Comprehensive Analysis of Germline Variants in Mexican Patients with Hereditary Breast and Ovarian Cancer Susceptibility


 , , , , , , ,
, , , , , , ,  , and add
Show full author list
, and add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Clinical and Epidemiological Description of Breast Cancer Cases
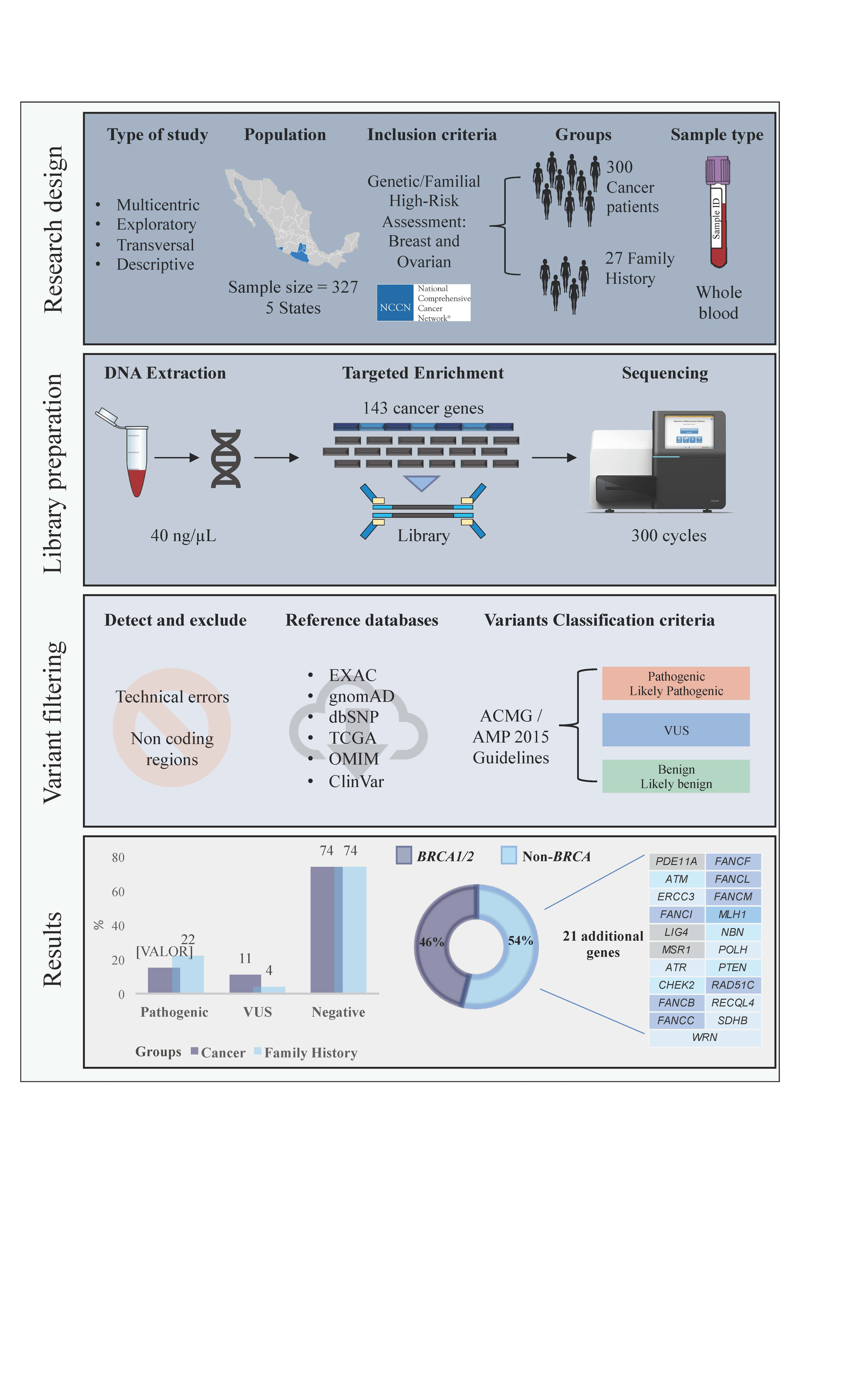
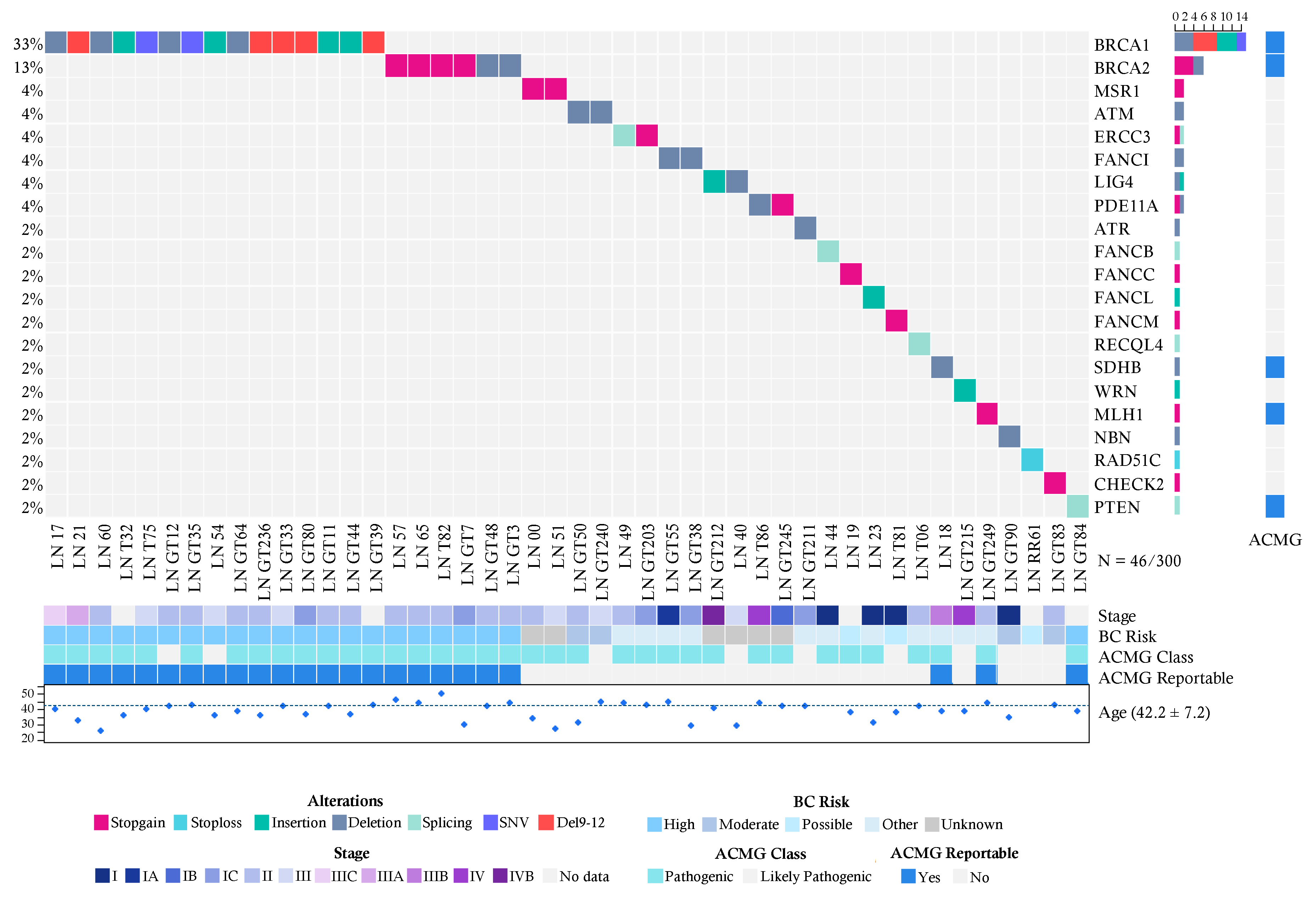
2.2. Pathogenic Variants in the Breast Cancer Cases
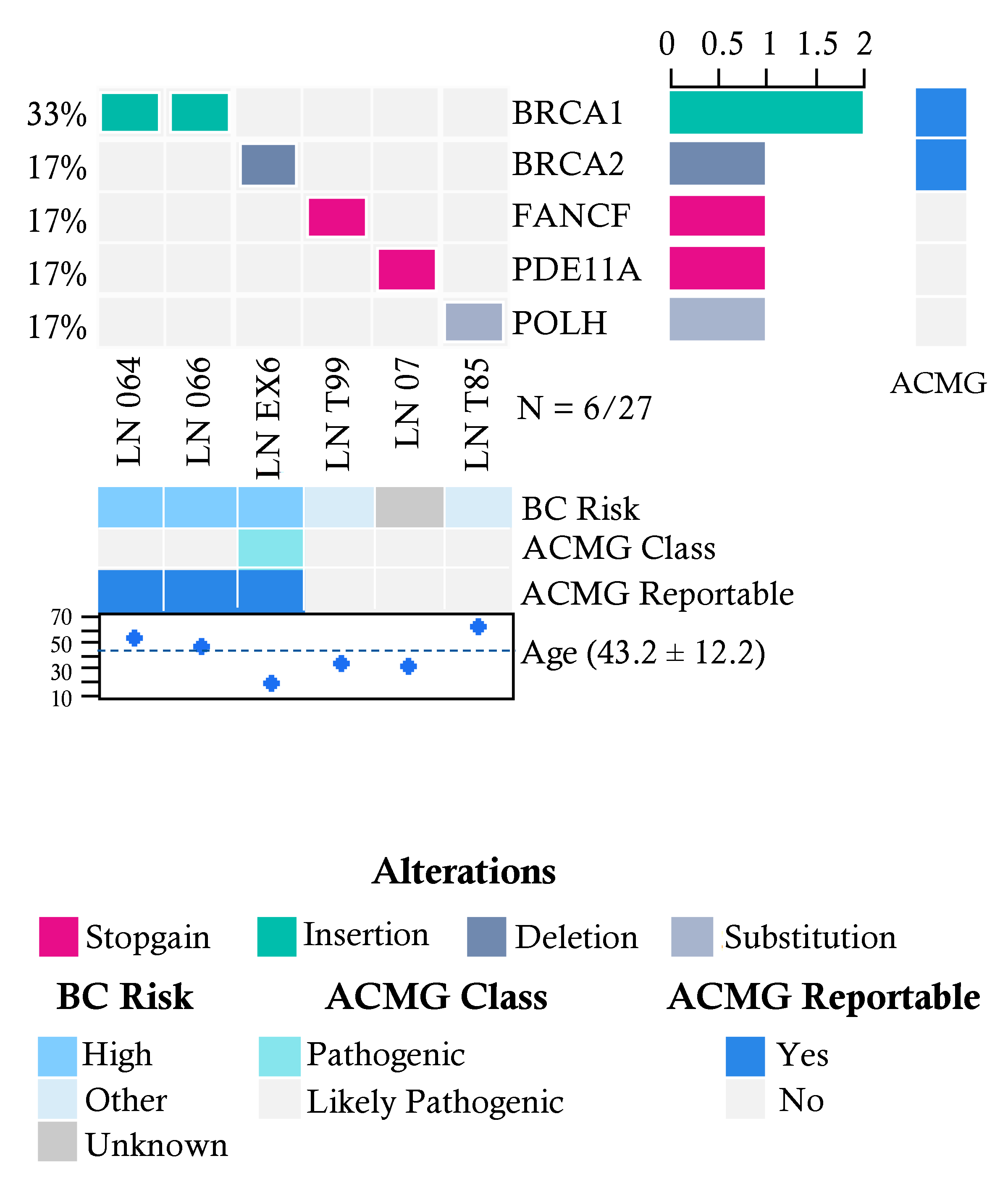
2.3. Pathogenic Variants in Familial Breast Cancer Risk Patients without Cancer Diagnosis
2.4. Recurrent Mutations in BRCA1 and BRCA2 in Both Groups
2.5. Pathogenic Variants in Genes with Unknown Risk in Breast Cancer
2.6. Description of Variants with Unknown Clinical Significance by Phosphorylation Site Disruption Analysis
3. Discussion
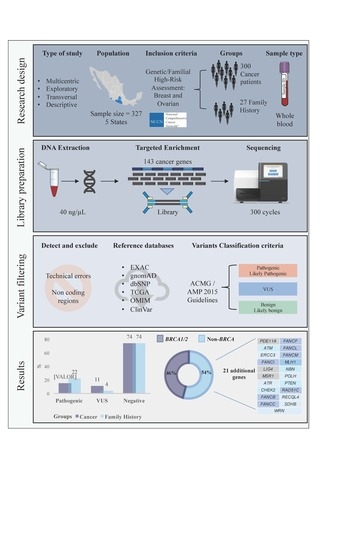
4. Materials and Methods
4.1. Study Population and Data Collection
4.2. Sample Preparation and DNA Extraction
4.3. Library Preparation and Massive Parallel Sequencing
4.4. Pathogenic Variant Detection
4.5. Detection of Exon 9-12 Deletion in BRCA1
4.6. Phosphorylation Site Disruption Analysis
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ervik, M.L.F.; Ferlay, J.; Mery, L.; Soerjomataram, I.; Bray, F. Cancer Today. Lyon: International Agency for Research on Cancer. 2016. Available online: http://gco.iarc.fr/today (accessed on 24 November 2016).
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Kast, K.; Rhiem, K.; Wappenschmidt, B.; Hahnen, E.; Hauke, J.; Bluemcke, B.; Zarghooniet, V.; Herold, N.; Ditsch, N.; Kiechle, M.; et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J. Med. Genet. 2016, 53, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Torres-Mejia, G.; Royer, R.; Llacuachaqui, M.; Akbari, M.R.; Giuliano, A.R.; Martinez-Matsushita, L.; Angeles-Llerenas, A.; Ortega-Olvera, C.; Ziv, E.; Lazcano-Ponce, E.; et al. Recurrent BRCA1 and BRCA2 mutations in Mexican women with breast cancer. Cancer Epidemiol. Biomark. Prev. 2015, 24, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Dutil, J.; Golubeva, V.A.; Pacheco-Torres, A.L.; Diaz-Zabala, H.J.; Matta, J.L.; Monteiro, A.N. The spectrum of BRCA1 and BRCA2 alleles in Latin America and the Caribbean: A clinical perspective. Breast Cancer Res. Treat. 2015, 154, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; van Overeem Hansen, T.; Sorensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xie, M.; Wendl, M.C.; Wang, J.; McLellan, M.D.; Leiserson, M.D.; Huang, K.-L.; Wyczalkowski, M.A.; Jayasinghe, R.; Banerjee, T.; et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 2015, 6, 10086. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Hart, S.N.; Vijai, J.; Schrader, K.A.; Slavin, T.P.; Thomas, T.; Wubbenhorst, B.; Ravichandran, V.; Moore, R.M.; Hu, C.; et al. Evaluation of ACMG-Guideline-Based Variant Classification of Cancer Susceptibility and Non-Cancer-Associated Genes in Families Affected by Breast Cancer. Am. J. Hum. Genet. 