Colorectal Cancers: An Update on Their Molecular Pathology


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Classification by Molecular Subtype
2.1. Integrated Molecular Characterization (TCGA Classification)
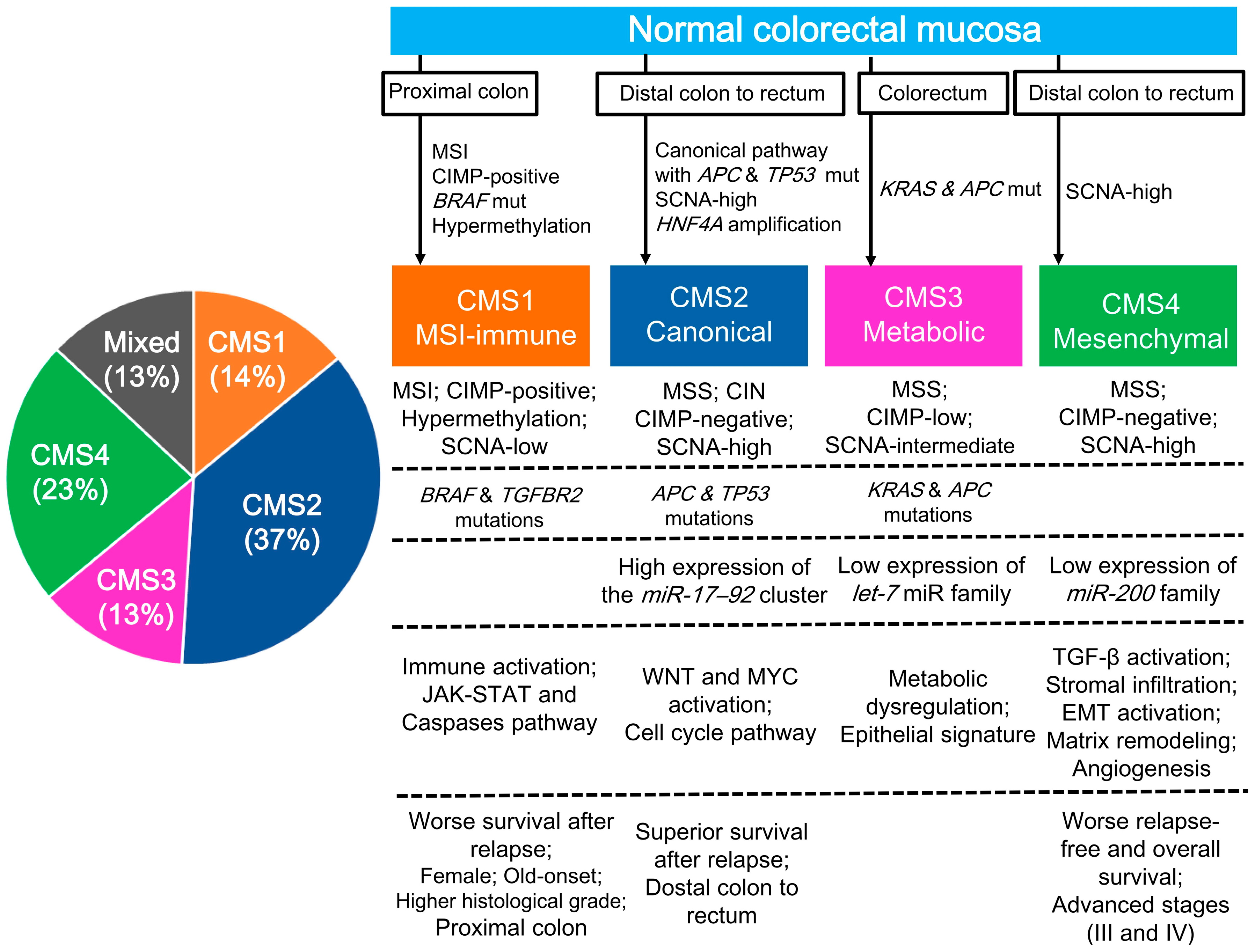
2.2. CRC Gene Expression Profiling (CMS Classification)
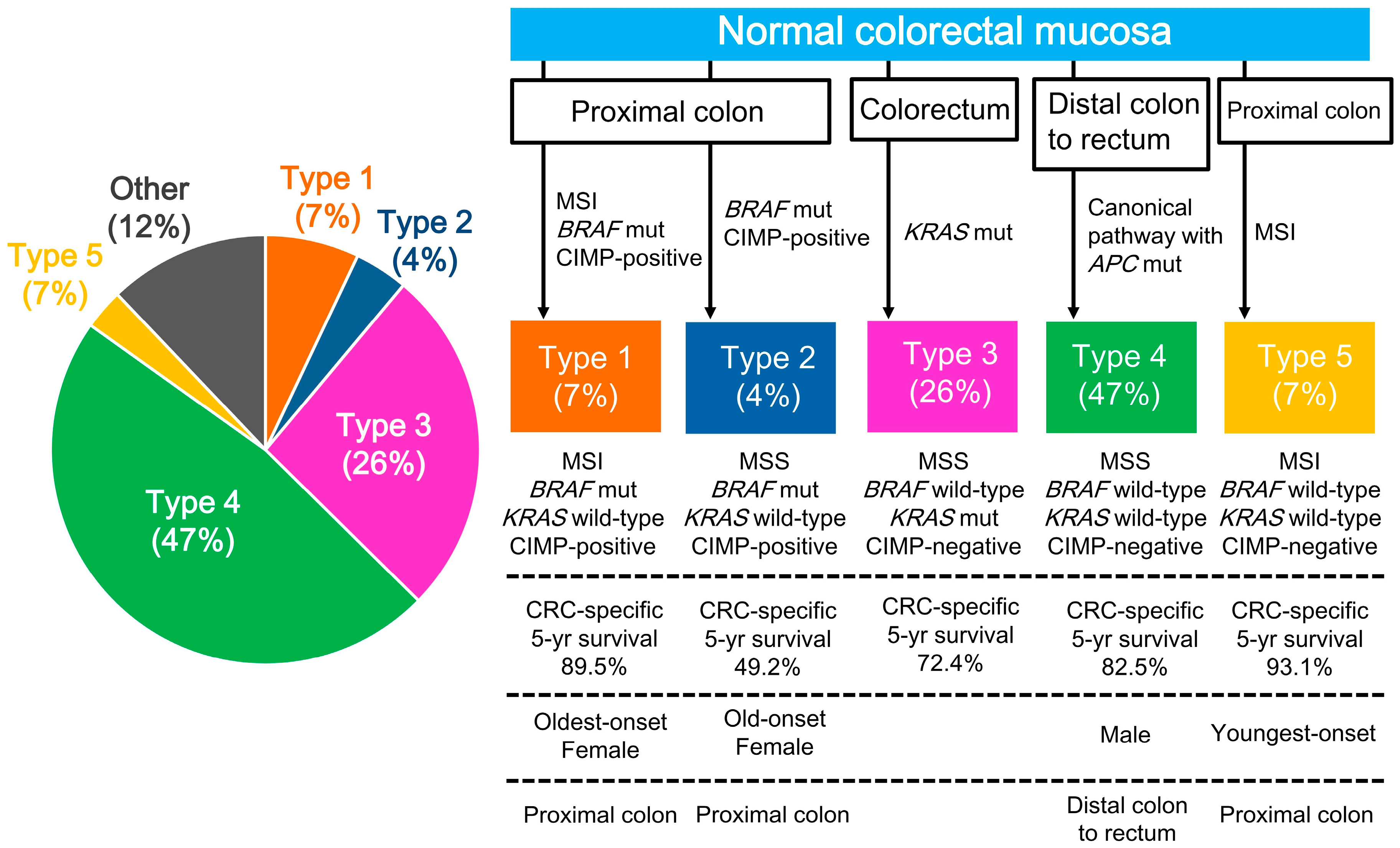
2.3. CRC Subtypes Classified by Key Molecular Features
3. Molecular Biomarkers
3.1. CIN
3.2. MSI
3.3. CIMP
3.4. POLE Mutations
3.5. LINE-1 Hypomethylation
3.6. RAS, BRAF, and PIK3CA Mutations in the MAPK/PIK3 Pathway
3.7. WNT/APC/CTNNB1/TGF-β Pathway
3.8. TP53 Mutations
3.9. Immune Biomarkers and the Microbiome
4. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| CIMP | CpG island methylator phenotype |
| CIN | chromosomal instability |
| CMS | consensus molecular subtype |
| CRC | colorectal cancer |
| EMT | epithelial–mesenchymal transition |
| FDA | Food and Drug Administration |
| LINE-1 | long interspersed nucleotide element-1 |
| mCRC | metastatic colorectal cancer |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| MSS | microsatellite stable |
| NCI | National Cancer Institute |
| NSAID | nonsteroidal anti-inflammatory drug |
| SCNA | somatic copy number alteration |
| SNP | single-nucleotide polymorphism |
| TCGA | The Cancer Genome Atlas |
References
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Shi, Q.; Smyrk, T.C.; Thibodeau, S.N.; Dienstmann, R.; Guinney, J.; Bot, B.M.; Tejpar, S.; Delorenzi, M.; Goldberg, R.M.; et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology 2015, 148, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.I.; Limburg, P.J.; Baron, J.A.; Burnett-Hartman, A.N.; Weisenberger, D.J.; Laird, P.W.; Sinicrope, F.A.; Rosty, C.; Buchanan, D.D.; Potter, J.D.; et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology 2015, 148, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Jung, B.H. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology 2015, 149, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Okamoto, K.; Kasi, P.M.; Kawakami, H. Molecular biomarkers in the personalized treatment of colorectal cancer. Clin. Gastroenterol. Hepatol. 2016, 14, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Han, S.W.; Cha, Y.; Bae, J.M.; Kim, H.P.; Lyu, J.; Han, H.; Kim, H.; Jang, H.; Bang, D.; et al. Association between mutations of critical pathway genes and survival outcomes according to the tumor location in colorectal cancer. Cancer 2017, 123, 3513–3523. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Datta, P.K. Regulation of EMT in colorectal cancer: A culprit in metastasis. Cancers (Basel) 2017, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, A.J.; Cadigan, K.M. The Role of the C-Clamp in Wnt-Related Colorectal Cancers. Cancers (Basel) 2016, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Boudjadi, S.; Bernatchez, G.; Senicourt, B.; Beausejour, M.; Vachon, P.H.; Carrier, J.C.; Beaulieu, J.F. Involvement of the Integrin alpha1beta1 in the Progression of Colorectal Cancer. Cancers (Basel) 2017, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, P.C.; Lin, C.C.; Lan, Y.T.; Lin, H.H.; Lin, C.H.; Yang, S.H.; Liang, W.Y.; Chen, W.S.; Jiang, J.K.; et al. Molecular and Clinicopathological Differences by Age at the Diagnosis of Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 1441. [Google Scholar] [CrossRef] [PubMed]
