Ovarian Tumor Microenvironment Signaling: Convergence on the Rac1 GTPase

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
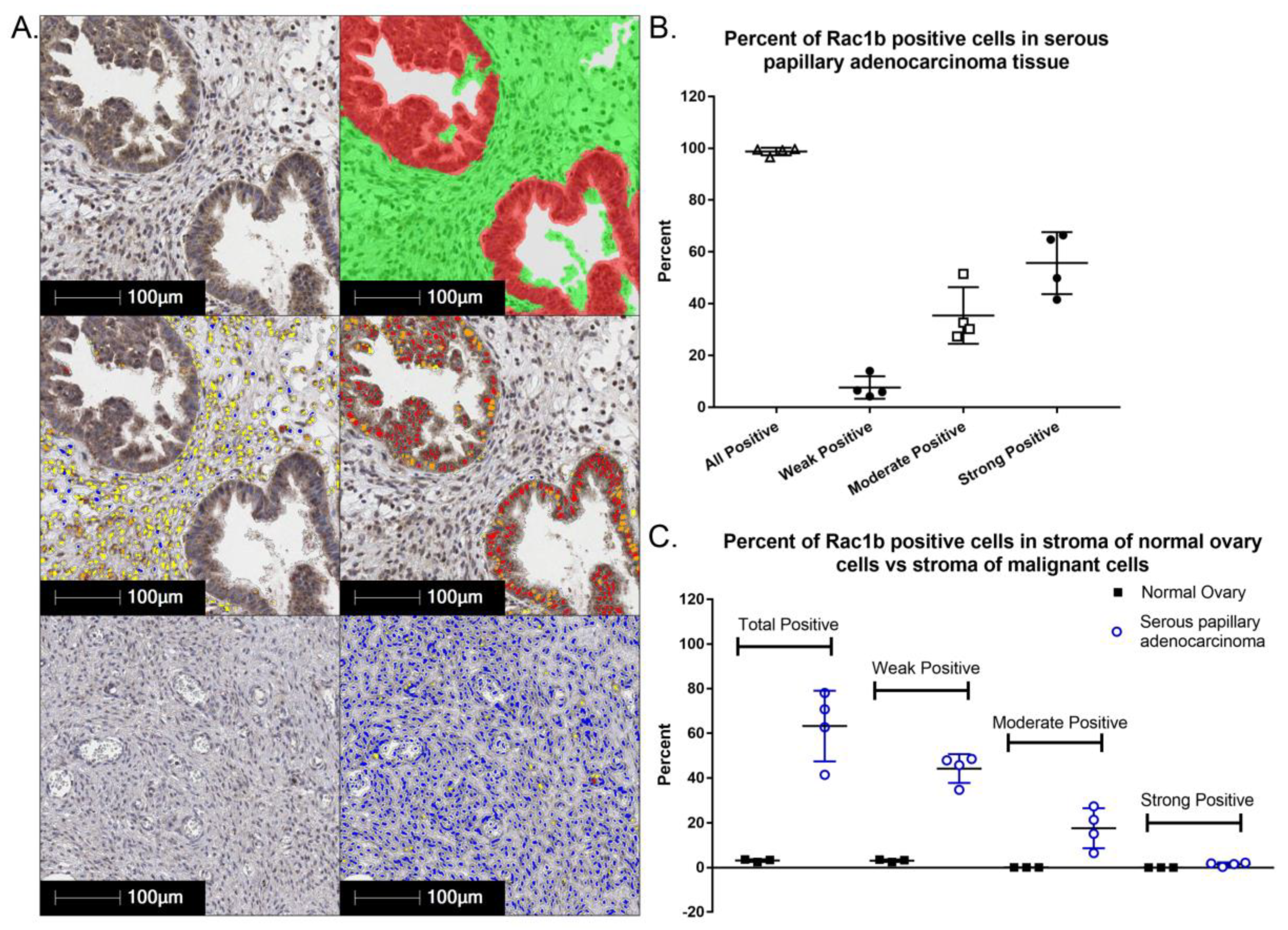{kind=link}
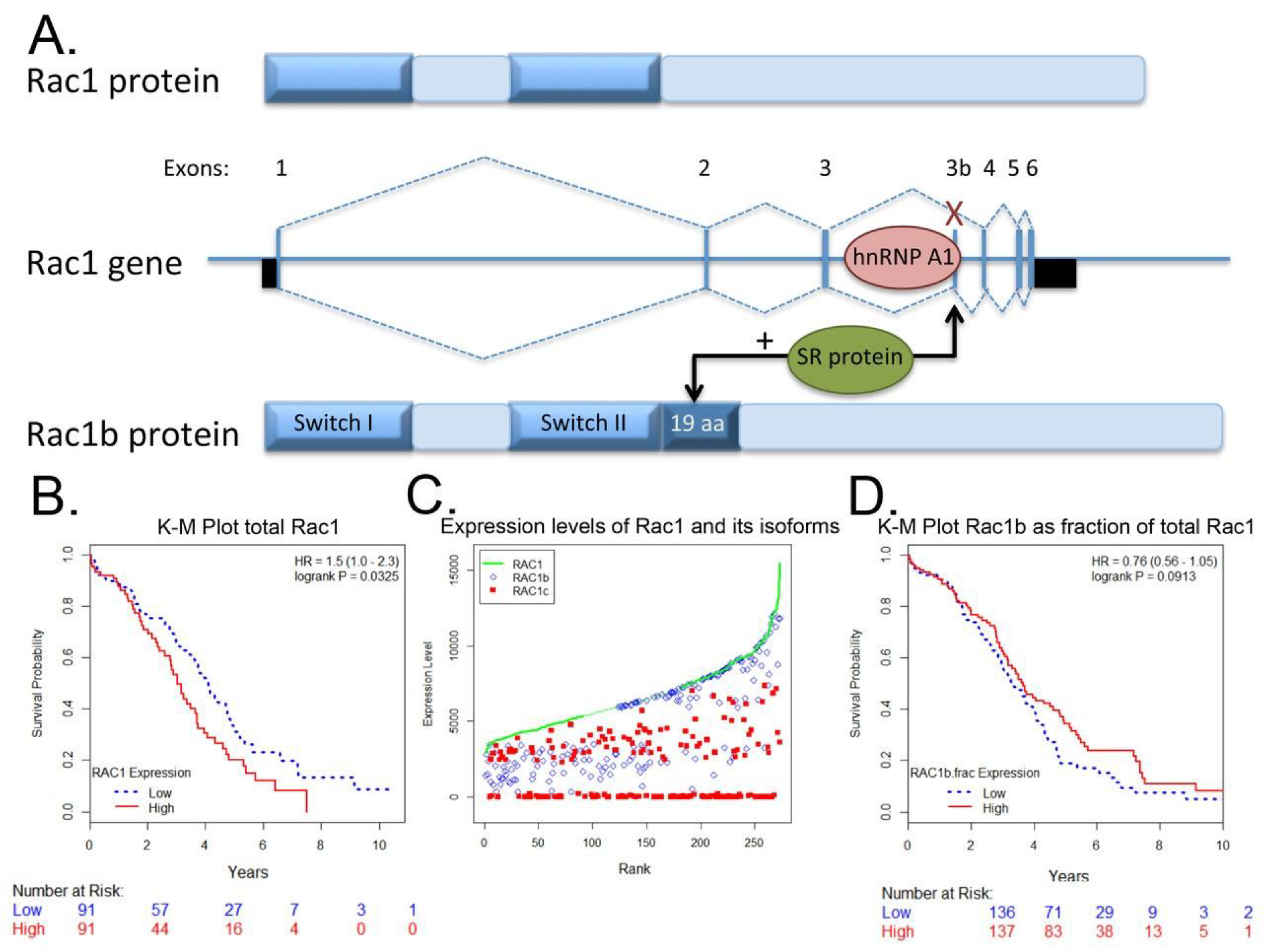{kind=link}
Abstract
1. Introduction
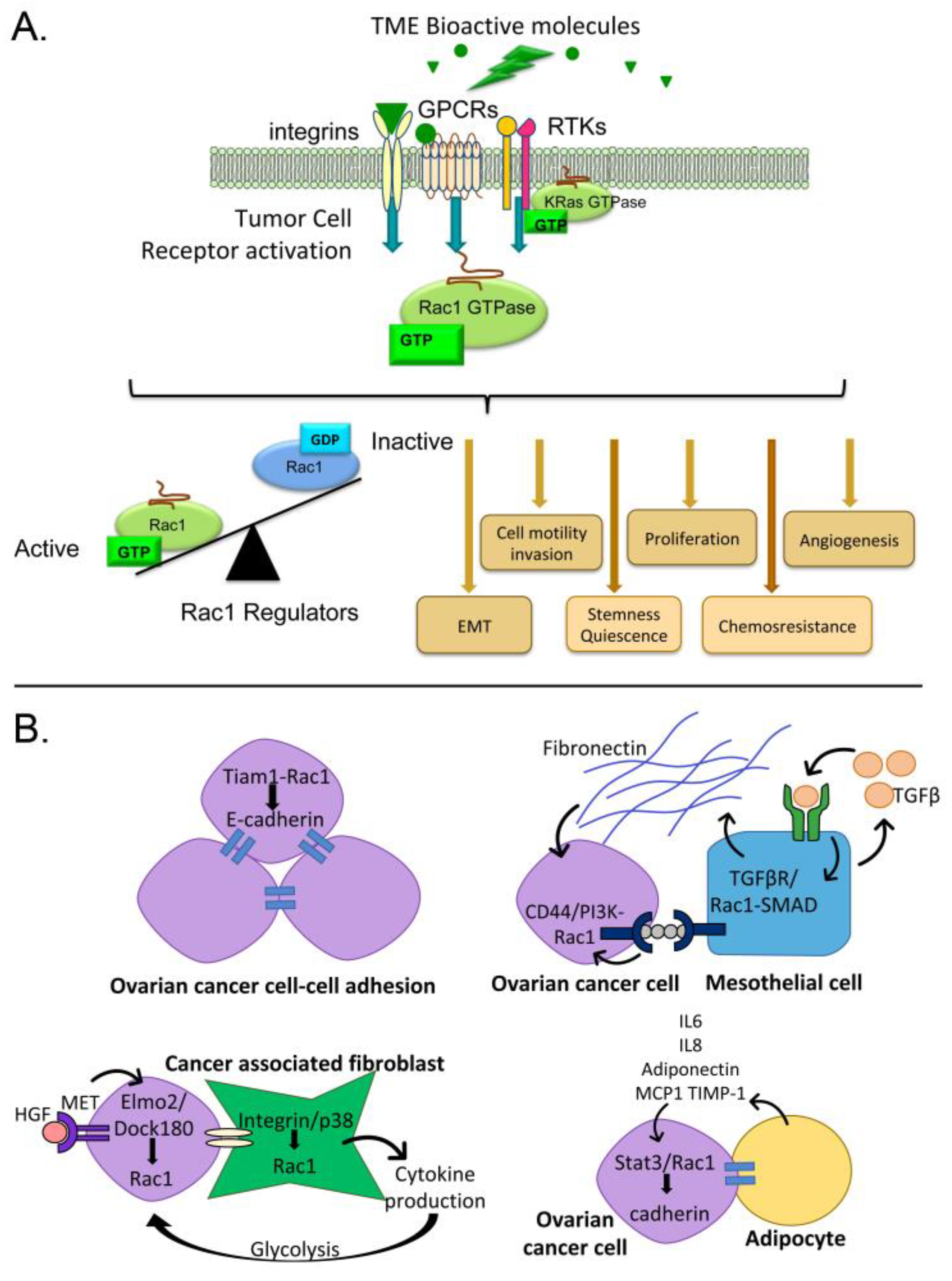
2. Consequences of Rac1 Activation in Cancer
3. Pathways for Rac1 Activation by the Ovarian Tumor Microenvironment
3.1. Activators of G-Protein Coupled Receptors and Rac1 Activity
3.2. Activators of Tyrosine Kinases and Rac1 Activity
3.3. Cell Interactions Leading to Rac1 Activation
3.3.1. Tumor Cell-Cell Adhesion
3.3.2. Mesothelial Cells
3.3.3. Fibroblasts
3.3.4. Adipocytes
4. Mechanisms of Rac1 Dysregulation and Evidence in Ovarian Cancer
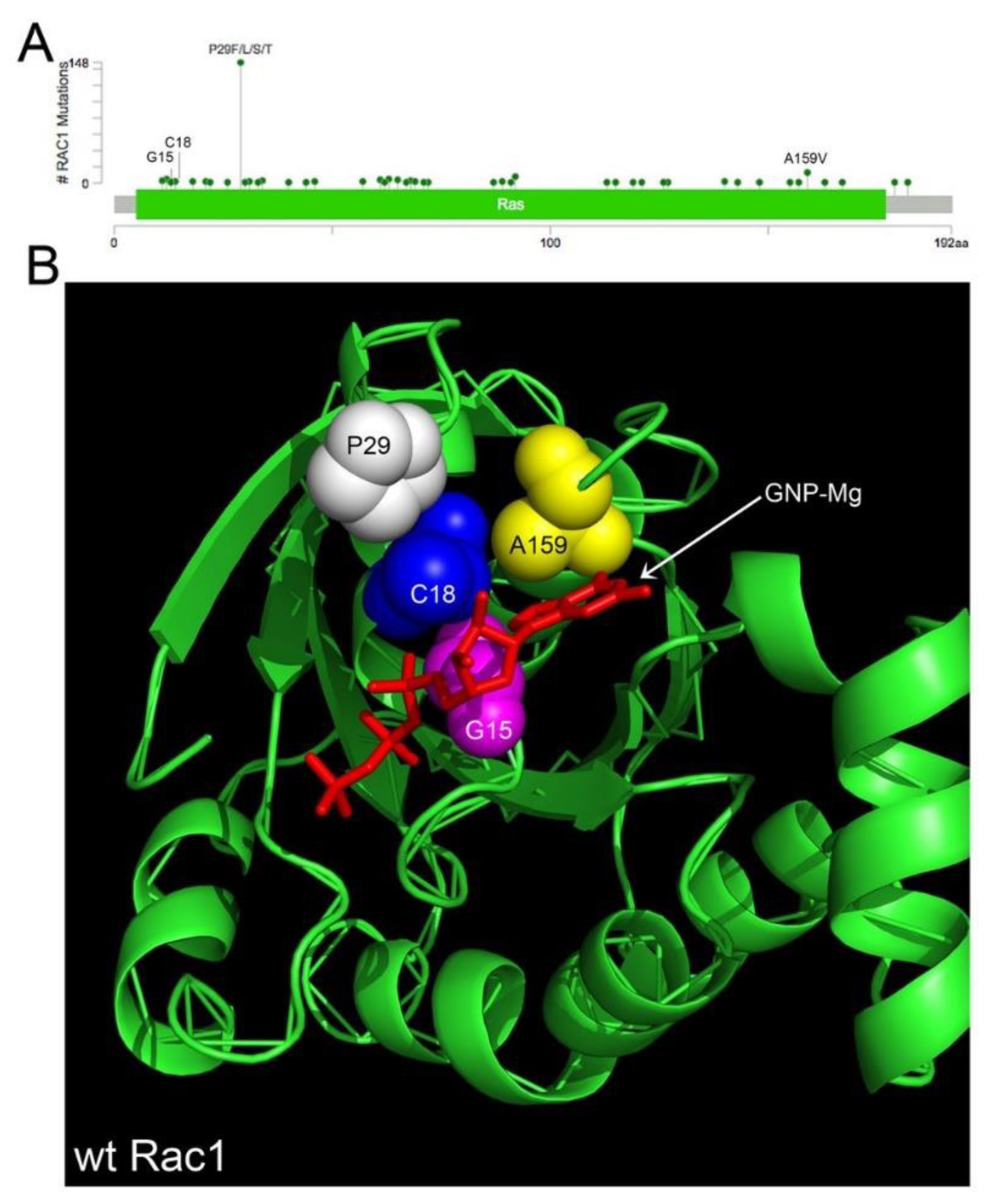
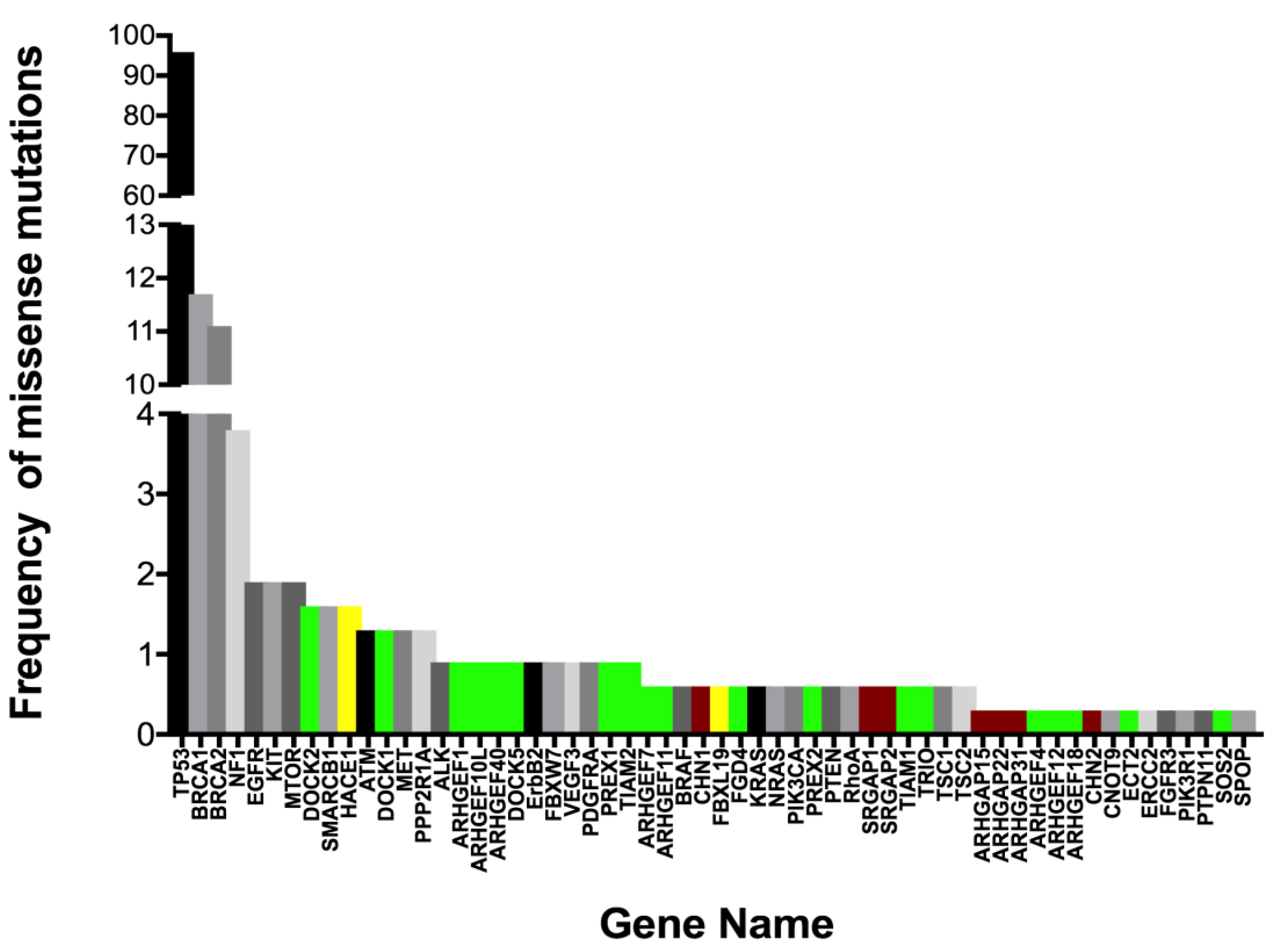
4.1. Rac1 Overexpression and Somatic Mutation
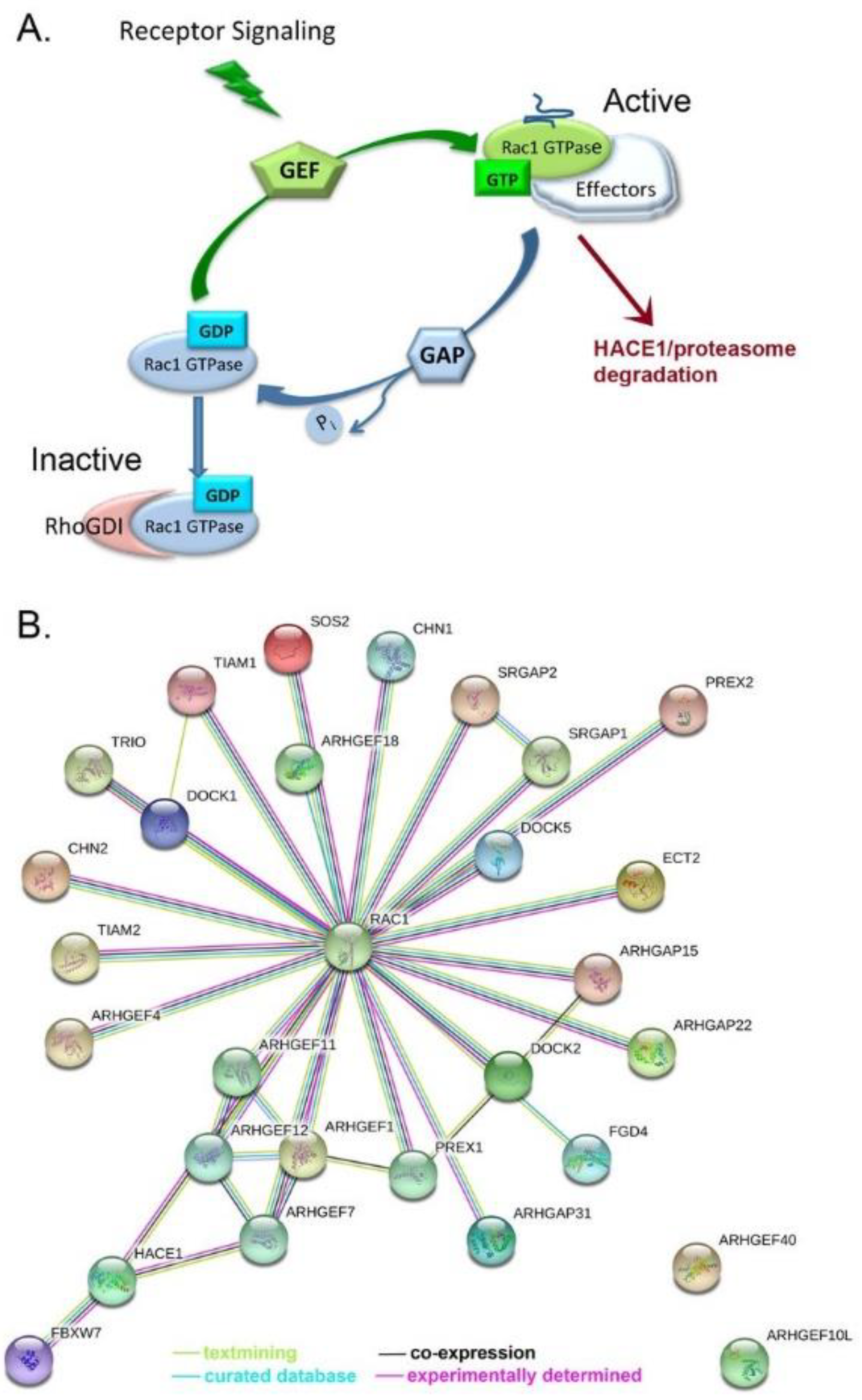
4.2. Rac1 Regulators
4.3. Rac1b Splice Variant
5. Potential Benefits of Targeting Aberrant Rac1 Activity in Ovarian Cancer
6. Other Ovarian Tumor Microenvironments: Extraperitoneal Dissemination and Bone Niche as a Sanctuary Site and Potential Reservoir for Relapse
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Christie, E.L.; Bowtell, D.D.L. Acquired chemotherapy resistance in ovarian cancer. Ann. Oncol. 2017, 28, viii13–viii15. [Google Scholar] [CrossRef] [PubMed]
- DiSilvestro, P.; Alvarez Secord, A. Maintenance treatment of recurrent ovarian cancer: Is it ready for prime time? Cancer Treat. Rev. 2018, 69, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, P.T., Jr.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Previs, R.A.; Sood, A.K.; Mills, G.B.; Westin, S.N. The rise of genomic profiling in ovarian cancer. Expert Rev. Mol. Diagn. 2016, 16, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, B.; Song, Y.S. Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer Sci. 2016, 107, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Thibault, B.; Castells, M.; Delord, J.P.; Couderc, B. Ovarian cancer microenvironment: Implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 2014, 33, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Afify, H.; Salih, Z.; Kelly, M.; Said, N. Role of tumor microenvironment in ovarian cancer pathobiology. Oncotarget 2018, 9, 22832–22849. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Afify, H.; Salih, Z.; Kelly, M.; Said, N. Role of tumor microenvironment in the pathobiology of ovarian cancer: Insights and therapeutic opportunities. Cancer Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rueger, R. Mechanisms and Targets Involved in Dissemination of Ovarian Cancer. Cancer Genom. Proteom. 2016, 13, 407–423. [Google Scholar] [CrossRef]
- Worzfeld, T.; Pogge von Strandmann, E.; Huber, M.; Adhikary, T.; Wagner, U.; Reinartz, S.; Muller, R. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front. Oncol. 2017, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Steinkamp, M.P.; Winner, K.K.; Davies, S.; Muller, C.; Zhang, Y.; Hoffman, R.M.; Shirinifard, A.; Moses, M.; Jiang, Y.; Wilson, B.S. Ovarian tumor attachment, invasion, and vascularization reflect unique microenvironments in the peritoneum: Insights from xenograft and mathematical models. Front. Oncol 2013, 3, 97. [Google Scholar] [CrossRef] [PubMed]
- Giridhar, P.V.; Funk, H.M.; Gallo, C.A.; Porollo, A.; Mercer, C.A.; Plas, D.R.; Drew, A.F. Interleukin-6 receptor enhances early colonization of the murine omentum by upregulation of a mannose family receptor, LY75, in ovarian tumor cells. Clin. Exp. Metastasis 2011, 28, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Chiang, C.Y.; White, E.A.; Schryver, E.M.; Habis, M.; Romero, I.L.; Ladanyi, A.; Penicka, C.V.; George, J.; Matlin, K.; et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Investig. 2014, 124, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Kieu, Q.M.; Dawson, M.R. The malignancy of metastatic ovarian cancer cells is increased on soft matrices through a mechanosensitive Rho-ROCK pathway. J. Cell Sci. 2014, 127, 2621–2626. [Google Scholar] [CrossRef] [PubMed]
- Lungchukiet, P.; Sun, Y.; Kasiappan, R.; Quarni, W.; Nicosia, S.V.; Zhang, X.; Bai, W. Suppression of epithelial ovarian cancer invasion into the omentum by 1alpha,25-dihydroxyvitamin D3 and its receptor. J. Steroid Biochem. Mol. Biol. 2015, 148, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.M.; Funk, H.M.; Thiolloy, S.; Lotan, T.L.; Hickson, J.; Prins, G.S.; Drew, A.F.; Rinker-Schaeffer, C.W. In vitro metastatic colonization of human ovarian cancer cells to the omentum. Clin. Exp. Metastasis 2010, 27, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.J.; Choi, H.J.; Bischoff, F.Z.; et al. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [PubMed]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.A.; Gonzalez, N.; Maggio, J.; Menna, P.L.; Gomez, D.E. Rho GTPases as therapeutic targets in cancer (Review). Int. J. Oncol. 