2016, 98, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Lee, S.T.; Nam, E.J.; Han, J.W.; Lee, J.Y.; Kim, J.; Kim, T.I.; Park, H.S. Variants of cancer susceptibility genes in Korean BRCA1/2 mutation-negative patients with high risk for hereditary breast cancer. BMC Cancer 2018, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Hoyer, J.; Vasileiou, G.; Wunderle, M.; Lux, M.P.; Fasching, P.A.; Krumbiegel, M.; Uebe, S.; Reuter, M.; Beckmann, M.W.; et al. Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int. J. Cancer 2017, 140, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Lolas Hamameh, S.; Renbaum, P.; Kamal, L.; Dweik, D.; Salahat, M.; Jaraysa, T.; Rayyan, A.A.; Casadei, S.; Mandell, J.B.; Gulsuner, S.; et al. Genomic analysis of inherited breast cancer among Palestinian women: Genetic heterogeneity and a founder mutation in TP53. Int. J. Cancer 2017, 141, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Shao, K.; Qin, Q.; Wang, X.; Song, S.; Wang, X. Clinical and genetic characterization of hereditary breast cancer in a Chinese population. Hered. Cancer Clin. Pract. 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Cock-Rada, A.M.; Ossa, C.A.; Garcia, H.I.; Gomez, L.R. A multi-gene panel study in hereditary breast and ovarian cancer in Colombia. Fam. Cancer 2017, 17, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.; Shin, V.Y.; Au, C.H.; Law, F.B.; Ho, D.N.; Ip, B.K.; Wong, A.T.C.; Lau, S.S.; To, R.M.Y.; Choy, G.; et al. Detection of Germline Mutation in Hereditary Breast and/or Ovarian Cancers by Next-Generation Sequencing on a Four-Gene Panel. J. Mol. Diagn. 2016, 18, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meeks, H.; Feng, B.J.; Healey, S.; Thorne, H.; Makunin, I.; Campbell, I.; Southey, M.; Mitchell, G.; Clouston, D.; et al. Targeted massively parallel sequencing of a panel of putative breast cancer susceptibility genes in a large cohort of multiple-case breast and ovarian cancer families. J. Med. Genet. 2016, 53, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Mannan, A.U.; Singh, J.; Lakshmikeshava, R.; Thota, N.; Singh, S.; Sowmya, T.S.; Mishra, A.; Sinha, A.; Deshwal, S.; Soni, M.R.; et al. Detection of high frequency of mutations in a breast and/or ovarian cancer cohort: Implications of embracing a multi-gene panel in molecular diagnosis in India. J. Hum. Genet 2016, 61, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Kuo, W.H.; Huang, A.C.; Lu, Y.S.; Lin, C.H.; Kuo, S.H.; Wang, M.-Y.; Liu, C.-Y.; Cheng, F.T.-F.; Yeh, M.-H.; et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016, 7, 8310–8320. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, S.E.; Kobayashi, Y.; Anderson, M.J.; Yang, S.; Desmond, A.J.; Mills, M.A.; Nilsen, G.B.; Jacobs, K.B.; Monzon, F.A.; Kurian, A.W.; et al. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More Than 1000 Patients. J. Mol. Diagn. 2015, 17, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Desmond, A.; Kurian, A.W.; Gabree, M.; Mills, M.A.; Anderson, M.J.; Kobayashi, Y.; Horick, N.; Yang, S.; Shannon, K.M.; Tung, N.; et al. Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol. 2015, 1, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.; Faust, U.; Sturm, M.; Hackmann, K.; Grundmann, K.; Harmuth, F.; Bosse, K.; Kehrer, M.; Benkert, T.; Klink, B.; et al. HBOC multi-gene panel testing: Comparison of two sequencing centers. Breast Cancer Res. Treat. 