- Marmol, I.; Sanchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Punt, C.J.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Lochhead, P.; Nishihara, R.; Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Imamura, Y.; Qian, Z.R.; Baba, Y.; Shima, K.; et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N. Engl. J. Med. 2012, 367, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.M.; Coyle, V.M.; Kennedy, R.D.; Wilson, R.H. Molecular subtypes and personalized therapy in metastatic colorectal cancer. Curr. Colorectal. Cancer Rep. 2016, 12, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Hamada, T.; Cao, Y.; Qian, Z.R.; Masugi, Y.; Nowak, J.A.; Yang, J.; Song, M.; Mima, K.; Kosumi, K.; Liu, L.; et al. Aspirin use and colorectal cancer survival according to tumor CD274 (programmed cell death 1 ligand 1) Expression Status. J. Clin. Oncol. 2017, 35, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Hayashi, N.; Kuroda, Y.; Ito, S.; Eguchi, H.; Mimori, K. MicroRNAs as Biomarkers in Colorectal Cancer. Cancers (Basel) 2017, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Shirasawa, S.; Lazarova, D.L. In Hyperthermia Increased ERK and WNT Signaling Suppress Colorectal Cancer Cell Growth. Cancers (Basel) 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Matikas, A.; Asimakopoulou, N.; Georgoulias, V.; Souglakos, J. The place of targeted agents in the treatment of elderly patients with metastatic colorectal cancer. Cancers (Basel) 2015, 7, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Suyama, K.; Baba, H. Recent Advances in Targeting the EGFR Signaling Pathway for the Treatment of Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 752. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Oliver, P.G.; Lu, W.; Pathak, V.; Sridharan, S.; Augelli-Szafran, C.E.; Buchsbaum, D.J.; Suto, M.J. SRI36160 is a specific inhibitor of Wnt/beta-catenin signaling in human pancreatic and colorectal cancer cells. Cancer Lett. 2017, 389, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, K.; Robertson, G.R.; Sladek, F.M. HNF4alpha: A new biomarker in colon cancer? Biomark. Med. 2012, 6, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, T.; Matsushima, S.; Tsunoda, T.; Tahara, H.; Nakamura, Y.; Furukawa, Y. Identification of TOMM34, which shows elevated expression in the majority of human colon cancers, as a novel drug target. Int. J. Oncol. 2006, 29, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Mao, W.; Coppola, D.; Kang, J.; Loubeau, J.M.; Trudeau, W.; Karl, R.; Fujita, D.J.; Jove, R.; Yeatman, T.J. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 1999, 21, 187–190. [Google Scholar] [PubMed]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; Di Narzo, A.F.; Yan, P.; Hodgson, J.G.; Weinrich, S.; et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reynies, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene expression classification of colon cancer into molecular subtypes: Characterization, validation, and prognostic value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, A.; Beran, G.; Chresta, C.M.; McWalter, G.; Pritchard, A.; Weston, S.; Runswick, S.; Davenport, S.; Heathcote, K.; Castro, D.A.; et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med. Genom. 2012, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Vedeld, H.M.; Merok, M.; Jeanmougin, M.; Danielsen, S.A.; Honne, H.; Presthus, G.K.; Svindland, A.; Sjo, O.H.; Hektoen, M.; Eknaes, M.; et al. CpG island methylator phenotype identifies high risk patients among microsatellite stable BRAF mutated colorectal cancers. Int. J. Cancer 2017, 141, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Kuipers, J.; Rosenberg, C.; Smits, R.; Kielman, M.; Gaspar, C.; van Es, J.H.; Breukel, C.; Wiegant, J.; Giles, R.H.; et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat. Cell Biol. 2001, 3, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009, 58, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.N.; Lai, J.C.W.; Ho, S.L.; Leung, W.K.; Law, W.L.; Lee, J.F.Y.; Chan, A.K.W.; Tsui, W.Y.; Chan, A.S.Y.; Lee, B.C.H.; et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2017, 66, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite instability testing and its role in the management of colorectal cancer. Curr. Treat. Options Oncol. 2015, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Gavin, P.G.; Paik, S.; Yothers, G.; Pogue-Geile, K.L. Colon cancer mutation: Prognosis/prediction–response. Clin. Cancer Res. 2013, 19, 1301. [Google Scholar] [CrossRef] [PubMed]
- Collura, A.; Lagrange, A.; Svrcek, M.; Marisa, L.; Buhard, O.; Guilloux, A.; Wanherdrick, K.; Dorard, C.; Taieb, A.; Saget, A.; et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil-based chemotherapy. Gastroenterology 2014, 146, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Dorard, C.; de Thonel, A.; Collura, A.; Marisa, L.; Svrcek, M.; Lagrange, A.; Jego, G.; Wanherdrick, K.; Joly, A.L.; Buhard, O.; et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat. Med. 2011, 17, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- American Association for Cancer Research. First tissue-agnostic drug approval issued. Cancer Discov. 2017, 7, 656. [Google Scholar]