2017, 51, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Gosens, R.; Wieland, T.; Schmidt, M. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol. Ther. 2018, 183, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Zandvakili, I.; Lin, Y.; Morris, J.C.; Zheng, Y. Rho GTPases: Anti- or pro-neoplastic targets? Oncogene 2017, 36, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Troeger, A.; Williams, D.A. Hematopoietic-specific Rho GTPases Rac2 and RhoH and human blood disorders. Exp. Cell Res. 2013, 319, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G.; Caloca, M.J. The Rac GTPase in Cancer: From Old Concepts to New Paradigms. Cancer Res. 2017, 77, 5445–5451. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [PubMed]
- Sadok, A.; Marshall, C.J. Rho GTPases: Masters of cell migration. Small GTPases 2014, 5, e29710. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Herraiz, C.; Sanz-Moreno, V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5, e29019. [Google Scholar] [CrossRef] [PubMed]
- Akunuru, S.; Palumbo, J.; Zhai, Q.J.; Zheng, Y. Rac1 targeting suppresses human non-small cell lung adenocarcinoma cancer stem cell activity. PLoS ONE 2011, 6, e16951. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Yu, P.; Chen, S.; Xing, H.; Chen, Y.; Wang, M.; Tang, K.; Tian, Z.; Rao, Q.; Wang, J. Activation of Rac1 GTPase promotes leukemia cell chemotherapy resistance, quiescence and niche interaction. Mol. Oncol. 2013, 7, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Hyun, K.H.; Kim, R.K.; Lee, H.; Lim, E.J.; Chung, H.Y.; An, S.; Park, M.J.; Suh, Y.; Kim, M.J.; et al. The small GTPase Rac1 is involved in the maintenance of stemness and malignancies in glioma stem-like cells. FEBS Lett. 2011, 585, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.A.; Alonso, D.F.; Gonzalez, N.; Maggio, J.; Gomez, D.E.; Rolfo, C.; Menna, P.L. Relevance of small GTPase Rac1 pathway in drug and radio-resistance mechanisms: Opportunities in cancer therapeutics. Crit. Rev. Oncol. Hematol. 2018, 124, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Wang, P.; Yang, J.; Ma, J.; Liu, C.; Zhou, M. Prognostic and Clinicopathological Value of Rac1 in Cancer Survival: Evidence from a Meta-Analysis. J. Cancer 2018, 9, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Nayak, R.C.; Chang, K.H.; Vaitinadin, N.S.; Cancelas, J.A. Rho GTPases control specific cytoskeleton-dependent functions of hematopoietic stem cells. Immunol. Rev. 2013, 256, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Reimer, D.; Boesch, M.; Wolf, D.; Marth, C.; Sopper, S.; Hatina, J.; Altevogt, P.; Parson, W.; Hackl, H.; Zeimet, A.G. Truncated isoform Vav3.1 is highly expressed in ovarian cancer stem cells and clinically relevant in predicting prognosis and platinum-response. Int. J. Cancer 2018, 142, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, L.; Chen, H.; Hao, J.; Ni, J.; Chang, L.; Duan, W.; Graham, P.; Li, Y. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 2016, 7, 55771–55788. [Google Scholar] [CrossRef] [PubMed]
- Barbolina, M.V.; Moss, N.M.; Westfall, S.D.; Liu, Y.; Burkhalter, R.J.; Marga, F.; Forgacs, G.; Hudson, L.G.; Stack, M.S. Microenvironmental regulation of ovarian cancer metastasis. Cancer Treat. Res. 2009, 149, 319–334. [Google Scholar] [PubMed]
- da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Munoz, P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Chen, H.; Zhu, J.Y.; Wang, W.; Teng, Y.; Ding, H.F.; Jing, Q.; Su, S.B.; Huang, S. Epithelial-mesenchymal transition of ovarian cancer cells is sustained by Rac1 through simultaneous activation of MEK1/2 and Src signaling pathways. Oncogene 2017, 36, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.; Liao, G.; Wang, H.; Kuang, J.; Tang, L. Rac1 expression in epithelial ovarian cancer: Effect on cell EMT and clinical outcome. Med. Oncol. 2015, 32, 329. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Peng, F.; Zhong, Y.; Chen, Y.; Tang, M.; Li, D. Rhein suppresses matrix metalloproteinase production by regulating the Rac1/ROS/MAPK/AP-1 pathway in human ovarian carcinoma cells. Int. J. Oncol. 2017, 50, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Romero-Laorden, N.; Olmos, D.; Fehm, T.; Garcia-Donas, J.; Diaz-Padilla, I. Circulating and disseminated tumor cells in ovarian cancer: A systematic review. Gynecol. Oncol. 2014, 133, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Fehm, T.; Banys, M.; Rack, B.; Janni, W.; Marth, C.; Blassl, C.; Hartkopf, A.; Trope, C.; Kimmig, R.; Krawczyk, N.; et al. Pooled analysis of the prognostic relevance of disseminated tumor cells in the bone marrow of patients with ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Fehm, T.; Becker, S.; Bachmann, C.; Beck, V.; Gebauer, G.; Banys, M.; Wallwiener, D.; Solomayer, E.F. Detection of disseminated tumor cells in patients with gynecological cancers. Gynecol. Oncol. 2006, 103, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Gasparri, M.L.; Savone, D.; Besharat, R.A.; Farooqi, A.A.; Bellati, F.; Ruscito, I.; Panici, P.B.; Papadia, A. Circulating tumor cells as trigger to hematogenous spreads and potential biomarkers to predict the prognosis in ovarian cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bian, B.; Yuan, X.; Xie, G.; Ma, Y.; Shen, L. Prognostic Value of Circulating Tumor Cells in Ovarian Cancer: A Meta-Analysis. PLoS ONE 2015, 10, e0130873. [Google Scholar] [CrossRef] [PubMed]