2015, 152, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Nakagomi, H.; Sakamoto, I.; Amemiya, K.; Oyama, T.; Mochizuki, H.; Omata, M. Multigene panel analysis identified germline mutations of DNA repair genes in breast and ovarian cancer. Mol. Genet. Genomic Med. 2015, 3, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Nathanson, K.L. Common breast cancer risk variants in the post-COGS era: A comprehensive review. Breast Cancer Res. 2013, 15, 212. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Caminsky, N.G.; Mucaki, E.J.; Perri, A.M.; Lu, R.; Knoll, J.H.; Rogan, P.K. Prioritizing Variants in Complete Hereditary Breast and Ovarian Cancer Genes in Patients Lacking Known BRCA Mutations. Hum. Mutat. 2016, 37, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Friebel, T.M.; Domchek, S.M.; Rebbeck, T.R. Modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: Systematic review and meta-analysis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, M.M.; Kuchenbaecker, K.B.; Vijai, J.; Klein, R.J.; Kirchhoff, T.; McGuffog, L.; Barrowdale, D.; Dunning, A.M.; Lee, A.; Dennis, J.; et al. Identification of a BRCA2-specific modifier locus at 6p24 related to breast cancer risk. PLoS Genet. 2013, 9, e1003173. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Antoniou, A.C. Modifiers of breast and ovarian cancer risks for BRCA1 and BRCA2 mutation carriers. Endocr. Relat. Cancer 2016, 23, T69–T84. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Neuhausen, S.L.; Robson, M.; Barrowdale, D.; McGuffog, L.; Mulligan, A.M.; Andrulis, I.L.; Spurdle, A.B.; Schmidt, M.K.; Schmutzler, R.K.; et al. Associations of common breast cancer susceptibility alleles with risk of breast cancer subtypes in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. 2014, 16, 3416. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Ciampa, J.; Yeager, M.; Amundadottir, L.; Jacobs, K.; Kraft, P.; Chung, C.; Wacholder, S.; Yu, K.; Wheeler, W.; Thun, M.J.; et al. Large-scale exploration of gene-gene interactions in prostate cancer using a multistage genome-wide association study. Cancer Res. 2011, 71, 3287–3295. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Li, Z.; Song, Z.; Chen, J.; Shi, Y. Genome-wide two-locus interaction analysis identifies multiple epistatic SNP pairs that confer risk of prostate cancer: A cross-population study. Int. J. Cancer 2017, 140, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Cherdyntseva, N.V.; Denisov, E.V.; Litviakov, N.V.; Maksimov, V.N.; Malinovskaya, E.A.; Babyshkina, N.N.; Slonimskaya, E.M.; Voevoda, M.I.; Choinzonov, E.L. Crosstalk between the FGFR2 and TP53 genes in breast cancer: Data from an association study and epistatic interaction analysis. DNA Cell Biol. 2012, 31, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Naushad, S.M.; Pavani, A.; Digumarti, R.R.; Gottumukkala, S.R.; Kutala, V.K. Epistatic interactions between loci of one-carbon metabolism modulate susceptibility to breast cancer. Mol. Biol. Rep. 2011, 38, 4893–4901. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; Furriol, J.; Tormo, E.; Ballester, S.; Lluch, A.; Eroles, P. Epistatic interaction of Arg72Pro TP53 and -710 C/T VEGFR1 polymorphisms in breast cancer: Predisposition and survival. Mol. Cell. Biochem. 