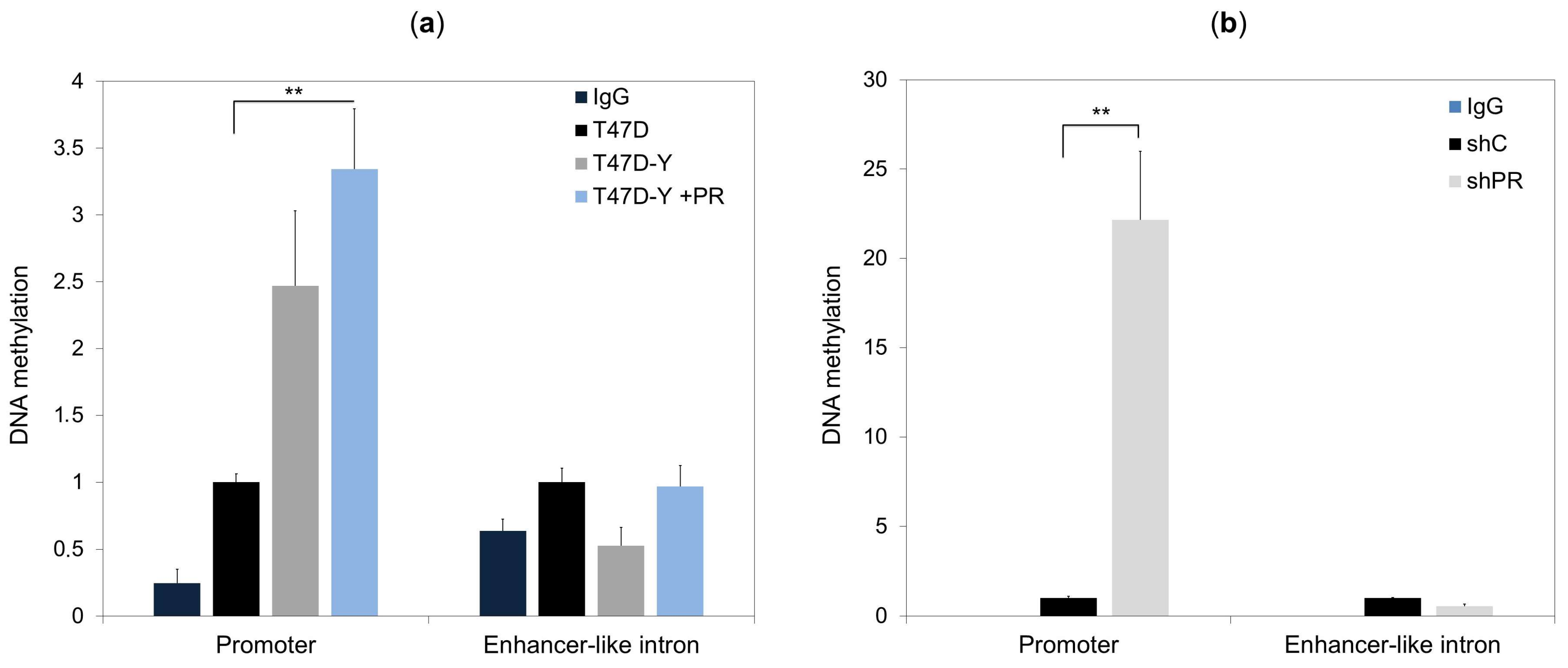
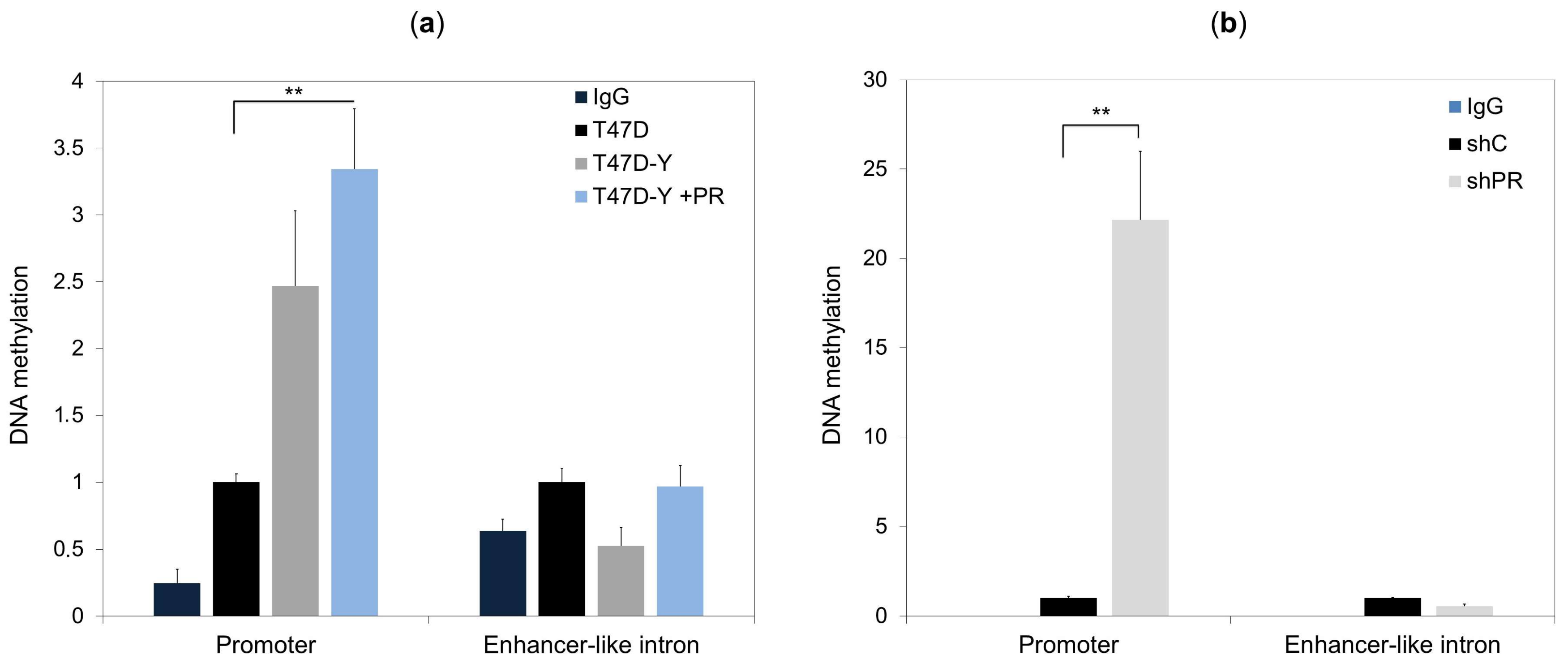
2.4. Lack of PR Affects DNA Methylation at the ESR1 Promoter
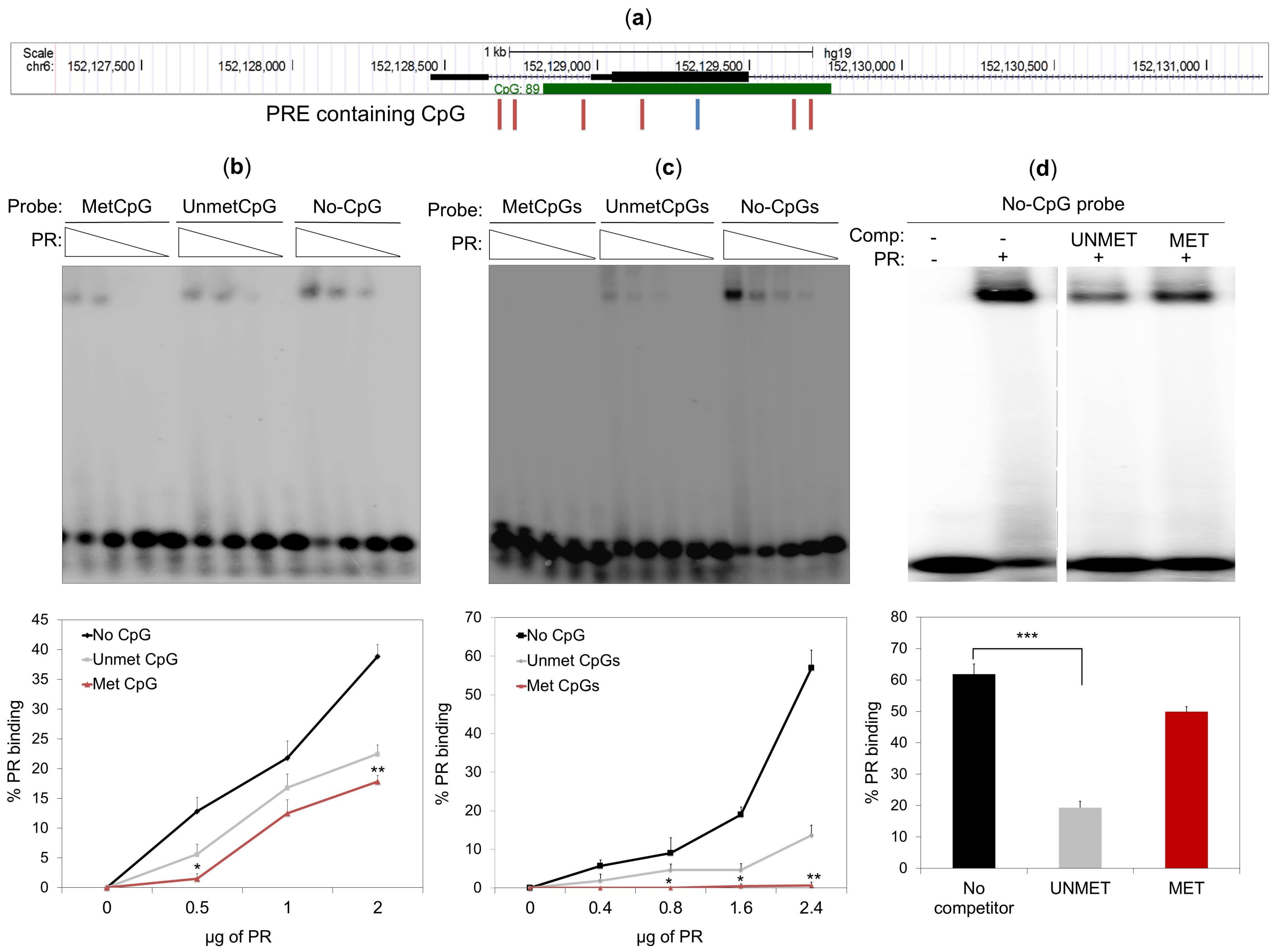
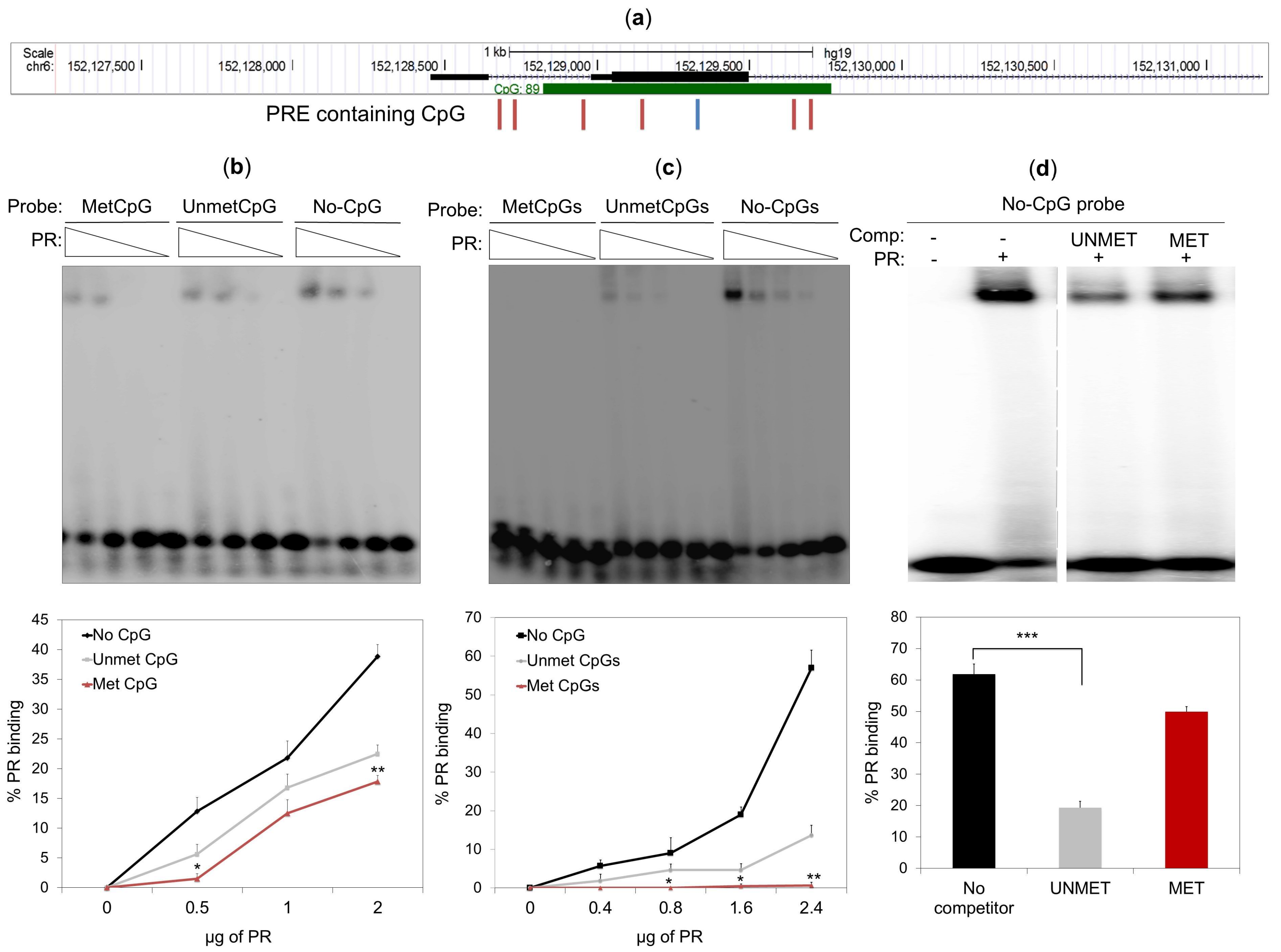
2.5. DNA Methylation Impedes PR Binding to Hormone Responsive Elements
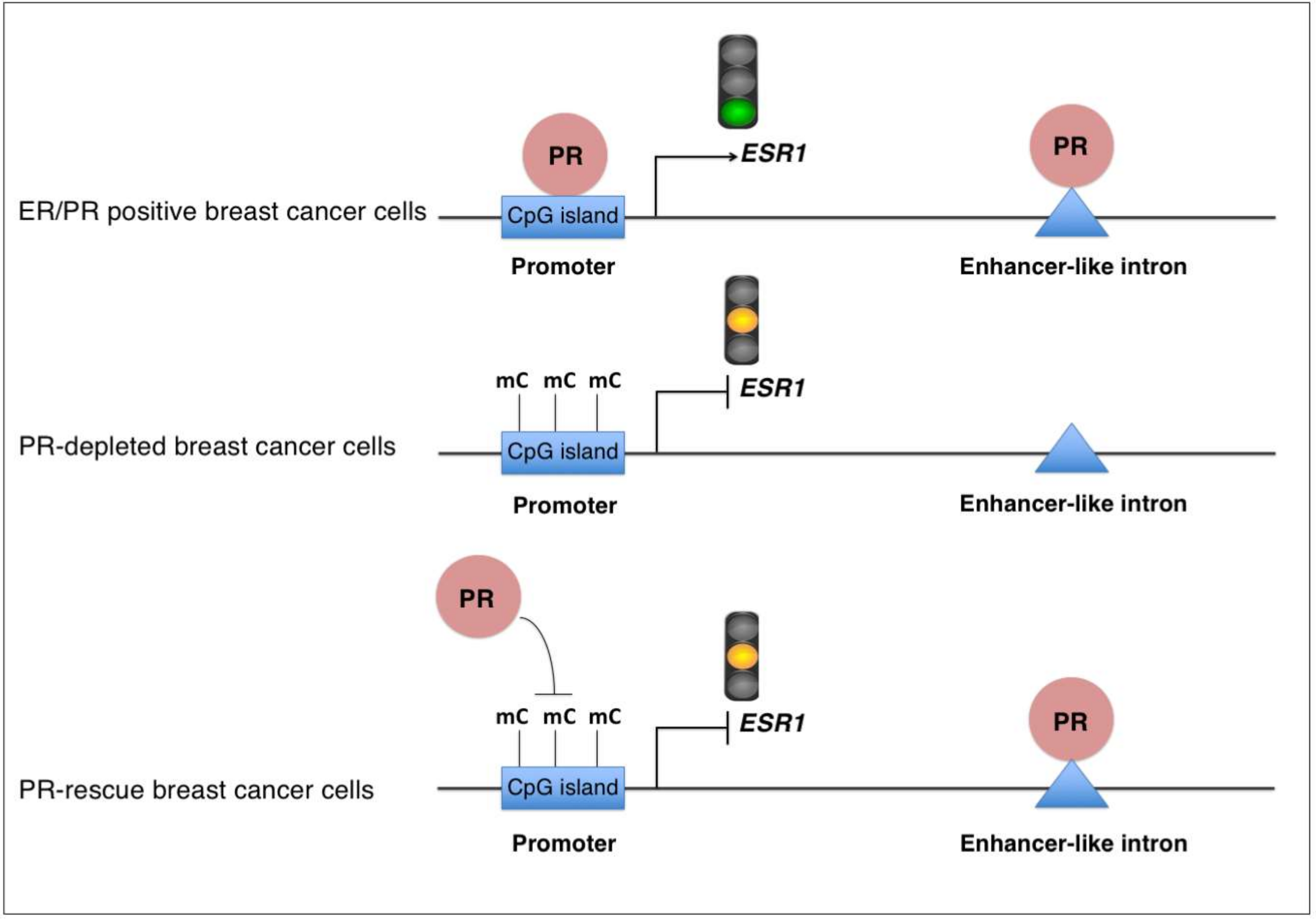
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemical Treatments
4.2. Lentivirus Preparation and Infection
4.3. Reverse Transcription and Quantitative PCR
4.4. Western Blotting
4.5. Chromatin Immunoprecipitation
4.6. DNA Methylation
4.7. Electrophoresis Mobility-Shift Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hilton, H.N.; Clarke, C.L.; Graham, J.D. Estrogen and progesterone signaling in the normal breast and its implications for cancer development. Mol. Cell. Endocrinol. 2018, 466, 2–14, pii:S0303-7207(17), 30433-1. [Google Scholar] [CrossRef] [PubMed]
- Ballaré, C.; Castellano, G.; Gaveglia, L.; Althammer, S.; González-Vallinas, J.; Eyras, E.; Le Dily, F.; Zaurin, R.; Soronellas, D.; Vicent, G.P.; et al. Nucleosome-Driven Transcription Factor Binding and Gene Regulation. Mol. Cell 2013, 49, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Wierer, M.; Verde, G.; Pisano, P.; Molina, H.; Font-Mateu, J.; Di Croce, L.; Beato, M. PLK1 signaling in breast cancer cells cooperates with estrogen receptor-dependent gene transcription. Cell Rep. 2013, 3, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Dunnwald, L.K.; Rossing, M.A.; Li, C.I. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007, 9, R6. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, C.K.; Lee, A.V. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef] [PubMed]
- Daniel, A.R.; Gaviglio, A.L.; Knutson, T.P.; Ostrander, J.H.; D’Assoro, A.B.; Ravindranathan, P.; Peng, Y.; Raj, G.V.; Yee, D.; Lange, C.A. Progesterone receptor-B enhances estrogen responsiveness of breast cancer cells via scaffolding PELP1- and estrogen receptor-containing transcription complexes. Oncogene 2015, 34, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Encarnacion, C.A.; Ciocca, S.R.; McGuire, W.L.; Clark, G.M.; Fuqua, S.A.W.; Osborne, C.K. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res. Treat. 1993, 26, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Lange, C.A. MAP kinases couple multiple functions of human progesterone receptors: Degradation, transcriptional synergy, and nuclear association. J. Steroid Biochem. Mol. Biol. 2003, 85, 147–157. [Google Scholar] [CrossRef]
- Daniel, A.R.; Lange, C.A. Protein kinases mediate ligand-independent derepression of sumoylated progesterone receptors in breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14287–14292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendrat, K.; Fritz, P.; Müller, S.; Brockmöller, S.; Debus, A.; Friedrichs, K.; Lindner, C.; Brinkmann, F.; Heidemann, E.; Niendorf, A. Improved Risk Stratification for Breast Cancer Samples Based on the Expression Ratio of the Estrogen and Progesterone Receptor. Anticancer Res. 2016, 36, 3855–3863. [Google Scholar] [PubMed]
- Sartorius, C.A.; Groshong, S.D.; Miller, L.A.; Powell, R.L.; Tung, L.; Takimoto, G.S.; Horwitz, K.B. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: Only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 1994, 54, 3868–3877. [Google Scholar] [PubMed]
- Quiles, I.; Millán-Ariño, L.; Subtil-Rodríguez, A.; Miñana, B.; Spinedi, N.; Ballaré, C.; Beato, M.; Jordan, A. Mutational analysis of progesterone receptor functional domains in stable cell lines delineates sets of genes regulated by different mechanisms. Mol. Endocrinol. 2009, 23, 809–826. [Google Scholar] [CrossRef] [PubMed]
- Alexander, I.E.; Shine, J.; Sutherland, R.L. Progestin regulation of estrogen receptor messenger RNA in human breast cancer cells. Mol. Endocrinol. 