- Blassl, C.; Kuhlmann, J.D.; Webers, A.; Wimberger, P.; Fehm, T.; Neubauer, H. Gene expression profiling of single circulating tumor cells in ovarian cancer—Establishment of a multi-marker gene panel. Mol. Oncol. 2016, 10, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Nwani, N.G.; Sima, L.E.; Nieves-Neira, W.; Matei, D. Targeting the Microenvironment in High Grade Serous Ovarian Cancer. Cancers 2018, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Minion, L.E.; Coleman, R.L. Anti-angiogenic agents in ovarian cancer: Past, present, and future. Ann. Oncol. 2016, 27, i33–i39. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Coleman, R.L.; Sood, A.K. Targeting the tumour microenvironment in ovarian cancer. Eur. J. Cancer 2016, 56, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Nohata, N.; Uchida, Y.; Stratman, A.N.; Adams, R.H.; Zheng, Y.; Weinstein, B.M.; Mukouyama, Y.S.; Gutkind, J.S. Temporal-specific roles of Rac1 during vascular development and retinal angiogenesis. Dev. Biol. 2016, 411, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Galan Moya, E.M.; Le Guelte, A.; Gavard, J. PAKing up to the endothelium. Cell. Signal. 2009, 21, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Scarcia, M.; Bagshaw, R.D.; McMahon, K.; Grant, G.; Harvey, T.; Yeo, M.; Esteves, F.O.; Thygesen, H.H.; Jones, P.F.; et al. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat. Commun. 2015, 6, 7286. [Google Scholar] [CrossRef] [PubMed]
- Fryer, B.H.; Field, J. Rho, Rac, Pak and angiogenesis: Old roles and newly identified responsibilities in endothelial cells. Cancer Lett. 2005, 229, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Soga, N.; Connolly, J.O.; Chellaiah, M.; Kawamura, J.; Hruska, K.A. Rac regulates vascular endothelial growth factor stimulated motility. Cell Commun. Adhes. 2001, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Bi, F.; Zhang, X.; Pan, Y.; Liu, N.; Zheng, Y.; Fan, D. Inhibition of endothelial cell proliferation by targeting Rac1 GTPase with small interference RNA in tumor cells. Biochem. Biophys. Res. Commun. 2004, 320, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Hernandez-O’Farrill, E.; Borrero-Garcia, L.D.; Forestier-Roman, I.; Gerena, Y.; Blanco, M.; Rivera-Robles, M.J.; Rodriguez-Medina, J.R.; Cubano, L.A.; et al. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther. 2017, 16, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac Inhibitor EHop-016 Inhibits Mammary Tumor Growth and Metastasis in a Nude Mouse Model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Ramachandran, S.; Rashid, O.M.; Takabe, K. Lymphangiogenesis: A new player in cancer progression. World J. Gastroenterol. 2010, 16, 4003–4012. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.M.; Reisen, F.; Zgraggen, S.; Fischer, S.; Yuen, D.; Kang, G.J.; Chen, L.; Schneider, G.; Detmar, M. Phenotype-based high-content chemical library screening identifies statins as inhibitors of in vivo lymphangiogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2665–E2674. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, E.W.; Gerber, S.A.; Sedlacek, A.L.; Rybalko, V.Y.; Chan, W.M.; Lord, E.M. Omental immune aggregates and tumor metastasis within the peritoneal cavity. Immunol. Res. 2009, 45, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Kuerti, S.; Oliveira-Ferrer, L.; Milde-Langosch, K.; Schmalfeldt, B.; Legler, K.; Woelber, L.; Prieske, K.; Mahner, S.; Trillsch, F. VEGF-C expression attributes the risk for lymphatic metastases to ovarian cancer patients. Oncotarget 2017, 8, 43218–43227. [Google Scholar] [CrossRef] [PubMed]
- Hisamatsu, T.; Mabuchi, S.; Sasano, T.; Kuroda, H.; Takahashi, R.; Matsumoto, Y.; Kawano, M.; Kozasa, K.; Takahashi, K.; Sawada, K.; et al. The significance of lymphatic space invasion and its association with vascular endothelial growth factor-C expression in ovarian cancer. Clin. Exp. Metastasis 2015, 32, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Luthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Y.; Qi, W.; Zhao, Y.; Huang, A.; Sheng, W.; Lei, B.; Lin, P.; Zhu, H.; Li, W.; et al. Expression of Tiam1 predicts lymph node metastasis and poor survival of lung adenocarcinoma patients. Diagn. Pathol. 2014, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Li, Y.; Peng, Q.; Zhou, J.; Zhou, Q.; Zhang, R.; Liang, H. Expression of Tiam1 and VEGF-C correlates with lymphangiogenesis in human colorectal carcinoma. Cancer Biol. Ther. 2009, 8, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.J.; Yang, K.; Taylor-Harding, B.; Wiedemeyer, W.R.; Buckanovich, R.J. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, S.W.; Krenn, P.W.; Ganghammer, S.; Asslaber, D.; Pichler, U.; Oberascher, K.; Henschler, R.; Wallner, M.; Kerschbaum, H.; Greil, R.; et al. Tiam1/Rac1 signals contribute to the proliferation and chemoresistance, but not motility, of chronic lymphocytic leukemia cells. Blood 2014, 123, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Lim, Y.; Baek, S.Y.; Jin, S.; Jeong, Y.H.; Kwak, J.Y.; Yoon, S. Co-targeting of Tiam1/Rac1 and Notch ameliorates chemoresistance against doxorubicin in a biomimetic 3D lymphoma model. Oncotarget 2018, 9, 2058–2075. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Chung, V.Y.; Thiery, J.P. Targeting pathways contributing to epithelial-mesenchymal transition (EMT) in epithelial ovarian cancer. Curr. Drug Targets 2012, 13, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets 2010, 10, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Castro-Castro, A. Rac-ing to the plasma membrane: The long and complex work commute of Rac1 during cell signaling. Small GTPases 2012, 3, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Prado, J.; Bracho-Valdes, I.; Cervantes-Villagrana, R.D.; Reyes-Cruz, G. Gbetagamma Pathways in Cell Polarity and Migration Linked to Oncogenic GPCR Signaling: Potential Relevance in Tumor Microenvironment. Mol. Pharmacol. 2016, 90, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment. Cancers 2018, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Devine, K.M.; Smicun, Y.; Hope, J.M.; Fishman, D.A. S1P induced changes in epithelial ovarian cancer proteolysis, invasion, and attachment are mediated by Gi and Rac. Gynecol. Oncol. 2008, 110, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, X.; Pan, Z.K.; Huang, S. Integrity of SOS1/EPS8/ABI1 tri-complex determines ovarian cancer metastasis. Cancer Res. 2010, 70, 9979–9990. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.D.; Dhanasekaran, D.N. LPA Stimulates the Phosphorylation of p130Cas via Galphai2 in Ovarian Cancer Cells. Genes Cancer 2012, 3, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.D.; Ha, J.H.; Jayaraman, M.; Dhanasekaran, D.N. LPA-mediated migration of ovarian cancer cells involves translocalization of Galphai2 to invadopodia and association with Src and beta-pix. Cancer Lett. 2015, 356, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Fritz, R.D.; Pertz, O. The dynamics of spatio-temporal Rho GTPase signaling: Formation of signaling patterns. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, Y.; Lemmon, M.A.; Kazanietz, M.G. Essential role for Rac in heregulin beta1 mitogenic signaling: A mechanism that involves epidermal growth factor receptor and is independent of ErbB4. Mol. Cell. Biol. 2006, 26, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Fan, Z.; Ding, M.; Zhang, H.; Mu, L.; Ding, Y.; Zhang, Y.; Jia, B.; Chen, L.; Chang, Z.; et al. An EGFR/PI3K/AKT axis promotes accumulation of the Rac1-GEF Tiam1 that is critical in EGFR-driven tumorigenesis. Oncogene 2015, 34, 5971–5982. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.; Holmes, A.; Lomo, L.; Steinkamp, M.P.; Kang, H.; Muller, C.Y.; Wilson, B.S. High incidence of ErbB3, ErbB4, and MET expression in ovarian cancer. Int. J. Gynecol. Pathol. 2014, 33, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Haber, C.; Barrio-Real, L.; Casado-Medrano, V.; Kazanietz, M.G. Heregulin/ErbB3 Signaling Enhances CXCR4-Driven Rac1 Activation and Breast Cancer Cell Motility via Hypoxia-Inducible Factor 1alpha. Mol. Cell. Biol. 2016, 36, 2011–2026. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xiong, X.; Abdalla, A.; Alejo, S.; Zhu, L.; Lu, F.; Sun, H. HGF-induced formation of the MET-AXL-ELMO2-DOCK180 complex promotes RAC1 activation, receptor clustering, and cancer cell migration and invasion. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Sklar, L.A.; Agola, J.O.; Guo, Y.; Silberberg, M.; Roxby, J.; Vestling, A.; Romero, E.; Surviladze, Z.; Waller, A.; et al. Novel Activities of Select NSAID R-Enantiomers against Rac1 and Cdc42 GTPases. PLoS ONE 2015, 10, e0142812. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Palmby, T.R.; Gavard, J.; Amornphimoltham, P.; Zheng, Y.; Gutkind, J.S. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008, 22, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.V.; Nagy, J.A.; Senger, D.R. Active Rac1 improves pathologic VEGF neovessel architecture and reduces vascular leak: Mechanistic similarities with angiopoietin-1. Blood 2011, 117, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.; Carpy, A.; Woroniuk, A.; Vennin, C.; White, G.; Timpson, P.; Macek, B.; Malliri, A. Differential Rac1 signalling by guanine nucleotide exchange factors implicates FLII in regulating Rac1-driven cell migration. Nat. Commun. 2016, 7, 10664. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yang, Y.; Dong, L.; Qiu, W.; Yang, L.; Wang, X.; Liu, L. Construction and characteristics of an E-cadherin-related three-dimensional suspension growth model of ovarian cancer. Sci. Rep. 2014, 4, 5646. [Google Scholar] [CrossRef] [PubMed]