2013, 379, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, Y.; Mackey, J.R.; Lai, R.; Franco-Villalobos, C.; Lupichuk, S.; Robson, P.J.; Kopciuk, K.; Cass, C.E.; Yasui, Y.; Damaraju, S. Assessing SNP-SNP interactions among DNA repair, modification and metabolism related pathway genes in breast cancer susceptibility. PLoS ONE 2014, 8, e64896. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cuevas, S.; Guisa-Hohenstein, F.; Labastida-Almendaro, S. First breast cancer mammography screening program in Mexico: Initial results 2005–2006. Breast J. 2009, 15, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Berwick, M.; Satagopan, J.M.; Ben-Porat, L.; Carlson, A.; Mah, K.; Henry, R.; Diotti, R.; Milton, K.; Pujara, K.; Landers, T.; et al. Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res. 2007, 67, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.J.; Fernandez, V.; Osorio, A.; Barroso, A.; Fernandez, F.; Urioste, M.; Benitez, J. Mutational analysis of FANCL, FANCM and the recently identified FANCI suggests that among the 13 known Fanconi Anemia genes, only FANCD1/BRCA2 plays a major role in high-risk breast cancer predisposition. Carcinogenesis 2009, 30, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Orloff, M.; Peterson, C.; He, X.; Ganapathi, S.; Heald, B.; Yang, Y.R.; Bebek, G.; Romigh, T.; Song, J.H.; Wu, W.; et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA 2011, 306, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zheng, S.L.; Komiya, A.; Mychaleckyj, J.C.; Isaacs, S.D.; Hu, J.J.; Sterling, D.; Lange, E.M.; Hawkins, G.A.; Turner, A.; et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat. Genet. 2002, 32, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Hope, Q.; Bullock, S.; Evans, C.; Meitz, J.; Hamel, N.; Edwards, S.M.; Severi, G.; Dearnaley, D.; Jhavar, S.; Southgate, C.; et al. Macrophage scavenger receptor 1 999C>T (R293X) mutation and risk of prostate cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; McDonnell, S.K.; Cunningham, J.M.; Hebbring, S.; Jacobsen, S.J.; Cerhan, J.R.; Slager, S.L.; Blute, M.L.; Schaid, D.J.; Thibodeau, S.N. No association of germline alteration of MSR1 with prostate cancer risk. Nat. Genet. 2003, 35, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, X.; Kamdar, R.P.; Wanotayan, R.; Sharma, M.K.; Adachi, N.; Matsumoto, Y. C-Terminal region of DNA ligase IV drives XRCC4/DNA ligase IV complex to chromatin. Biochem. Biophys. Res. Commun. 2013, 439, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Leeksma, O.C.; de Miranda, N.F.; Veelken, H. Germline mutations predisposing to diffuse large B.-cell lymphoma. Blood Cancer J. 2017, 7, e532. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Cerosaletti, K.M.; Girard, P.M.; Dai, Y.; Stumm, M.; Kysela, B.; Hirsch, B.; Gennery, A.; Palmer, S.E.; Seidel, J.; et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell 2001, 8, 1175–1185. [Google Scholar] [CrossRef]
