The Unfolded Protein Response in Breast Cancer

 , , ,
, , ,  , and
, and 
Abstract
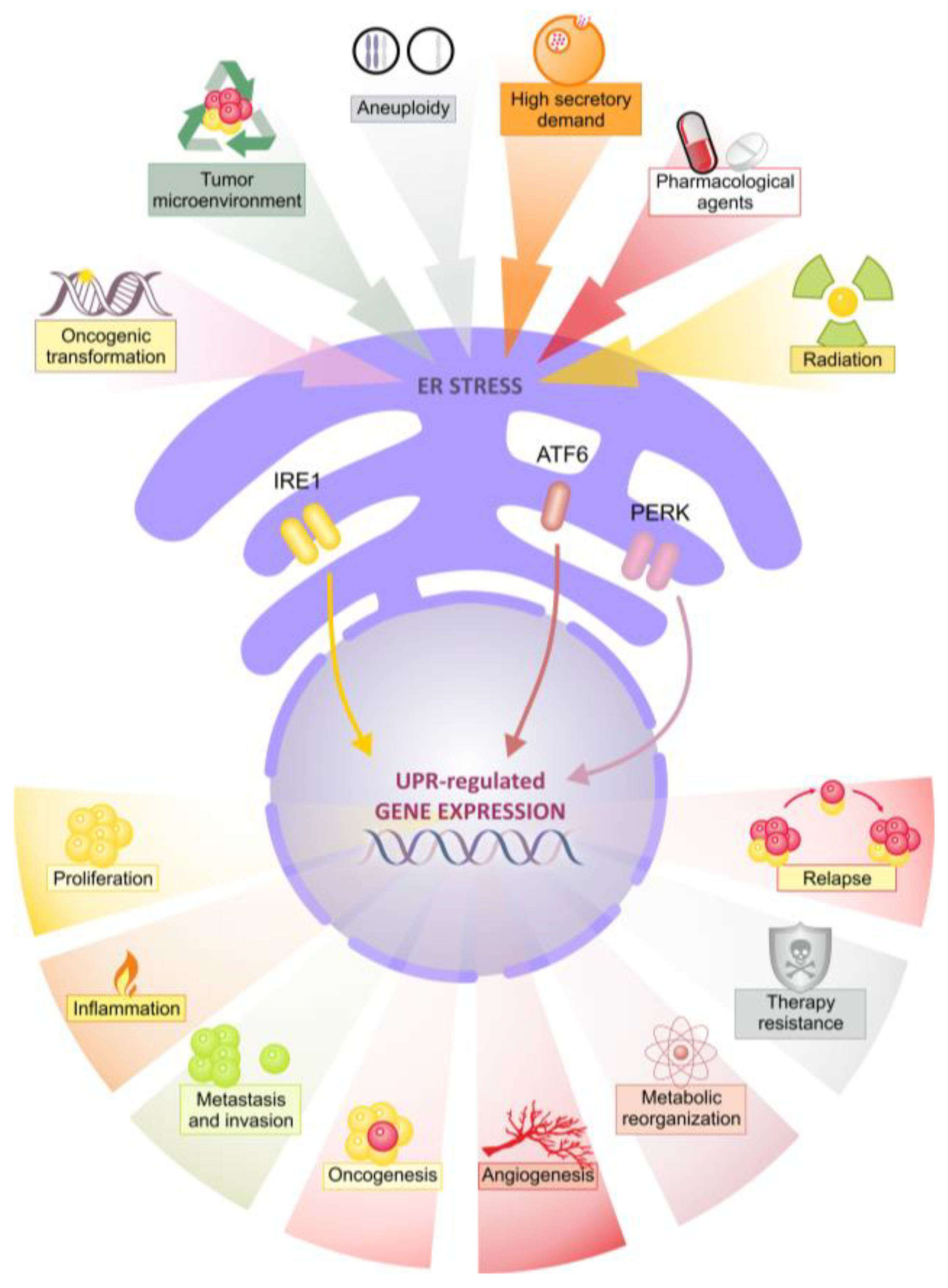
1. Introduction
2. The Unfolded Protein Reponses
3. Aberrant UPR Signaling in Breast Cancer
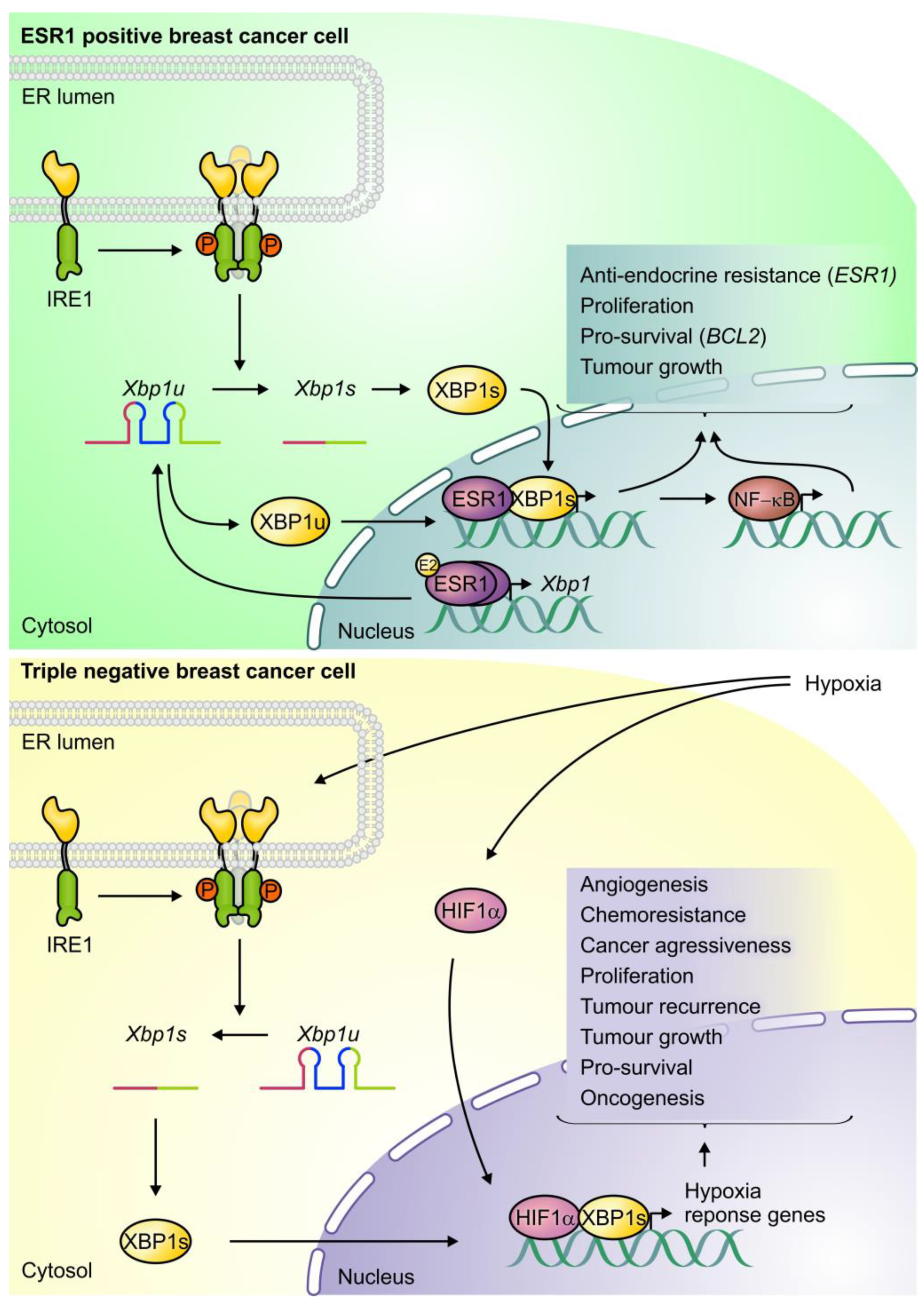
3.1. IRE1/XBP1s
3.2. PERK
3.3. ATF6
3.4. GRP78
4. UPR Signalling Promotes Therapy Resistance in Breast Cancer
4.1. IRE1/XBP1
4.2. PERK
4.3. ATF6
4.4. GRP78
5. UPR-Targeting Drugs: Stand-Alone and Combination Therapies
6. Future Perspectives and Challenges
7. Conclusions
Funding
Conflicts of Interest
References
- Sotiriou, C.; Pusztai, L. Gene-expression signatures in breast cancer. N. Engl. J. Med. 2009, 360, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Reis-Filho, J.S. Histological and molecular types of breast cancer: Is there a unifying taxonomy? Nat. Rev. Clin. Oncol. 2009, 6, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell. 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the hif1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Yanagitani, K.; Imagawa, Y.; Iwawaki, T.; Hosoda, A.; Saito, M.; Kimata, Y.; Kohno, K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell 2009, 34, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Pytel, D.; Majsterek, I.; Diehl, J.A. Tumor progression and the different faces of the PERK kinase. Oncogene 2016, 35, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.J.; van Hoef, V.; Jobava, R.; Elroy-Stein, O.; Valasek, L.S.; Cargnello, M.; Gao, X.H.; Krokowski, D.; Merrick, W.C.; Kimball, S.R.; et al. A unique ISR program determines cellular responses to chronic stress. Mol. Cell 2017, 68, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N.; Nakanishi, K.; Nakano, A. Activating transcription factor-6 (ATF6) mediates apoptosis with reduction of myeloid cell leukemia sequence 1 (MCL-1) protein via induction of WW domain binding protein 1. J. Biol. Chem. 2011, 286, 35227–35235. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumour cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Banh, A.; Papandreou, I.; Cao, H.; Galvez, M.G.; Gurtner, G.C.; Denko, N.C.; Le, Q.T.; Koong, A.C. Imaging the unfolded protein response in primary tumours reveals microenvironments with metabolic variations that predict tumour growth. Cancer Res. 2010, 70, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Xi, L.; Boye, S.L.; Hauswirth, W.W.; Han, S.; Chen, Z.J.; Lewin, A.S.; Boulton, M.E.; Law, B.K.; Jiang, W.G.; et al. Development of an anti-angiogenic therapeutic model combining scAAV2-delivered siRNAs and noninvasive photoacoustic imaging of tumour vasculature development. Cancer Lett. 2013, 332, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Andruska, N.; Zheng, X.; Yang, X.; Helferich, W.G.; Shapiro, D.J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene 2015, 34, 3760–3769. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Leclercq, G. About GATA3, HNF3A, AND XBP1, three genes co-expressed with the oestrogen receptor-alpha gene (ESR1) in breast cancer. Mol. Cell. Endocrinol. 