- Shishido, A.; Mori, S.; Yokoyama, Y.; Hamada, Y.; Minami, K.; Qian, Y.; Wang, J.; Hirose, H.; Wu, X.; Kawaguchi, N.; et al. Mesothelial cells facilitate cancer stemlike properties in spheroids of ovarian cancer cells. Oncol. Rep. 2018, 40, 2105–2114. [Google Scholar] [PubMed]
- Ween, M.P.; Oehler, M.K.; Ricciardelli, C. Role of versican, hyaluronan and CD44 in ovarian cancer metastasis. Int. J. Mol. Sci. 2011, 12, 1009–1029. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Tang, H.; Xu, L.; Wang, X.; Yang, C.; Ruan, S.; Guo, J.; Hu, S.; Wang, Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 2012, 33, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell MeTable 2018. [Google Scholar] [CrossRef] [PubMed]
- Raptis, L.; Arulanandam, R.; Geletu, M.; Turkson, J. The R(h)oads to Stat3: Stat3 activation by the Rho GTPases. Exp. Cell Res. 2011, 317, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Savant, S.S.; Sriramkumar, S.; O’Hagan, H.M. The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer. Cancers 2018, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.D.; Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014, 5, e27958. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.T.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Bohm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Pearce, O.; Maniati, E.; Vincent, B.G.; Bixby, L.; Bohm, S.; Dowe, T.; Wilkes, E.H.; Chakravarty, P.; Thompson, R.; et al. A Strong B-cell Response Is Part of the Immune Landscape in Human High-Grade Serous Ovarian Metastases. Clin. Cancer Res. 2017, 23, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Flies, D.B.; Higuchi, T.; Harris, J.C.; Jha, V.; Gimotty, P.A.; Adams, S.F. Immune checkpoint blockade reveals the stimulatory capacity of tumor-associated CD103(+) dendritic cells in late-stage ovarian cancer. Oncoimmunology 2016, 5, e1185583. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kenney, S.R.; Cook, L.; Adams, S.F.; Rutledge, T.; Romero, E.; Oprea, T.I.; Sklar, L.A.; Bedrick, E.; Wiggins, C.L.; et al. A Novel Pharmacologic Activity of Ketorolac for Therapeutic Benefit in Ovarian Cancer Patients. Clin. Cancer Res. 2015, 21, 5064–5072. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. Molecular dynamic simulation reveals damaging impact of RAC1 F28L mutation in the switch I region. PLoS ONE 2013, 8, e77453. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chang, M.T.; Johnsen, H.C.; Gao, S.P.; Sylvester, B.E.; Sumer, S.O.; Zhang, H.; Solit, D.B.; Taylor, B.S.; Schultz, N.; et al. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 2017, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.; Malliri, A. GEFs: Dual regulation of Rac1 signaling. Small GTPases 2017, 8, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.; Brazao, R.; Boavida, M.G.; Gespach, C.; Chastre, E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999, 18, 6835–6839. [Google Scholar] [CrossRef] [PubMed]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Jordan, P. Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp. Cell Res. 2005, 305, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Orlichenko, L.; Geyer, R.; Yanagisawa, M.; Khauv, D.; Radisky, E.S.; Anastasiadis, P.Z.; Radisky, D.C. The 19-amino acid insertion in the tumor-associated splice isoform Rac1b confers specific binding to p120 catenin. J. Biol. Chem. 2010, 285, 19153–19161. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Carmo, F.; Bugalho, M.J. RAC1b overexpression in papillary thyroid carcinoma: A role to unravel. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2013, 168, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Licciulli, S.; Avila, J.L.; Cho, M.; Troutman, S.; Jiang, P.; Kossenkov, A.V.; Showe, L.C.; Liu, Q.; Vachani, A.; et al. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene 2013, 32, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ying, L.; Wang, H.; Wei, S.S.; Chen, J.; Chen, Y.H.; Xu, W.P.; Jie, Q.Q.; Zhou, Q.; Li, Y.G.; et al. Rac1b enhances cell survival through activation of the JNK2/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways. Oncotarget 2016, 7, 17970–17985. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D. Both sides of the same coin: Rac1 splicing regulating by EGF signaling. Cell Res. 2017, 27, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fu, X.; Chen, P.; Wu, P.; Fan, X.; Li, N.; Zhu, H.; Jia, T.T.; Ji, H.; Wang, Z.; et al. SPSB1-mediated HnRNP A1 ubiquitylation regulates alternative splicing and cell migration in EGF signaling. Cell Res. 2017, 27, 540–558. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. PyMOL: An Open-Source Molecular Graphics Tool; Delano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Kawazu, M.; Ueno, T.; Kontani, K.; Ogita, Y.; Ando, M.; Fukumura, K.; Yamato, A.; Soda, M.; Takeuchi, K.; Miki, Y.; et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.R.; Li, L.; Cabeceiras, P.K.; Mahdavi, M.; Gutschner, T.; Genovese, G.; Wang, G.; Fang, Z.; Tepper, J.M.; Stemke-Hale, K.; et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014, 74, 4845–4852. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Rusin, S.; Scott, J.A.; East, A.; Ali, L.D.; Lizotte, P.H.; Wong, T.C.; et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12372–12377. [Google Scholar] [CrossRef] [PubMed]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E.; et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Zoughlami, Y.; van Stalborgh, A.M.; van Hennik, P.B.; Hordijk, P.L. Nucleophosmin1 is a negative regulator of the small GTPase Rac1. PLoS ONE 2013, 8, e68477. [Google Scholar] [CrossRef] [PubMed]
- Payapilly, A.; Malliri, A. Compartmentalisation of RAC1 signalling. Curr. Opin. Cell Biol. 2018, 54, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mettouchi, A.; Lemichez, E. Ubiquitylation of active Rac1 by the E3 ubiquitin-ligase HACE1. Small GTPases 2012, 3, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Finn, A.J.; Pedone, K.H.; Thomas, N.E.; Der, C.J.; Cox, A.D. ERK/MAPK Signaling Drives Overexpression of the Rac-GEF, PREX1, in BRAF- and NRAS-Mutant Melanoma. Mol. Cancer Res. MCR 2016, 14, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Misek, S.A.; Chen, J.; Schroeder, L.; Rattanasinchai, C.; Sample, A.; Sarkaria, J.N.; Gallo, K.A. EGFR Signals through a DOCK180-MLK3 Axis to Drive Glioblastoma Cell Invasion. Mol. Cancer Res. MCR 2017, 15, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Lissanu Deribe, Y.; Shi, Y.; Rai, K.; Nezi, L.; Amin, S.B.; Wu, C.C.; Akdemir, K.C.; Mahdavi, M.; Peng, Q.; Chang, Q.E.; et al. Truncating PREX2 mutations activate its GEF activity and alter gene expression regulation in NRAS-mutant melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E1296–E1305. [Google Scholar] [CrossRef] [PubMed]
- Earp, M.; Tyrer, J.P.; Winham, S.J.; Lin, H.Y.; Chornokur, G.; Dennis, J.; Aben, K.K.H.; Anton-Culver, H.; Antonenkova, N.; Bandera, E.V.; et al. Variants in genes encoding small GTPases and association with epithelial ovarian cancer susceptibility. PLoS ONE 2018, 13, e0197561. [Google Scholar] [CrossRef] [PubMed]
- Winkler, S.; Mohl, M.; Wieland, T.; Lutz, S. GrinchGEF—A novel Rho-specific guanine nucleotide exchange factor. Biochem. Biophys. Res. Commun. 2005, 335, 1280–1286. [Google Scholar] [CrossRef] [PubMed]
- Zoughlami, Y.; Voermans, C.; Brussen, K.; van Dort, K.A.; Kootstra, N.A.; Maussang, D.; Smit, M.J.; Hordijk, P.L.; van Hennik, P.B. Regulation of CXCR4 conformation by the small GTPase Rac1: Implications for HIV infection. Blood 2012, 119, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kim, D.; Kim, Y.; Choi, K.; Miller, J.E.; Kim, D.; Lee, Y. CAS-viewer: Web-based tool for splicing-guided integrative analysis of multi-omics cancer data. BMC Med Genom. 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.S.; Son, H.; Kim, S.; Kim, S. ISOexpresso: A web-based platform for isoform-level expression analysis in human cancer. BMC Genom. 2016, 17, 631. [Google Scholar] [CrossRef] [PubMed]
- Mehner, C.; Miller, E.; Khauv, D.; Nassar, A.; Oberg, A.L.; Bamlet, W.R.; Zhang, L.; Waldmann, J.; Radisky, E.S.; Crawford, H.C.; et al. Tumor cell-derived MMP3 orchestrates Rac1b and tissue alterations that promote pancreatic adenocarcinoma. Mol. Cancer Res. MCR 2014, 12, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Mehner, C.; Miller, E.; Nassar, A.; Bamlet, W.R.; Radisky, E.S.; Radisky, D.C. Tumor cell expression of MMP3 as a prognostic factor for poor survival in pancreatic, pulmonary, and mammary carcinoma. Genes Cancer 2015, 6, 480–489. [Google Scholar] [PubMed]
- Stallings-Mann, M.L.; Waldmann, J.; Zhang, Y.; Miller, E.; Gauthier, M.L.; Visscher, D.W.; Downey, G.P.; Radisky, E.S.; Fields, A.P.; Radisky, D.C. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci. Transl. Med. 2012, 4, 142ra195. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Espinaco, V.; Cuatrecasas, M.; Alonso, V.; Escudero, P.; Marmol, M.; Horndler, C.; Ortego, J.; Gallego, R.; Codony-Servat, J.; Garcia-Albeniz, X.; et al. RAC1b overexpression correlates with poor prognosis in KRAS/BRAF WT metastatic colorectal cancer patients treated with first-line FOLFOX/XELOX chemotherapy. Eur. J. Cancer 2014, 50, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Huff, L.P.; Decristo, M.J.; Trembath, D.; Kuan, P.F.; Yim, M.; Liu, J.; Cook, D.R.; Miller, C.R.; Der, C.J.; Cox, A.D. The Role of Ect2 Nuclear RhoGEF Activity in Ovarian Cancer Cell Transformation. Genes Cancer 2013, 4, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [PubMed]