- Faucz, F.R.; Horvath, A.; Rothenbuhler, A.; Almeida, M.Q.; Libe, R.; Raffin-Sanson, M.L.; Bertherat, J.; Carraro, D.M.; Soares, F.A.; Molina, G.d.C.; et al. Phosphodiesterase 11A (PDE11A) genetic variants may increase susceptibility to prostatic cancer. J. Clin. Endocrinol. Metab. 2011, 96. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Korde, L.; Greene, M.H.; Libe, R.; Osorio, P.; Faucz, F.R.; Raffin-Sanson, M.L.; Tsang, K.M.; Drori-Herishanu, L.; Patronas, Y.; et al. Functional phosphodiesterase 11A mutations may modify the risk of familial and bilateral testicular germ cell tumors. Cancer Res. 2009, 69, 5301–5306. [Google Scholar] [CrossRef] [PubMed]
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tuppurainen, K.; Ebeling, T.M.L.; Salmela, P.I.; Paschke, R.; et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006, 312, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Formosa, R.; Vassallo, J. Aryl Hydrocarbon Receptor-Interacting Protein (AIP) N-Terminus Gene Mutations Identified in Pituitary Adenoma Patients Alter Protein Stability and Function. Horm. Cancer 2017, 8, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Deka, J.; Kuhlmann, J.; Muller, O. A domain within the tumor suppressor protein APC shows very similar biochemical properties as the microtubule-associated protein tau. Eur. J. Biochem. 1998, 253, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, J.N.; Lagos, V.I.; Herzog, J.S.; Judkins, T.; Hendrickson, B.; Ho, J.S.; Ricker, C.N.; Lowstuter, K.J.; Blazer, K.R.; Tomlinson, G.; et al. Evidence for common ancestral origin of a recurring BRCA1 genomic rearrangement identified in high-risk Hispanic families. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Ewald, I.P.; Ribeiro, P.L.; Palmero, E.I.; Cossio, S.L.; Giugliani, R.; Ashton-Prolla, P. Genomic rearrangements in BRCA1 and BRCA2: A. literature review. Genet. Mol. Biol. 2009, 32, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yao, F.; Luan, H.; Wang, Y.; Dong, X.; Zhou, W.; Wang, Q. Three novel functional polymorphisms in the promoter of FGFR2 gene and breast cancer risk: A HuGE review and meta-analysis. Breast Cancer Res. Treat. 2012, 136, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Cassa, C.A.; Tong, M.Y.; Jordan, D.M. Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum. Mutat. 2013, 34, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012, 335, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gardner, S.A.; Hovhannisyan, H.; Natalizio, A.; Weymouth, K.S.; Chen, W.; Thibodeau, I.; Bogdanova, E.; Letovsky, S.; Willis, A.; et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: Insights of relevance to variant classification. Genet. Med. 2016, 18, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Genes | Sample | Country | Methods | BRCA Frequency 1 | Non-BRCA Frequency 3 | Non–BRCA Genes | Ref. |
|---|---|---|---|---|---|---|---|---|
| Proportion 2 | Proportion 4 | |||||||
| 2018 | 143 | 327 | Mexico | GeneRead (Qiagen) | 7.3% (24/327) | 8.5% (28/327) | MSR1, ATM, ERCC3, FANCI, LIG4, PDE11A, ATR, FANCB, FANCC, FANCL, FANCM, RECQL4, SDHB, WRN, MLH1, NBN, RAD51C, CHEK2, FANCF, POLH and PTEN | This study |
| 46.1% (24/52) | 53.8% (28/52) | |||||||
| 2018 | 35 | 120 | Korea | OncoRisk (Celemics) | Negative | 7.5% (9/120) | TP53, PALB2, BARD1 and MRE11A | [9] |