2004, 219, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yan, J.; Zhu, J.; Zhong, H.; Lu, Q.; Wang, Z.; Huang, C.; Ye, Q. Ligand-independent activation of estrogen receptor alpha by XBP-1. Nucleic Acids Res. 2003, 31, 5266–5274. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yan, J.; Ding, L.; Liu, Y.; Zhu, J.; Huang, C.; Zhao, H.; Lu, Q.; Zhang, X.; Yang, X.; et al. XBP-1 increases ERalpha transcriptional activity through regulation of large-scale chromatin unfolding. Biochem. Biophys. Res. Commun. 2004, 323, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Warri, A.; Jin, L.; Zwart, A.; Riggins, R.B.; Fang, H.B.; Clarke, R. NF-κB signaling is required for XBP1 (unspliced and spliced)-mediated effects on antiestrogen responsiveness and cell fate decisions in breast cancer. Mol. Cell Biol. 2015, 35, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Sharma, C.G.N.; Jordan, V.C. Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells. Mol. Biol. Clin. Investig. 2010, 2, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-differentiation confers multidrug resistance via noncanonical PERK-NrF2 signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.F.; Mao, X.Y.; Wang, E.H. Elevated p-CREB-2 (ser 245) expression is potentially associated with carcinogenesis and development of breast carcinoma. Mol. Med. Rep. 2012, 5, 357–362. [Google Scholar] [PubMed]
- Ameri, K.; Lewis, C.E.; Raida, M.; Sowter, H.; Hai, T.; Harris, A.L. Anoxic induction of ATF-4 through HIF-1-independent pathways of protein stabilization in human cancer cells. Blood 2004, 103, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Z.; Cao, Z.G.; Hu, X.; Shao, Z.M. The endoplasmic reticulum stress markers GRP78 and CHOP predict disease-free survival and responsiveness to chemotherapy in breast cancer. Breast Cancer Res. Treat. 2014, 145, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumour growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.R.; Singleton, D.C.; Buffa, F.; Abramczyk, O.; Phadwal, K.; Li, J.L.; Simon, A.K.; Murray, J.T.; Harris, A.L. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J. 2013, 449, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Cojocari, D.; Keulers, T.G.; Jutten, B.; Starmans, M.H.; de Jong, M.C.; Begg, A.C.; Savelkouls, K.G.; Bussink, J.; Vooijs, M.; et al. The autophagy associated gene, ULK1, promotes tolerance to chronic and acute hypoxia. Radiother. Oncol. 2013, 108, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of Grp78/BiP by translational block: Activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Tai, W.C.; Bai, Y.; Poizat, C.; Lee, A.S. In vivo regulation of Grp78/BiP transcription in the embryonic heart: Role of the endoplasmic reticulum stress response element and GATA-4. J. Biol. Chem. 2006, 281, 8877–8887. [Google Scholar] [CrossRef] [PubMed]