- Pajic, M.; Herrmann, D.; Vennin, C.; Conway, J.R.; Chin, V.T.; Johnsson, A.K.; Welch, H.C.; Timpson, P. The dynamics of Rho GTPase signaling and implications for targeting cancer and the tumor microenvironment. Small GTPases 2015, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Smithers, C.C.; Overduin, M. Structural Mechanisms and Drug Discovery Prospects of Rho GTPases. Cells 2016, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Nassar, N.; Cancelas, J.; Zheng, J.; Williams, D.A.; Zheng, Y. Structure-function based design of small molecule inhibitors targeting Rho family GTPases. Curr. Top. Med. Chem. 2006, 6, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Dharmawardhane, S.; Hernandez, E.; Vlaar, C. Development of EHop-016: A small molecule inhibitor of Rac. Enzymes 2013, 33 Pt A, 117–146. [Google Scholar]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Kenney, S.R.; Phillips, G.K.; Simpson, D.; Schroeder, C.E.; Noth, J.; Romero, E.; Swanson, S.; Waller, A.; Strouse, J.J.; et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Biol. Chem. 2013, 288, 8531–8543. [Google Scholar] [CrossRef] [PubMed]
- Forget, P.; Bentin, C.; Machiels, J.P.; Berliere, M.; Coulie, P.G.; De Kock, M. Intraoperative use of ketorolac or diclofenac is associated with improved disease-free survival and overall survival in conservative breast cancer surgery. Br. J. Anaesth. 2014. [Google Scholar] [CrossRef] [PubMed]
- Retsky, M.; Demicheli, R.; Hrushesky, W.J.; Forget, P.; De Kock, M.; Gukas, I.; Rogers, R.A.; Baum, M.; Sukhatme, V.; Vaidya, J.S. Reduction of breast cancer relapses with perioperative non-steroidal anti-inflammatory drugs: New findings and a review. Curr. Med. Chem. 2013, 20, 4163–4176. [Google Scholar] [CrossRef] [PubMed]
- Forget, P.; Vandenhende, J.; Berliere, M.; Machiels, J.P.; Nussbaum, B.; Legrand, C.; De Kock, M. Do intraoperative analgesics influence breast cancer recurrence after mastectomy? A retrospective analysis. Anesth. Analg. 2010, 110, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Yang, C.; Tan, Q.; Song, W.; Lu, M.; Zhao, W.; Lou, G.; Li, Z.; Li, K.; Hou, Y. Sites of distant metastases and overall survival in ovarian cancer: A study of 1481 patients. Gynecol. Oncol. 2018, 150, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Obermayr, E.; Bednarz-Knoll, N.; Orsetti, B.; Weier, H.U.; Lambrechts, S.; Castillo-Tong, D.C.; Reinthaller, A.; Braicu, E.I.; Mahner, S.; Sehouli, J.; et al. Circulating tumor cells: Potential markers of minimal residual disease in ovarian cancer? a study of the OVCAD consortium. Oncotarget 2017, 8, 106415–106428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, H.; Yu, X.; Li, S.; Lei, Z.; Li, C.; Zhang, Q.; Han, Q.; Li, Y.; Zhang, K.; et al. Analysis of Circulating Tumor Cells in Ovarian Cancer and Their Clinical Value as a Biomarker. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Keyver-Paik, M.D.; Arden, J.M.; Luders, C.; Thiesler, T.; Abramian, A.; Hoeller, T.; Hecking, T.; Ayub, T.H.; Doeser, A.; Kaiser, C.; et al. Impact of Chemotherapy on Retroperitoneal Lymph Nodes in Ovarian Cancer. Anticancer Res. 2016, 36, 1815–1824. [Google Scholar] [PubMed]
- Hjerpe, E.; Staf, C.; Dahm-Kahler, P.; Stalberg, K.; Bjurberg, M.; Holmberg, E.; Borgfeldt, C.; Tholander, B.; Hellman, K.; Kjolhede, P.; et al. Lymph node metastases as only qualifier for stage IV serous ovarian cancer confers longer survival than other sites of distant disease—A Swedish Gynecologic Cancer Group (SweGCG) study. Acta Oncol. 2018, 57, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Sehouli, J.; Olschewski, J.; Schotters, V.; Fotopoulou, C.; Pietzner, K. Prognostic role of early versus late onset of bone metastasis in patients with carcinoma of the ovary, peritoneum and fallopian tube. Ann. Oncol. 2013, 24, 3024–3028. [Google Scholar] [CrossRef] [PubMed]
- Chebouti, I.; Blassl, C.; Wimberger, P.; Neubauer, H.; Fehm, T.; Kimmig, R.; Kasimir-Bauer, S. Analysis of disseminated tumor cells before and after platinum based chemotherapy in primary ovarian cancer. Do stem cell like cells predict prognosis? Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabieres, C. Bone marrow as a reservoir for disseminated tumor cells: A special source for liquid biopsy in cancer patients. BoneKEy Rep. 2014, 3, 584. [Google Scholar] [CrossRef] [PubMed]
- Banys, M.; Solomayer, E.F.; Becker, S.; Krawczyk, N.; Gardanis, K.; Staebler, A.; Neubauer, H.; Wallwiener, D.; Fehm, T. Disseminated tumor cells in bone marrow may affect prognosis of patients with gynecologic malignancies. Int. J. Gynecol. Cancer 2009, 19, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Wimberger, P.; Heubner, M.; Otterbach, F.; Fehm, T.; Kimmig, R.; Kasimir-Bauer, S. Influence of platinum-based chemotherapy on disseminated tumor cells in blood and bone marrow of patients with ovarian cancer. Gynecol. Oncol. 2007, 107, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wimberger, P.; Roth, C.; Pantel, K.; Kasimir-Bauer, S.; Kimmig, R.; Schwarzenbach, H. Impact of platinum-based chemotherapy on circulating nucleic acid levels, protease activities in blood and disseminated tumor cells in bone marrow of ovarian cancer patients. Int. J. Cancer 2011, 128, 2572–2580. [Google Scholar] [CrossRef] [PubMed]