| 2017 | 21 | 65,057 | USA Multicentric | Multiple | 2.8% (1874/65057) | 5.3% (3422/65057) | CDH1, PTEN, TP53, ATM, BARD1, CHEK2, PALB2, and RAD51D. | [10] |
| 35% (1874/5296) | 64% (3422/5296) | |||||||
| 2017 | 10 | 581 | Germany | TruSight Cancer | 12.4% (72/581) | 5.5% (32/581) | CHEK2, PALB2, NBN, RAD51C, ATM, TP53, RAD51D and MSH6 | [11] |
| 69% (72/104) | 30% (32/104) | |||||||
| 2017 | 16 | 453 | Palestine | SureSelect (Agilent) | 6.8% (31/453) | 6.6 (30/453) | TP53 (founder mutation), ATM, CHEK2, BARD1, BRIP1, PALB2, MRE11A, PTEN, and XRCC2 | [12] |
| 50.8% (31/61) | 49.1% (30/61) | |||||||
| 2017 | 94 | 255 | Italy | Trusight Cancer (Illumina) | 22.3% (57/255) | 6.6% (17/255) | PALB2, ATM, BRIP1, RAD51D, MSH6, PPM1D, RECQL4, ERCC3, TSC2, SLX4 and other Fanconi anemia genes | [13] |
| 77% (57/74) | 22.9% (17/74) | |||||||
| 2017 | 27 | 240 120 = BC 120 = High-risk | China | BGI chip (Blackbird platform) | 5.8% (14/240) | 9.6% (23/240) | MUTYH, CHEK2, PALB2, ATM, BARD1, NBN, RAD51C, TP53 and BRIP1 | [14] |
| 38% (14/37) | 62% (23/37) | |||||||
| 2017 | 25 | 85 | Colombia | MyRisk (Myriad) | 17.6% (15/85) | 4.7% (4/85) | PALB2, ATM, MSH2 and PMS2 | [15] |
| 79% (15/19) | 21% (4/19) | |||||||
| 2016 | 29 | 10,030 | USA | SureSelect targeted capture | 2.54% (255/10,030) | 6.7% (682/10,030) | MLH1, MSH2, MSH6, PMS2, EPCAM, APC, MUTYH, CDH1, PTEN, STK11, and TP53 | [16] |
| 27% (255/937) | 73% (682/937) | |||||||
| 2016 | 4 | 1427 479 = Sanger 948 = NGS | China | PCR design | 8.8% (126/1427) | 0.49% (7/1427) | TP53 and PTEN | [17] |
| 95% (126/133) | 5% (7/133) | |||||||
| 2016 | 19 | 684 BRCA negative patients | Australia | Agilent Target Enrichment | Negative | 11.1% (76/684) | TP53, PALB2, ATM, CHEK2, CDH1, PTEN and STK11 Segregation study: CDH1, CHEK2, PALB2 and TP53 | [18] |
| 2016 | 13 | 141 | India | Trusight Cancer | 4.9% (7/141) | 9.9% (14/141) | ATM, BRIP1, CHEK2, PALB2, RAD51C and TP53 | [19] |
| 33% (7/21) | 66% (14/21) | |||||||
| 2016 | 68 | 133 | Taiwan | NimblGen capture (Roche) | 15% (20/133) | 7.5% (10/133) | RAD50, TP53, ATM, BRIP1, FANCI, MSH2, MUTYH, and RAD51C | [20] |
| 66% (20/30) | 33% (10/30) | |||||||
| 2015 | 25 | 2158 Cohort 1 = 1781 (BRCA1/2) Cohort 2 = 377 negative BRCA) | USA | RainDance Thunderstorm emulsion polymerase chain reaction (PCR) system | Cohort 1 9.3% (165/1781) Cohort 2 NA | Cohort 1 4.2% (15/377) Cohort 2 3.7% (14/377) | CHEK2, ATM and PALB2 | [21] |
| 2015 | 29 | Total: 1062 735-clinically representative | USA | SureSelect and Integrated DNA Technologies | 9% (66/735) | 3.9% (26/735) | ATM, PALB2, CHEK2, MLH1, MSH2, MSH6, and PMS2 | [22] |
| 72% (66/92) | 28% (26/92) | |||||||
| 2015 | 29 (Invitae) 25 (Myriad) | 1046 BRCA negative patients | USA | Hereditary Cancer Syndromes test (Invitae) MyRisk test (Myriad Genetics) | Negative | 3.8% (40/1046) | CHEK2, ATM, PALB2 | [23] |
| 2015 | 94 genes and 284 SNPs | 620 | Germany | TruSight (Illumina) and Haloplex | 9.2% (57/620) | 2.9% (18/620) | CHEK2, ATM, CDH1, NBN, PALB2 and TP53 | [24] |
| 76% (57/75) | 24% (18/75) | |||||||
| 2015 | 25 | 155 | Japan | AmpliSeq Library Kit 2.0 | 7% (11/155) | 1.9% (3/155) | ATM, MRE11A and MSH6 | [25] |
| 78.5% (11/14) | 21.5% (3/14) |
| Epidemiological and Clinical Characteristics | n | (%) |