- Kamagate, A.; Kim, D.H.; Zhang, T.; Slusher, S.; Gramignoli, R.; Strom, S.C.; Bertera, S.; Ringquist, S.; Dong, H.H. FoxO1 links hepatic insulin action to endoplasmic reticulum stress. Endocrinology 2010, 151, 3521–3535. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Wang, F.; Pizzo, S.V. Transcription factor TFII-I causes transcriptional upregulation of GRP78 synthesis in prostate cancer cells. J. Cell. Biochem. 2009, 106, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.G.; Ciznadija, D.; Mantamadiotis, T.; Anderson, R.; Pearson, R. Expression of stress response protein glucose regulated protein-78 mediated by c-Myb. Int. J. Biochem. Cell Biol. 2005, 37, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Park, Y.K.; Lee, J.H.; Park, K. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumour cells through a protein kinase C-epsilon/ERK/AP-1 signaling cascade. Cancer Res. 2001, 61, 8322–8330. [Google Scholar] [PubMed]
- Odani, N.; Negishi, M.; Takahashi, S.; Kitano, Y.; Kozutsumi, Y.; Ichikawa, A. Regulation of BIP gene expression by cyclopentenone prostaglandins through unfolded protein response element. J. Biol. Chem. 1996, 271, 16609–16613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tai, L.K.; Wong, L.L.; Putti, T.C.; Sethi, S.K.; Teh, M.; Koay, E.S. Proteomic characterization of differentially expressed proteins in breast cancer: Expression of hnRNP H1, RKIP and GRP78 is strongly associated with HER-2/neu status. Proteom. Clin. Appl. 2008, 2, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Gazit, G.; Lu, J.; Lee, A.S. De-regulation of grp stress protein expression in human breast cancer cell lines. Breast Cancer Res. Treat. 1999, 54, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Nami, B.; Ghasemi-Dizgah, A.; Vaseghi, A. Overexpression of molecular chaperons GRP78 and GRP94 in CD44(hi)/CD24(lo) breast cancer stem cells. BioImpacts 2016, 6, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.C.; Vale, W. Cripto/GRP78 modulation of the TGF-β pathway in development and oncogenesis. FEBS Lett. 2012, 586, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Kelber, J.; Panopoulos, A.; Shani, G.; Booker, E.; Belmonte, J.; Vale, W.; Gray, P. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Macias, A.T.; Williamson, D.S.; Allen, N.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Francis, G.L.; Graham, C.J.; et al. Adenosine-derived inhibitors of 78 kDa glucose regulated protein (GRP78) ATPase: Insights into isoform selectivity. J. Med. Chem. 2011, 54, 4034–4041. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Tseng, C.F.; Wang, M.Y.; Chang, W.C.; Lee, C.C.; Chen, L.T.; Hung, M.C.; Su, J.L. Deacetylation of HSPA5 by HDAC6 leads to GP78-mediated HSPA5 ubiquitination at K447 and suppresses metastasis of breast cancer. Oncogene 2016, 35, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Chen, H.A.; Tseng, C.F.; Hong, C.C.; Ma, J.T.; Hung, M.C.; Wu, C.H.; Huang, M.T.; Su, J.L. De-acetylation and degradation of HSPA5 is critical for E1A metastasis suppression in breast cancer cells. Oncotarget 2014, 5, 10558–10570. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. Cosmic: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Moretti, L.; Mitchell, L.R.; Jung, D.K.; Lu, B. Endoplasmic reticulum stress mediates radiation-induced autophagy by PERK-eiF2alpha in caspase-3/7-deficient cells. Oncogene 2010, 29, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- Notte, A.; Rebucci, M.; Fransolet, M.; Roegiers, E.; Genin, M.; Tellier, C.; Watillon, K.; Fattaccioli, A.; Arnould, T.; Michiels, C. Taxol-induced