- Gunjal, P.M.; Schneider, G.; Ismail, A.A.; Kakar, S.S.; Kucia, M.; Ratajczak, M.Z. Evidence for induction of a tumor metastasis-receptive microenvironment for ovarian cancer cells in bone marrow and other organs as an unwanted and underestimated side effect of chemotherapy/radiotherapy. J. Ovarian Res. 2015, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Afrin, F.; Satija, N.; Tripathi, R.P.; Gangenahalli, G.U. Stromal-derived factor-1/CXCR4 signaling: Indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev. 2011, 20, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Duda, D.G. Role of stromal cell-derived factor 1alpha pathway in bone metastatic prostate cancer. J. Biomed. Res. 2016, 30, 181–185. [Google Scholar] [PubMed]
- Lapidot, T.; Kollet, O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia 2002, 16, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Petit, I.; Kollet, O.; Magid, M.; Ponomaryov, T.; Byk, T.; Nagler, A.; Ben-Hur, H.; Many, A.; Shultz, L.; et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 1999, 283, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra373. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.F.; Liu, S.Y.; Min, X.Y.; Ji, Y.Y.; Wang, N.; Liu, D.; Ma, N.; Li, Z.F.; Li, K. The prognostic value of CXCR4 in ovarian cancer: A meta-analysis. PLoS ONE 2014, 9, e92629. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Gao, B.L.; Zhang, X.J.; Liu, G.C.; Xu, F.; Fan, Q.Y.; Zhang, S.J.; Yang, B.; Wu, X.H. CXCL12-CXCR4 Axis Promotes Proliferation, Migration, Invasion, and Metastasis of Ovarian Cancer. Oncol. Res. 2015, 22, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, K.; Qiu, X.; Li, M.; Hao, Q.; Wei, L.; Zhang, W.; Chen, B.; Xin, X. Overexpression of CXCR4 is significantly associated with cisplatin-based chemotherapy resistance and can be a prognostic factor in epithelial ovarian cancer. BMB Rep. 2014, 47, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Ino, K.; Nawa, A.; Kikkawa, F. Involvement of SDF-1alpha/CXCR4 axis in the enhanced peritoneal metastasis of epithelial ovarian carcinoma. Int. J. Cancer 2008, 122, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Figueras, A.; Alsina-Sanchis, E.; Lahiguera, A.; Abreu, M.; Muinelo-Romay, L.; Moreno-Bueno, G.; Casanovas, O.; Graupera, M.; Matias-Guiu, X.; Vidal, A.; et al. A Role for CXCR4 in Peritoneal and Hematogenous Ovarian Cancer Dissemination. Mol. Cancer Ther. 2018, 17, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Cui, Z.M.; Zhang, J.; Huang, Y. Chemokine axes CXCL12/CXCR4 and CXCL16/CXCR6 correlate with lymph node metastasis in epithelial ovarian carcinoma. Chin. J. Cancer 2011, 30, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.P.; Vallee, A.; Robert, G.; Bonneau, J.; Leroy, C.; Varin-Blank, N.; Rio, A.G.; Troadec, M.B.; Galibert, M.D.; Gandemer, V. CD9, a key actor in the dissemination of lymphoblastic leukemia, modulating CXCR4-mediated migration via RAC1 signaling. Blood 2015, 126, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Mao, T.L.; Fan, K.F.; Liu, C.L. Targeting the CXCR4/CXCL12 axis in treating epithelial ovarian cancer. Gene Ther. 2017, 24, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.; Lewin, S.A.; Mihalko, L.A.; Schmidt, B.T.; Luker, K.E.; Luker, G.D. Noninvasive imaging reveals inhibition of ovarian cancer by targeting CXCL12-CXCR4. Neoplasia 2011, 13, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Righi, E.; Kashiwagi, S.; Yuan, J.; Santosuosso, M.; Leblanc, P.; Ingraham, R.; Forbes, B.; Edelblute, B.; Collette, B.; Xing, D.; et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 2011, 71, 5522–5534. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudson, L.G.; Gillette, J.M.; Kang, H.; Rivera, M.R.; Wandinger-Ness, A. Ovarian Tumor Microenvironment Signaling: Convergence on the Rac1 GTPase. Cancers 2018, 10, 358. https://doi.org/10.3390/cancers10100358
Hudson LG, Gillette JM, Kang H, Rivera MR, Wandinger-Ness A. Ovarian Tumor Microenvironment Signaling: Convergence on the Rac1 GTPase. Cancers. 2018; 10(10):358. https://doi.org/10.3390/cancers10100358
Chicago/Turabian StyleHudson, Laurie G., Jennifer M. Gillette, Huining Kang, Melanie R. Rivera, and Angela Wandinger-Ness. 2018. "Ovarian Tumor Microenvironment Signaling: Convergence on the Rac1 GTPase" Cancers 10, no. 10: 358. https://doi.org/10.3390/cancers10100358
APA StyleHudson, L. G., Gillette, J. M., Kang, H., Rivera, M. R., & Wandinger-Ness, A. (2018). Ovarian Tumor Microenvironment Signaling: Convergence on the Rac1 GTPase. Cancers, 10(10), 358. https://doi.org/10.3390/cancers10100358