|---|---|---|
| 300 | (100) | |
| Age | ||
| <40 years | 125 | (41.7) |
| 41–50 years | 135 | (45.0) |
| >50 years | 24 | (8.0) |
| Missing | 16 | (5.3) |
| BMI | ||
| Underweight (<18.5) | 1 | (0.3) |
| Normal weight (18.5 < 25) | 107 | (35.7) |
| Overweight (25.0 < 30) | 118 | (39.3) |
| Obese (30.0 < 40) | 66 | (22.0) |
| Extreme obese (>40) | 3 | (1.0) |
| Missing | 5 | (1.7) |
| Current Alcohol Drinker | ||
| No | 278 | (92.7) |
| Yes | 16 | (5.3) |
| Missing | 6 | (2.0) |
| Current Tobacco Smoker | ||
| No | 84 | (28.0) |
| Yes | 74 | (24.7) |
| Missing | 142 | (47.3) |
| Pregnancy | ||
| Yes | 256 | (85.4) |
| No | 43 | (14.3) |
| Missing | 1 | (0.3) |
| Ever Use of Oral Contraceptives | ||
| Yes | 115 | (38.3) |
| No | 179 | (59.7) |
| Missing | 6 | (2.0) |
| Family History of Cancer | ||
| Yes | 214 | (71.3) |
| No | 80 | (26.7) |
| Missing | 6 | (2.0) |
| Histopathological Subtype | ||
| DCIS | 43 | (14.4) |
| LCIS | 18 | (6.0) |
| IDC | 189 | (63.0) |
| ILC | 16 | (5.3) |
| MC | 3 | (1.0) |
| Missing | 31 | (10.3) |
| Stage | ||
| I | 51 | (17.0) |
| II | 115 | (38.3) |
| III | 86 | (28.7) |
| IV | 10 | (3.3) |
| Missing | 38 | (12.7) |
| ER Status | ||
| Negative | 38 | (12.7) |
| Positive | 20 | (6.6) |
| Missing | 242 | (80.7) |
| PR Status | ||
| Negative | 135 | (45.0) |
| Positive | 22 | (7.3) |
| Missing | 143 | (47.7) |
| HER2 Status | ||
| Negative | 7 | (2.3) |
| Positive | 45 | (15.0) |
| Missing | 248 | (82.7) |
| Mutational Status * | ||
| Non-mutated | 254 | (84.7) |
| Mutated | 46 | (15.3) |
| Gene | Frequency | Syndromes (OMIM) | Breast Cancer Risk | Inherited Pattern | Signaling Pathways | Reportable in ACMG * |
|---|---|---|---|---|---|---|
| BRCA1 | 17 | Hereditary Breast and Ovarian Cancer | High | AD | Double strand damage (HR) | Yes |
| BRCA2 | 11 | Fanconi Anemia/Hereditary Breast and Ovarian Cancer/Familiar Pancreatic Cancer/Hereditary Prostate Cancer | High | AD/AR | Double strand damage (HR) | Yes |
| PDE11A | 3 | Pigmented nodular adrenocortical disease | Novel | AD | Catalyze the hydrolysis of cAMP and cGMP, Metabolism of purines | No |
| ATM | 2 | Susceptibility to breast cancer/Ataxia Telangiectasia | Moderate | AD/AR | Double strand damage (HR) | No |
| ERCC3 | 2 | Xeroderma Pigmentosum | Not established | AR | Transcription initiation of RNA Pol II | No |
| FANCI | 2 | Fanconi Anemia | Not established | AR | Anemia Fanconi Pathway and Double strand damage response | No |
| LIG4 | 2 | LIG4 Syndrome | Novel | AR | Nucleotide excision DNA repair | No |
| MSR1 | 2 | Hereditary Barret Esophagus/Esophagus carcinoma/Hereditary prostate cancer | Novel | AD | Vesicle-mediated transport and AGE/RAGE pathway | No |
| ATR | 1 | Cutaneous telangiectasia and familial cancer syndrome/Seckel syndrome 1 | Not established | AD/AR | Cell cycle checkpoint regulator | No |
| CHEK2 | 1 | Li-Fraumeni syndrome/Susceptibility to breast, colorectal and prostate cancer | Moderate | AD | Cell cycle checkpoint regulator | No |
| FANCB | 1 | Fanconi Anemia | Not established | XLR | Anemia Fanconi Pathway and Double strand damage response | No |
| FANCC | 1 | Fanconi Anemia | Not established | AD/AR | Anemia Fanconi Pathway | No |
