Protein Kinase A Distribution Differentiates Human Glioblastoma from Brain Tissue


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
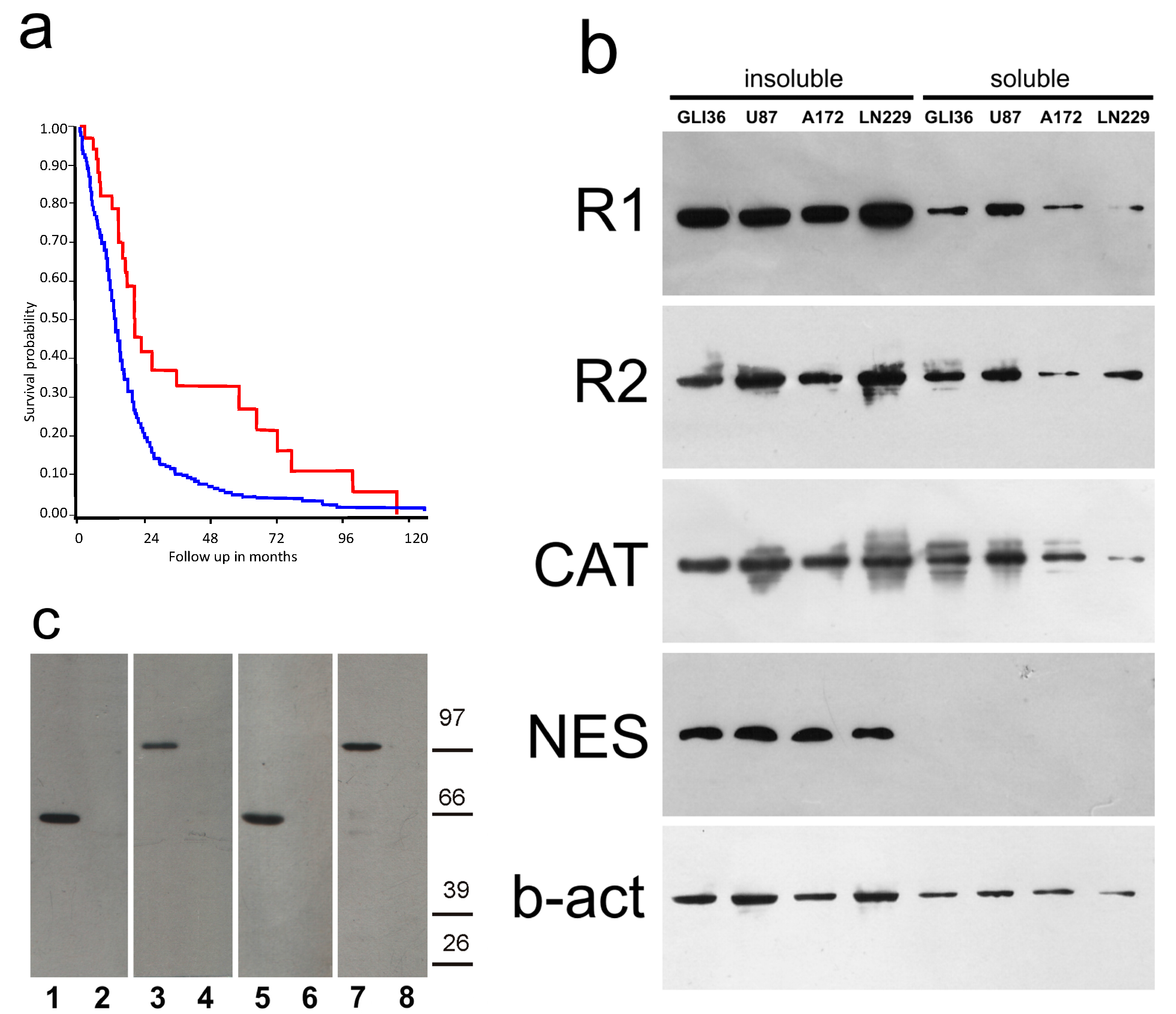