- Gelsomino, F.; Barbolini, M.; Spallanzani, A.; Pugliese, G.; Cascinu, S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat. Rev. 2016, 51, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, P.; Della Pepa, C.; Berardi, S.; Califano, D.; Scala, S.; Buonaguro, L.; Ciliberto, G.; Brauchli, P.; Pignata, S. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated Endometrial Cancers: New candidates for checkpoint blockade immunotherapy? Cancer Treat. Rev. 2016, 48, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Jansen, L.; Walter, V.; Tagscherer, K.; Roth, W.; Herpel, E.; Kloor, M.; Blaker, H.; Chang-Claude, J.; Brenner, H.; et al. No association of CpG island methylator phenotype and colorectal cancer survival: Population-based study. Br. J. Cancer 2016, 115, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Gao, X.; Zhang, Y.; Hoffmeister, M.; Brenner, H. Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: A systematic review. Clin. Epigenet. 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- East, J.E.; Atkin, W.S.; Bateman, A.C.; Clark, S.K.; Dolwani, S.; Ket, S.N.; Leedham, S.J.; Phull, P.S.; Rutter, M.D.; Shepherd, N.A.; et al. British Society of Gastroenterology position statement on serrated polyps in the colon and rectum. Gut 2017, 66, 1181–1196. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, R.; Wu, K.; Lochhead, P.; Morikawa, T.; Liao, X.; Qian, Z.R.; Inamura, K.; Kim, S.A.; Kuchiba, A.; Yamauchi, M.; et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N. Engl. J. Med. 2013, 369, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Soler, M.; Perez-Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barbera, V.M.; Juarez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 2013, 144, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Popanda, O.; Zheng, C.; Magdeburg, J.R.; Buttner, J.; Flohr, T.; Hagmuller, E.; Thielmann, H.W. Mutation analysis of replicative genes encoding the large subunits of DNA polymerase alpha and replication factors A and C in human sporadic colorectal cancers. Int. J. Cancer 2000, 86, 318–324. [Google Scholar] [CrossRef]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Stenzinger, A.; Pfarr, N.; Endris, V.; Penzel, R.; Jansen, L.; Wolf, T.; Herpel, E.; Warth, A.; Klauschen, F.; Kloor, M.; et al. Mutations in POLE and survival of colorectal cancer patients—Link to disease stage and treatment. Cancer Med. 2014, 3, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.C.; Lin, M.T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J. Natl. Compr. Cancer Netw. 2017, 15, 142–147. [Google Scholar] [CrossRef]
- Chen, R.Z.; Pettersson, U.; Beard, C.; Jackson-Grusby, L.; Jaenisch, R. DNA hypomethylation leads to elevated mutation rates. Nature 1998, 395, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Rodic, N.; Burns, K.H. Long interspersed element-1 (LINE-1): Passenger or driver in human neoplasms? PLoS Genet. 2013, 9, e1003402. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Estecio, M.R.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.P. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Aschacher, T.; Wolf, B.; Bergmann, M. A role of LINE-1 in telomere regulation. Front. Biosci. 2018, 23, 1310–1319. [Google Scholar]