| FANCF | 1 | Fanconi Anemia | Not established | AR | Anemia Fanconi Pathway | No |
| FANCL | 1 | Fanconi Anemia | Not established | AR | Anemia Fanconi Pathway, DNA damage, Cell cycle checkpoint regulator | No |
| FANCM | 1 | Fanconi Anemia | Not established | AD/AR | Anemia Fanconi Pathway, Double strand DNA damage | No |
| MLH1 | 1 | Hereditary nonpolyposis colorectal cancer, type 2/Mismatch repair cancer syndrome/Muir-Torre syndrome | Not established | AD/AR | Mismatch repair system | Yes |
| NBN | 1 | Aplastic Anemia/Acute lymphoblastic Leukemia/Nijmegen breakage syndrome | Moderate | AD/AR | Double strand damage respond in DNA | No |
| POLH | 1 | Xeroderma pigmentosum | Not established | AR | Homologous DNA recombination and strand interchange | No |
| PTEN | 1 | Bannayan-Riley-Ruvalcaba syndrome/Cowden syndrome | High risk | AD | Antagonizes the PI3K signaling pathway and negatively regulates the MAPK pathway | Yes |
| RAD51C | 1 | Fanconi Anemia/Susceptibility to breast and ovarian cancer | Not established | AD/AR | Double strand damage (HR) | No |
| RECQL4 | 1 | Rothmund-Thompson Syndrome | Not established | AR | DNA Damage response | No |
| SDHB | 1 | Carney-Stratakis Syndrome | Not established | AD | Metabolism (Krebs Cycle) | Yes |
| WRN | 1 | Werner Syndrome | Not established | AR | C strand synthesis in telomere and cell cycle checkpoint | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quezada Urban, R.; Díaz Velásquez, C.E.; Gitler, R.; Rojo Castillo, M.P.; Sirota Toporek, M.; Figueroa Morales, A.; Moreno García, O.; García Esquivel, L.; Torres Mejía, G.; Dean, M.; et al. Comprehensive Analysis of Germline Variants in Mexican Patients with Hereditary Breast and Ovarian Cancer Susceptibility. Cancers 2018, 10, 361. https://doi.org/10.3390/cancers10100361
Quezada Urban R, Díaz Velásquez CE, Gitler R, Rojo Castillo MP, Sirota Toporek M, Figueroa Morales A, Moreno García O, García Esquivel L, Torres Mejía G, Dean M, et al. Comprehensive Analysis of Germline Variants in Mexican Patients with Hereditary Breast and Ovarian Cancer Susceptibility. Cancers. 2018; 10(10):361. https://doi.org/10.3390/cancers10100361
Chicago/Turabian StyleQuezada Urban, Rosalía, Clara Estela Díaz Velásquez, Rina Gitler, María Patricia Rojo Castillo, Max Sirota Toporek, Andrea Figueroa Morales, Oscar Moreno García, Lizbeth García Esquivel, Gabriela Torres Mejía, Michael Dean, and et al. 2018. "Comprehensive Analysis of Germline Variants in Mexican Patients with Hereditary Breast and Ovarian Cancer Susceptibility" Cancers 10, no. 10: 361. https://doi.org/10.3390/cancers10100361
APA StyleQuezada Urban, R., Díaz Velásquez, C. E., Gitler, R., Rojo Castillo, M. P., Sirota Toporek, M., Figueroa Morales, A., Moreno García, O., García Esquivel, L., Torres Mejía, G., Dean, M., Delgado Enciso, I., Ochoa Díaz López, H., Rodríguez León, F., Jan, V., Garzón Barrientos, V. H., Ruiz Flores, P., Espino Silva, P. K., Haro Santa Cruz, J., Martínez Gregorio, H., ... Vaca Paniagua, F. (2018). Comprehensive Analysis of Germline Variants in Mexican Patients with Hereditary Breast and Ovarian Cancer Susceptibility. Cancers, 10(10), 361. https://doi.org/10.3390/cancers10100361