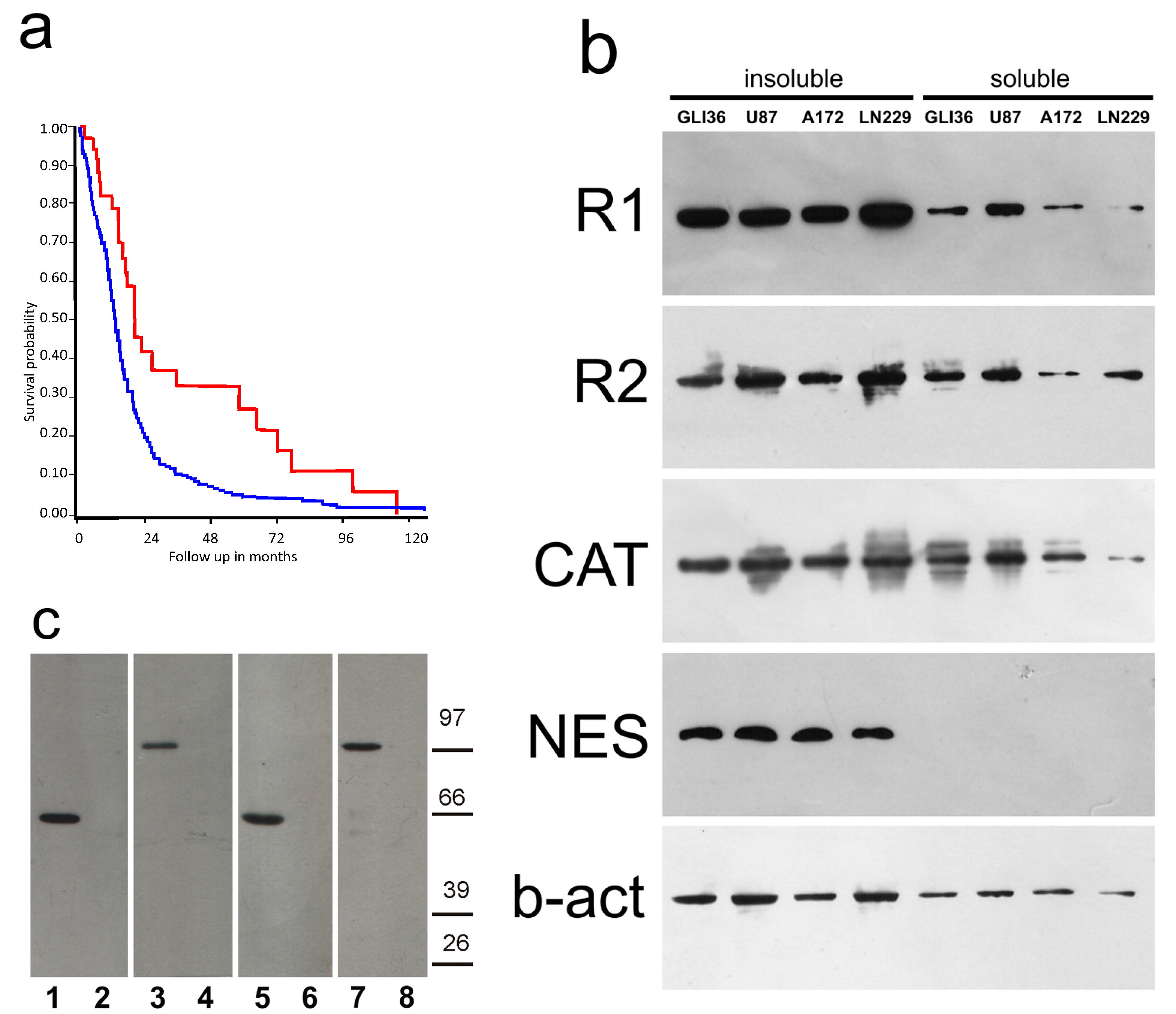
2.1. PKA Gene Expression