1990, 6, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Vicent, G.P.; Nacht, A.S.; Zaurin, R.; Font-Mateu, J.; Soronellas, D.; Le Dily, F.; Reyes, D.; Beato, M. Unliganded progesterone receptor mediated targeting of an RNA-containing repressive complex silences a subset of hormone-inducible genes. Genes Dev. 2013, 27, 1179–1197. [Google Scholar] [CrossRef] [PubMed]
- Le Dily, F.; Baù, D.; Pohl, A.; Vicent, G.P.; Serra, F.; Soronellas, D.; Castellano, G.; Wright, R.H.; Ballaré, C.; Filion, G.; et al. Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev. 2014, 28, 2151–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, How, and Why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Galán, J.; Torres-Torres, B.; Núñez, M.I.; López-Peñalver, J.; Del Moral, R.; Ruiz De Almodóvar, J.M.; Menjón, S.; Concha, A.; Chamorro, C.; Ríos, S.; et al. ESR1 gene promoter region methylation in free circulating DNA and its correlation with estrogen receptor protein expression in tumor tissue in breast cancer patients. BMC Cancer 2014, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Siegmund, K.D.; Müller, H.M.; Fiegl, H.; Marth, C.; Müller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. pii:eaaj2239. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.T.; Lapidus, R.G.; Baylin, S.B.; Davidson, N.E. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 1995, 55, 2279–2283. [Google Scholar] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Richer, J.K.; Jacobsen, B.M.; Manning, N.G.; Abel, M.G.; Wolf, D.M.; Horwitz, K.B. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J. Biol. Chem. 2002, 277, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.Y.; Kim, S.; Lee, J.H.; Lee, H.C.; Lee, S.K.; Kil, W.H.; Kim, S.W.; Lee, J.E.; Nam, S.J. Poor prognosis of single hormone receptor-positive breast cancer: similar outcome as triple-negative breast cancer. BMC Cancer 2015, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Dressing, G.E.; Knutson, T.P.; Schiewer, M.J.; Daniel, A.R.; Hagan, C.R.; Diep, C.H.; Knudsen, K.E.; Lange, C.A. Progesterone receptor-cyclin D1 complexes induce cell cycle-dependent transcriptional programs in breast cancer cells. Mol. Endocrinol. 2014, 28, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Strutt, H.; Paro, R. Mapping DNA target sites of chromatin proteins in vivo by formaldehyde crosslinking. Methods Mol. Biol. 1999, 119, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karolchik, D.; Barber, G.P.; Casper, J.; Clawson, H.; Cline, M.S.; Diekhans, M.; Dreszer, T.R.; Fujita, P.A.; Guruvadoo, L.; Haeussler, M.; et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014, 42, D764–D770. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Schübeler, D.; Roloff, T.C. Methylated DNA immunoprecipitation (MeDIP). Methods Mol. Biol. 2009, 507, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, T.E. Python for scientific computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef]