- Baba, Y.; Huttenhower, C.; Nosho, K.; Tanaka, N.; Shima, K.; Hazra, A.; Schernhammer, E.S.; Hunter, D.J.; Giovannucci, E.L.; Fuchs, C.S.; et al. Epigenomic diversity of colorectal cancer indicated by LINE-1 methylation in a database of 869 tumors. Mol. Cancer 2010, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Yamauchi, M.; Nishihara, R.; Lochhead, P.; Qian, Z.R.; Kuchiba, A.; Kim, S.A.; Mima, K.; Sukawa, Y.; Jung, S.; et al. Tumor LINE-1 methylation level and microsatellite instability in relation to colorectal cancer prognosis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.C.; Gardner, E.J.; Masood, A.; Chuang, N.T.; Vertino, P.M.; Devine, S.E. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res. 2016, 26, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Sunami, E.; Yamamoto, Y.; Hata, K.; Okada, S.; Murono, K.; Yasuda, K.; Otani, K.; Nishikawa, T.; Tanaka, T.; et al. LINE-1 hypomethylation status of circulating cell-free DNA in plasma as a biomarker for colorectal cancer. Oncotarget 2017, 8, 11906–11916. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Yamauchi, M.; Nishihara, R.; Kim, S.A.; Mima, K.; Sukawa, Y.; Li, T.; Yasunari, M.; Zhang, X.; Wu, K.; et al. Prognostic significance and molecular features of signet-ring cell and mucinous components in colorectal carcinoma. Ann. Surg. Oncol. 2015, 22, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.P.; Sutton, P.A.; Evans, J.P.; Clifford, R.; McAvoy, A.; Lewis, J.; Rousseau, A.; Mountford, R.; McWhirter, D.; Malik, H.Z. Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br. J. Cancer 2017, 116, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Takane, K.; Akagi, K.; Fukuyo, M.; Yagi, K.; Takayama, T.; Kaneda, A. DNA methylation epigenotype and clinical features of NRAS-mutation(+) colorectal cancer. Cancer Med. 2017, 6, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Van Brummelen, E.M.J.; de Boer, A.; Beijnen, J.H.; Schellens, J.H.M. BRAF mutations as predictive biomarker for response to anti-EGFR monoclonal antibodies. Oncologist 2017, 22, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.M.; Wang, Y.; Wang, Y.L.; Wang, Y.; Liu, T.; Ni, M.; Li, M.S.; Lin, L.; Ge, F.J.; Gong, C.; et al. PIK3CA Mutations Contribute to Acquired Cetuximab Resistance in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 4602–4616. [Google Scholar] [CrossRef] [PubMed]
- Mouradov, D.; Domingo, E.; Gibbs, P.; Jorissen, R.N.; Li, S.; Soo, P.Y.; Lipton, L.; Desai, J.; Danielsen, H.E.; Oukrif, D.; et al. Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am. J. Gastroenterol. 