unfolded protein response activation in breast cancer cells exposed to hypoxia: ATF4 activation regulates autophagy and inhibits apoptosis. Int. J. Biochem. Cell Biol. 2015, 62, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Ni, J.; Song, D.; Ding, M.; Huang, J. Activation of unfolded protein response protects osteosarcoma cells from cisplatin-induced apoptosis through NF-κB pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 10204–10215. [Google Scholar] [PubMed]
- Wang, J.; Yin, Y.; Hua, H.; Li, M.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Zhang, Y.; Jiang, Y. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J. Cell. Mol. Med. 2009, 13, 3888–3897. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, X.A.; Hu, J.; Jiang, J.; Li, Y.; Chan-Salis, K.Y.; Gu, Y.; Chen, G.; Thomas, C.; Pugh, B.F.; et al. ATF4 gene network mediates cellular response to the anticancer PAD inhibitor YW3-56 in triple negative breast cancer cells. Mol. Cancer Ther. 2015, 14, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, N.; Tang, Y.; Booth, L.; Hamed, H.; Grant, S.; Dent, P. Lapatinib and obatoclax kill breast cancer cells through reactive oxygen species-dependent endoplasmic reticulum stress. Mol. Pharmacol. 2012, 82, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.P.; Riggins, R.B.; Shajahan, A.N.; Klimach, U.; Wang, A.; Crawford, A.C.; Zhu, Y.; Zwart, A.; Wang, M.; Clarke, R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007, 21, 4013–4027. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; van der Kogel, A.J.; Sweep, F.C.; Span, P.N. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother. Oncol. 2013, 108, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ko, B.; Baumeister, P.; Swenson, S.; Costa, F.; Markland, F.; Stiles, C.; Patterson, J.B.; Bates, S.E.; Lee, A.S. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumour microenvironment. Cancer Res. 2005, 65, 5785–5791. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Tao, Z.H.; Zhao, J.; Li, T.; Wu, Z.H.; Zhang, J.F.; Zhang, J.; Hu, X.C. Glucose regulated protein 78 (GRP78) inhibits apoptosis and attentinutes chemosensitivity of gemcitabine in breast cancer cell via AKT/mitochondrial apoptotic pathway. Biochem. Biophys. Res. Commun. 2016, 474, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Soto-Pantoja, D.R.; Clarke, P.A.G.; Cruz, M.I.; Zwart, A.; Wärri, A.; Hilakivi-Clarke, L.; Roberts, D.D.; Clarke, R. Endoplasmic reticulum stress protein GRP78 modulates lipid metabolism to control drug sensitivity and anti-tumour immunity in breast cancer. Cancer Res. 2016, 76, 5657–5670. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; McGrath, E.P.; Mnich, K.; Healy, S.; Chevet, E.; Samali, A. Controlling the unfolded protein response-mediated life and death decisions in cancer. Semin. Cancer Biol. 2015, 33, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Lynch, C.; Medeiros, B.C.; Liedtke, M.; Bam, R.; Tam, A.B.; Yang, Z.; Alagappan, M.; Abidi, P.; Le, Q.T.; et al. Identification of doxorubicin as an inhibitor of the ire1α-xbp1 axis of the unfolded protein response. Sci. Rep. 2016, 6, 33353. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Ruan, S.; Wang, M.; Ye, D.; Fan, N.; Meng, Q.; Tian, B.; Huang, T. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget 2015, 6, 40692–40703. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Romeril, S.P.; Shu, A.; Ralph, J.; Medina, J.R.; Feng, Y.; Li, W.H.H.; Grant, S.W.; Heerding, D.A.; Minthorn, E.; et al. Discovery of GSK2656157: An optimized PERK inhibitor. ACS Med. Chem. Lett. 2013, 4, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Andrews, K.L.; Beckmann, H.; Bellon, S.F.; Beltran, P.J.; Booker, S.; Chen, H.; Chung, Y.A.; D’Angelo, N.D.; Dao, J.; et al. Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2015, 58, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-mesenchymal transition activates PERK-eiF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.H.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the atf6α branch. eLife 2016, 5, e11878. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, L.; Brown, N.J.; Christian, S.; Hornby, D. Quantum dot-conjugated anti-GRP78 scFv inhibits cancer growth in mice. Molecules 2012, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Duell, J.; Castro, I.C.; Dubljevic, V.; Chatterjee, M.; Knop, S.; Hensel, F.; Rosenwald, A.; Einsele, H.; Topp, M.S.; et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma-results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica 2015, 100, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Trondl, R.; Heffeter, P.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, a first-in-class anticancer drug showing mild side effects and activity in patients suffering from advanced refractory cancer. BMC Pharmacol. Toxicol. 2012, 13, A82. [Google Scholar] [CrossRef]
- Kawiak, A.; Domachowska, A.; Jaworska, A.; Lojkowska, E. Plumbagin sensitizes breast cancer cells to tamoxifen-induced cell death through GRP78 inhibition and Bik upregulation. Sci. Rep. 2017, 7, 43781. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; Ding, L.W.; Sun, Q.Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.L.; Zhang, Y.; Lee, K.P.; Harding, H.P.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol. Cell 2010, 38, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Jha, B.K.; Polyakova, I.; Kessler, P.; Dong, B.; Dickerman, B.; Sen, G.C.; Silverman, R.H. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J. Biol. Chem. 2011, 286, 26319–26326. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Perera, B.G.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schurer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the ire1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Villalta, S.A.; Feldman, H.C.; Register, A.C.; Rosenthal, W.; Hoffmann-Petersen, I.T.; Mehdizadeh, M.; Ghosh, R.; Wang, L.; Colon-Negron, K.; et al. Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab. 2017, 25, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Waller, D.D.; Jansen, G.; Golizeh, M.; Martel-Lorion, C.; Dejgaard, K.; Shiao, T.C.; Mancuso, J.; Tsantrizos, Y.S.; Roy, R.; Sebag, M.; et al. A covalent cysteine-targeting kinase inhibitor of IRE1 permits allosteric control of endoribonuclease activity. Chembiochem 2016, 17, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Concha, N.O.; Smallwood, A.; Bonnette, W.; Totoritis, R.; Zhang, G.; Federowicz, K.; Yang, J.; Qi, H.; Chen, S.; Campobasso, N.; et al. Long-range inhibitor-induced conformational regulation of human IRE1α endoribonuclease activity. Mol. Pharmacol. 2015, 88, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Bouchecareilh, M.; Higa, A.; Fribourg, S.; Moenner, M.; Chevet, E. Peptides derived from the bifunctional kinase/RNase enzyme IRE1α modulate IRE1α activity and protect cells from endoplasmic reticulum stress. FASEB J. 2011, 25, 3115–3129. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.M.; Galson, D.L.; Roodman, G.D.; Ouyang, H. Resveratrol triggers the pro-apoptotic endoplasmic reticulum stress response and represses pro-survival XBP1 signaling in human multiple myeloma cells. Exp. Hematol. 2011, 39, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J.; et al. Acridine derivatives as inhibitors of the IRE1α-XBP1 pathway are cytotoxic to human multiple myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7h-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumour and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Pariollaud, M.; Wixted, W.E.; Chitnis, N.; Fornwald, J.; Truong, M.; Pao, C.; Liu, Y.; Ames, R.S.; Callahan, J.; et al. Identification and characterization of PERK activators by phenotypic screening and their effects on NRF2 activation. PLoS ONE 2015, 10, e0119738. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Chen, T.; Ming, J.; Sun, H.; Cao, P.; Fusco, D.N.; Chung, R.T.; Chorev, M.; Jin, Q.; Aktas, B.H. Dual activators of protein kinase R (PKR) and protein kinase R-like kinase PERK identify common and divergent catalytic targets. ChemBioChem 2013, 14, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Zyryanova, A.F.; Weis, F.; Faille, A.; Alard, A.A.; Crespillo-Casado, A.; Sekine, Y.; Harding, H.P.; Allen, F.; Parts, L.; Fromont, C.; et al. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eiF2B. Science 2018, 359, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eiF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Butkevich, A.N.; Yamada, R.; Zhou, Y.; Debnath, B.; Duncan, R.; Zandi, E.; Petasis, N.A.; Neamati, N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc. Natl. Acad. Sci. USA 2012, 109, 16348–16353. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Pace, N.J.; Brown, D.R.; Weerapana, E. 1,3,5-triazine as a modular scaffold for covalent inhibitors with streamlined target identification. J. Am. Chem. Soc. 2013, 135, 2497–2500. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Zhang, C.J.; Li, L.; Chong, L.M.; Wu, X.; Hao, P.; Sze, S.K.; Yao, S.Q. Small molecule probe suitable for in situ profiling and inhibition of protein disulfide isomerase. ACS Chem. Biol. 2013, 8, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Haze, K.; Nadanaka, S.; Yoshida, H.; Seidah, N.G.; Hirano, Y.; Sato, R.; Negishi, M.; Mori, K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem. 2003, 278, 31024–31032. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.J.; Yu, H.Q.; Fan, L.L.; Li, X.Q.; Wang, F.; Liu, J.T.; Zhong, F.; Zhang, C.J.; Wei, W.; Wang, H.; et al. Melatonin, a novel selective ATF-6 inhibitor, induces human hepatoma cell apoptosis through COX-2 downregulation. World J. Gastroenterol. 2017, 23, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.J.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Cash, D.R.; Tavallai, S.; Jean, S.; Fidanza, A.; Cruz-Luna, T.; Siembiba, P.; Cycon, K.A.; Cornelissen, C.N.; et al. GRP78/BiP/HSPA5/Dna K is a universal therapeutic target for human disease. J. Cell Physiol. 2015, 230, 1661–1676. [Google Scholar] [CrossRef] [PubMed]
- Choo, S.J.; Park, H.R.; Ryoo, I.J.; Kim, J.P.; Yun, B.S.; Kim, C.J.; Shin-ya, K.; Yoo, I.D. Deoxyverrucosidin, a novel GRP78/BiP down-regulator, produced by Penicillium sp. J. Antibiot. 2005, 58, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Fasano, E.; Serini, S.; Piccioni, E.; Toesca, A.; Monego, G.; Cittadini, A.R.; Ranelletti, F.O.; Calviello, G. DHA induces apoptosis by altering the expression and cellular location of GRP78 in colon cancer cell lines. Biochim. Biophys. Acta 2012, 1822, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, X.; Wang, C.; Nawaz, A.; Wei, W.; Li, J.; Wang, L.; Yu, D.H. Arctigenin suppresses unfolded protein response and sensitizes glucose deprivation-mediated cytotoxicity of cancer cells. Planta Med. 2011, 77, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Lhomond, S.; Avril, T.; Dejeans, N.; Voutetakis, K.; Doultsinos, D.; McMahon, M.; Pineau, R.; Obacz, J.; Papadodima, O.; Jouan, F.; et al. Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 2018, 10, e7929. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Okreglak, V.; Peschek, J.; Kimmig, P.; Zubradt, M.; Weissman, J.S.; Walter, P. Engineering ER-stress dependent non-conventional mRNA splicing. eLife 2018, 7, e35388. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumour cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Bevan, M.J. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J. Immunol. 2008, 181, 5433–5441. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Tavernier, S.J.; Hoffmann, E.; Saeys, Y.; Martens, L.; Vetters, J.; Delrue, I.; De Rycke, R.; Parthoens, E.; Pouliot, P.; et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat. Immunol. 2014, 15, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER stress sensor XBP1 controls anti-tumour immunity by disrupting dendritic cell homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Bettigole, S.E.; Lis, R.; Adoro, S.; Lee, A.H.; Spencer, L.A.; Weller, P.F.; Glimcher, L.H. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat. Immunol. 2015, 16, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Inositol-requiring enzyme (IRE1) | |
| Luminal domain | p.P75Q, p.A371A, p.H386fs*8 |
| Transmembrane domain | p.L454L |
| Cytoplasmic domain | p.Q495_L496insQ |
| Kinase domain | p.G703D, p.L714L, p.V767A, p.R806C, p.A823V, p.F937F |
| X-box binding protein 1 (XBP1) | |
| bZIP/nuclear localization signal | p.R81fs*16, p.R90P |
| bZIP/leucine zipper | p.E108delE, p.E121D |
| Translational pausing of own mRNA | p.L236fs*16, p.L238fs*13 |