- Di Croce, L.; Koop, R.; Venditti, P.; Westphal, H.M.; Nightingale, K.P.; Corona, D.F.; Becker, P.B.; Beato, M. Two-step synergism between the progesterone receptor and the DNA-binding domain of nuclear factor 1 on MMTV minichromosomes. Mol. Cell 1999, 4, 45–54. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verde, G.; De Llobet, L.I.; Wright, R.H.G.; Quilez, J.; Peiró, S.; Le Dily, F.; Beato, M. Unliganded Progesterone Receptor Governs Estrogen Receptor Gene Expression by Regulating DNA Methylation in Breast Cancer Cells. Cancers 2018, 10, 371. https://doi.org/10.3390/cancers10100371
Verde G, De Llobet LI, Wright RHG, Quilez J, Peiró S, Le Dily F, Beato M. Unliganded Progesterone Receptor Governs Estrogen Receptor Gene Expression by Regulating DNA Methylation in Breast Cancer Cells. Cancers. 2018; 10(10):371. https://doi.org/10.3390/cancers10100371
Chicago/Turabian StyleVerde, Gaetano, Lara I. De Llobet, Roni H.G. Wright, Javier Quilez, Sandra Peiró, François Le Dily, and Miguel Beato. 2018. "Unliganded Progesterone Receptor Governs Estrogen Receptor Gene Expression by Regulating DNA Methylation in Breast Cancer Cells" Cancers 10, no. 10: 371. https://doi.org/10.3390/cancers10100371