2013, 108, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Passiglia, F.; Bronte, G.; Bazan, V.; Galvano, A.; Vincenzi, B.; Russo, A. Can KRAS and BRAF mutations limit the benefit of liver resection in metastatic colorectal cancer patients? A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2017, 99, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Mahoney, M.R.; Yoon, H.H.; Smyrk, T.C.; Thibodeau, S.N.; Goldberg, R.M.; Nelson, G.D.; Sargent, D.J.; Alberts, S.R. Analysis of Molecular Markers by Anatomic Tumor Site in Stage III Colon Carcinomas from Adjuvant Chemotherapy Trial NCCTG N0147 (Alliance). Clin. Cancer Res. 2015, 21, 5294–5304. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Le Malicot, K.; Shi, Q.; Penault Lorca, F.; Bouche, O.; Tabernero, J.; Mini, E.; Goldberg, R.M.; Folprecht, G.; Luc Van Laethem, J.; et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J. Natl. Cancer Inst. 2016, 109. [Google Scholar] [CrossRef]
- Inamura, K.; Song, M.; Jung, S.; Nishihara, R.; Yamauchi, M.; Lochhead, P.; Qian, Z.R.; Kim, S.A.; Mima, K.; Sukawa, Y.; et al. Prediagnosis plasma adiponectin in relation to colorectal cancer risk according to KRAS mutation status. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Summers, M.G.; Smith, C.G.; Maughan, T.S.; Kaplan, R.; Escott-Price, V.; Cheadle, J.P. BRAF and NRAS locus-specific variants have different outcomes on survival to colorectal cancer. Clin. Cancer Res. 2017, 23, 2742–2749. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med. 2007, 356, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin use and survival after diagnosis of colorectal cancer. JAMA 2009, 302, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.T.; Cantwell, M.M.; Coleman, H.G.; Loughrey, M.B.; Bankhead, P.; McQuaid, S.; O’Neill, R.F.; Arthur, K.; Bingham, V.; McGready, C.; et al. Evaluation of PTGS2 Expression, PIK3CA Mutation, Aspirin Use and Colon Cancer Survival in a Population-Based Cohort Study. Clin. Transl. Gastroenterol. 2017, 8, e91. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Phipps, A.I.; Burnett-Hartman, A.N.; Adams, S.V.; Hardikar, S.; Cohen, S.A.; Kocarnik, J.M.; Ahnen, D.J.; Lindor, N.M.; Baron, J.A.; et al. Timing of Aspirin and Other Nonsteroidal Anti-Inflammatory Drug Use Among Patients With Colorectal Cancer in Relation to Tumor Markers and Survival. J. Clin. Oncol. 2017, 35, 2806–2813. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Hutter, C.M.; Lin, Y.; Jacobs, E.J.; Ulrich, C.M.; White, E.; Baron, J.A.; Berndt, S.I.; Brenner, H.; Butterbach, K.; et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA 2015, 313, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.P.; Yamauchi, M.; Nishihara, R.; Jung, S.; Kuchiba, A.; Wu, K.; Cho, E.; Giovannucci, E.; Fuchs, C.S.; Ogino, S.; et al. Aspirin and the risk of colorectal cancer in relation to the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD). Sci. Transl. Med. 2014, 6, 233re2. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pasche, B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007, 16, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.H.; Likhter, M.; Jogunoori, W.; Belkin, M.; Ohshiro, K.; Mishra, L. TGF-beta signaling in liver and gastrointestinal cancers. Cancer Lett. 2016, 379, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Poleszczuk, J.; Suarez-Carmona, M.; Krisam, J.; Charoentong, P.; Valous, N.A.; Weis, C.A.; Tavernar, L.; Leiss, F.; Herpel, E.; et al. In silico modeling of immunotherapy and stroma-targeting therapies in human colorectal cancer. Cancer Res. 2017, 77, 6442–6452. [Google Scholar] [CrossRef] [PubMed]