| Other regions | p.P8P, p.P37A, p.Q43E, p.E97delE, p.S187fs*6, p.S190fs*1, p.P213fs*45, p.L232fs*22 |
| PKR-like ER Kinase (PERK) | |
| Luminal domain | p.R114I, p.S385R |
| Cytoplasmic domain | p.T537T, p.R588P, p.D1081fs*31, p.L1088L, p.S1098L |
| Cytoplasmic/kinase domain | p.S686F, p.C788C, p.R797T, p.R1027G, p.E1050D |
| Activating transcription factor 6 (ATF6) | |
| Cytoplasmic/transcription activation | p.E25Q |
| Cytoplasmic domain | p.Q237 * |
| Cytoplasmic/basic motif | p.R309K, p.K327N, |
| Cytoplasmic/bZIP | p.E365Q |
| Luminal domain | p.A450fs*7, p.C467fs*1, p.L477F, p.R484Q, p.S592S, p.R624S, p.S631L |
| Glucose-regulated protein 78 kDa (GRP78) | |
| Signal peptide | p.L13L |
| Nucleotide-binding domain | p.I132T, p.K138N, p.T166T, p.E243K |
| ATP-binding | p.A295fs*28 |
| Other regions | p.E308Q, p.E514Q, p.E603E |
| Inositol-Requiring Enzyme 1 (IRE1) | |
| RNase domain inhibition | Toyocamycin [93], MKC3946 [94], 4μ8c [95], 3-Methoxy-6-bromosalicyl-aldehyde [96], STF083010 [97], Doxorubicin [81], MKC8866 [24,30], B-H09 [98], 2-hydroxy-1-naphthaldehyde [99] |
| Q-site | Quercetin [100] |
| Kinase domain inhibition | APY29 [26], Sunitinib [101], Compound 3 [102], KIRA6 [26], KIRA8 [103], UPRM8 [104], GSK2850163 [105], FIRE [106] |
| Not determined | Resveratrol [107], 3,6-DMAD [108] |
| PKR-like ER Kinase (PERK) | |
| Kinase inhibition | GSK2606414 [109], GSK2656157 [110], AMG PERK 44 [84] |
| Kinase activation | Compounds A, B, C [111], DHBDC [112] |
| Inhibit downstream effect of EIF2A | ISRIB [113] |
| Promotes maintenance of EIF2A phosphorylation | Salubrinal [114], Guanlabenz [115] |
| Activating transcription factor 6 (ATF6) | |
| Inhibit nuclear translocation | CEAPIN Class 1 [87] |
| Inhibit transcriptional activity | CEAPIN Class 2 [87], |
| PDI inhibitor | PACMA 31 [116], RB11-ca [117], P1 [118], 16F16 [119] |
| Prevent AFT6 cleavage (Serine protease inhibitor) | AEBSF [120] |
| Not determined | Melatonin [121], Compounds 147, 263 [122] |
| Glucose-regulated protein 78 kDa (GRP78) | |
| Reduce GRP78 levels | OSU-03012 (AR-12) [123], Deoxyverrucosidin [124] Plumbagin [92], HA15 [88], DHA [125], |
| Inhibit GRP78 activity | PAT-SM6 [90] |
| Block GRP78 transcriptional induction | Arctigenin [126] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jäger, R.; Gorman, A.M.; Samali, A. The Unfolded Protein Response in Breast Cancer. Cancers 2018, 10, 344. https://doi.org/10.3390/cancers10100344
McGrath EP, Logue SE, Mnich K, Deegan S, Jäger R, Gorman AM, Samali A. The Unfolded Protein Response in Breast Cancer. Cancers. 2018; 10(10):344. https://doi.org/10.3390/cancers10100344
Chicago/Turabian StyleMcGrath, Eoghan P., Susan E. Logue, Katarzyna Mnich, Shane Deegan, Richard Jäger, Adrienne M. Gorman, and Afshin Samali. 2018. "The Unfolded Protein Response in Breast Cancer" Cancers 10, no. 10: 344. https://doi.org/10.3390/cancers10100344
APA StyleMcGrath, E. P., Logue, S. E., Mnich, K., Deegan, S., Jäger, R., Gorman, A. M., & Samali, A. (2018). The Unfolded Protein Response in Breast Cancer. Cancers, 10(10), 344. https://doi.org/10.3390/cancers10100344