- Nebot-Bral, L.; Brandao, D.; Verlingue, L.; Rouleau, E.; Caron, O.; Despras, E.; El-Dakdouki, Y.; Champiat, S.; Aoufouchi, S.; Leary, A.; et al. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur. J. Cancer 2017, 84, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Boland, P.M.; Ma, W.W. Immunotherapy for Colorectal Cancer. Cancers (Basel) 2017, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Roelands, J.; Kuppen, P.J.K.; Vermeulen, L.; Maccalli, C.; Decock, J.; Wang, E.; Marincola, F.M.; Bedognetti, D.; Hendrickx, W. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int. J. Mol. Sci. 2017, 18, 2229. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Di Caro, G.; Marchesi, F.; Laghi, L.; Grizzi, F. Immune cells: Plastic players along colorectal cancer progression. J. Cell. Mol. Med. 2013, 17, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Nishihara, R.; Qian, Z.R.; Tabung, F.K.; Nevo, D.; Zhang, X.; Song, M.; Cao, Y.; Mima, K.; Masugi, Y.; et al. Association Between Inflammatory Diet Pattern and Risk of Colorectal Carcinoma Subtypes Classified by Immune Responses to Tumor. Gastroenterology 2017. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Nishihara, R.; Yang, J.; Mima, K.; da Silva, A.; Shi, Y.; Inamura, K.; Cao, Y.; Song, M.; Nowak, J.A.; et al. Tumour CD274 (PD-L1) expression and T cells in colorectal cancer. Gut 2017, 66, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Barnich, N.; Nguyen, H.T.T. Microbiota, Inflammation and Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 1310. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.V.; Visnovska, M.; Biggs, P.J.; Schmeier, S.; Frizelle, F.A. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci. Rep. 2017, 7, 11590. [Google Scholar] [CrossRef] [PubMed]
- Alwers, E.; Jia, M.; Kloor, M.; Blaker, H.; Brenner, H.; Hoffmeister, M. Associations between molecular classifications of colorectal cancer and patient survival: A systematic review. Clin. Gastroenterol. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inamura, K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers 2018, 10, 26. https://doi.org/10.3390/cancers10010026
Inamura K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers. 2018; 10(1):26. https://doi.org/10.3390/cancers10010026
Chicago/Turabian StyleInamura, Kentaro. 2018. "Colorectal Cancers: An Update on Their Molecular Pathology" Cancers 10, no. 1: 26. https://doi.org/10.3390/cancers10010026
APA StyleInamura, K. (2018). Colorectal Cancers: An Update on Their Molecular Pathology. Cancers, 10(1), 26. https://doi.org/10.3390/cancers10010026