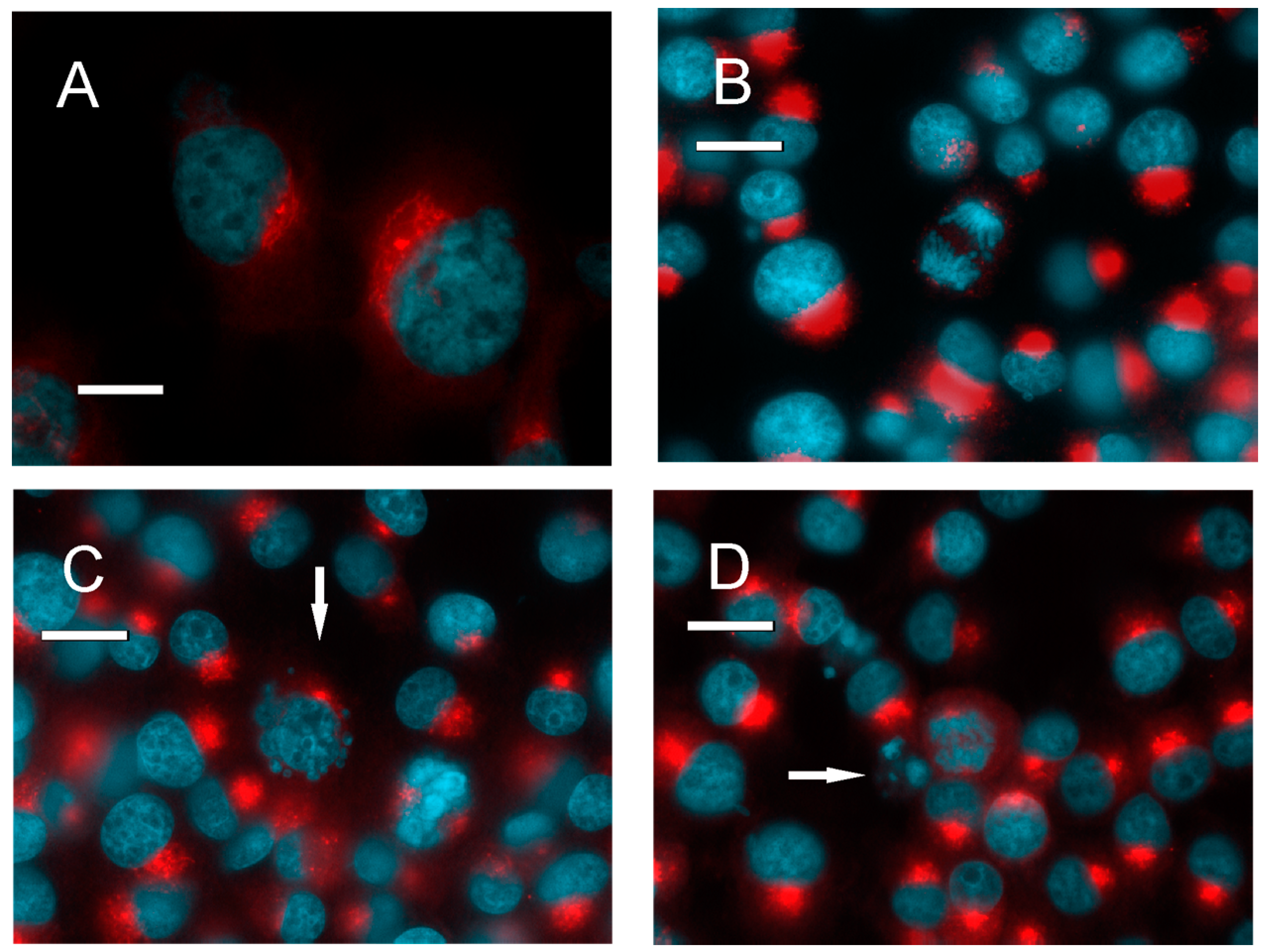
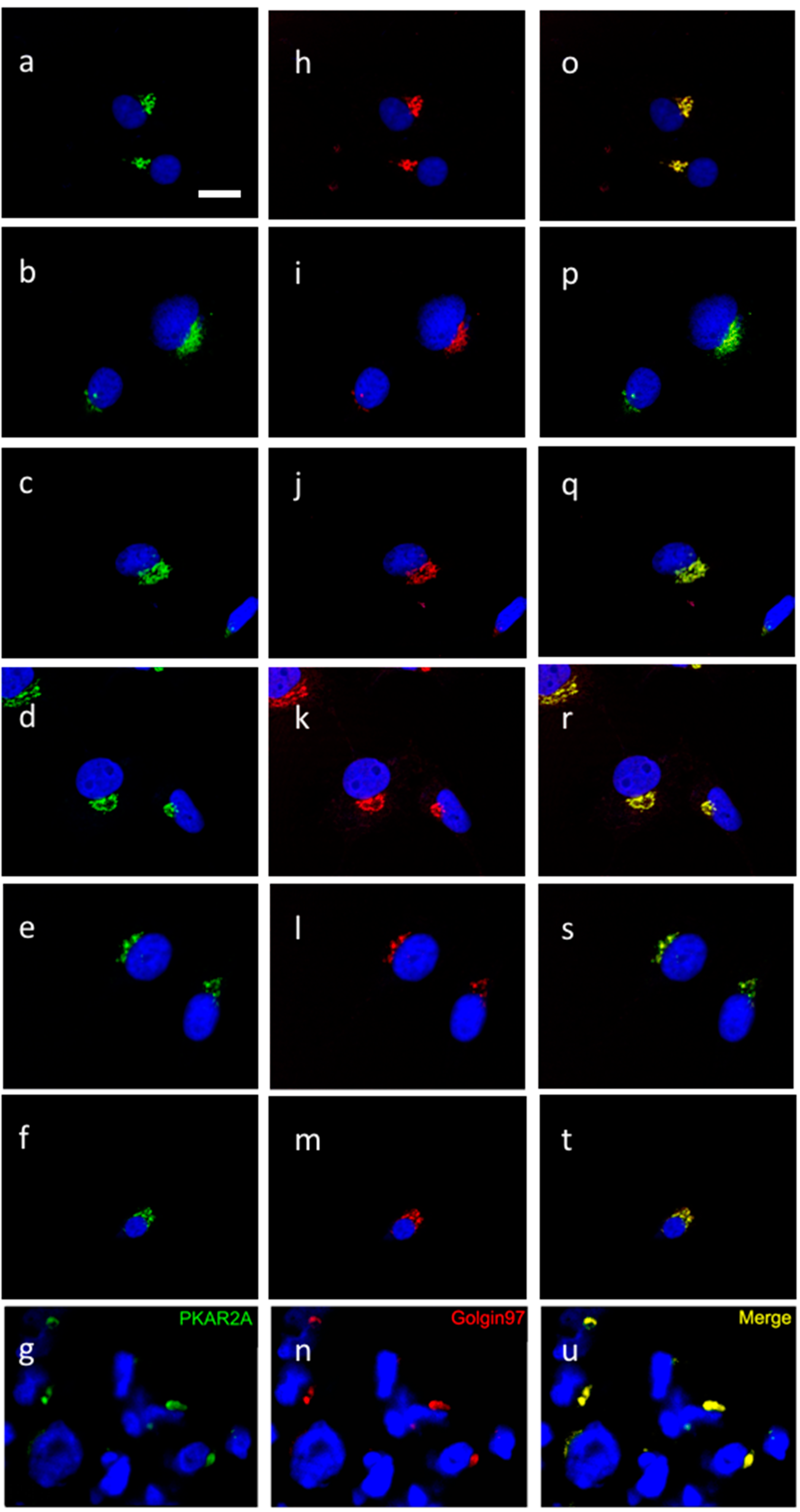
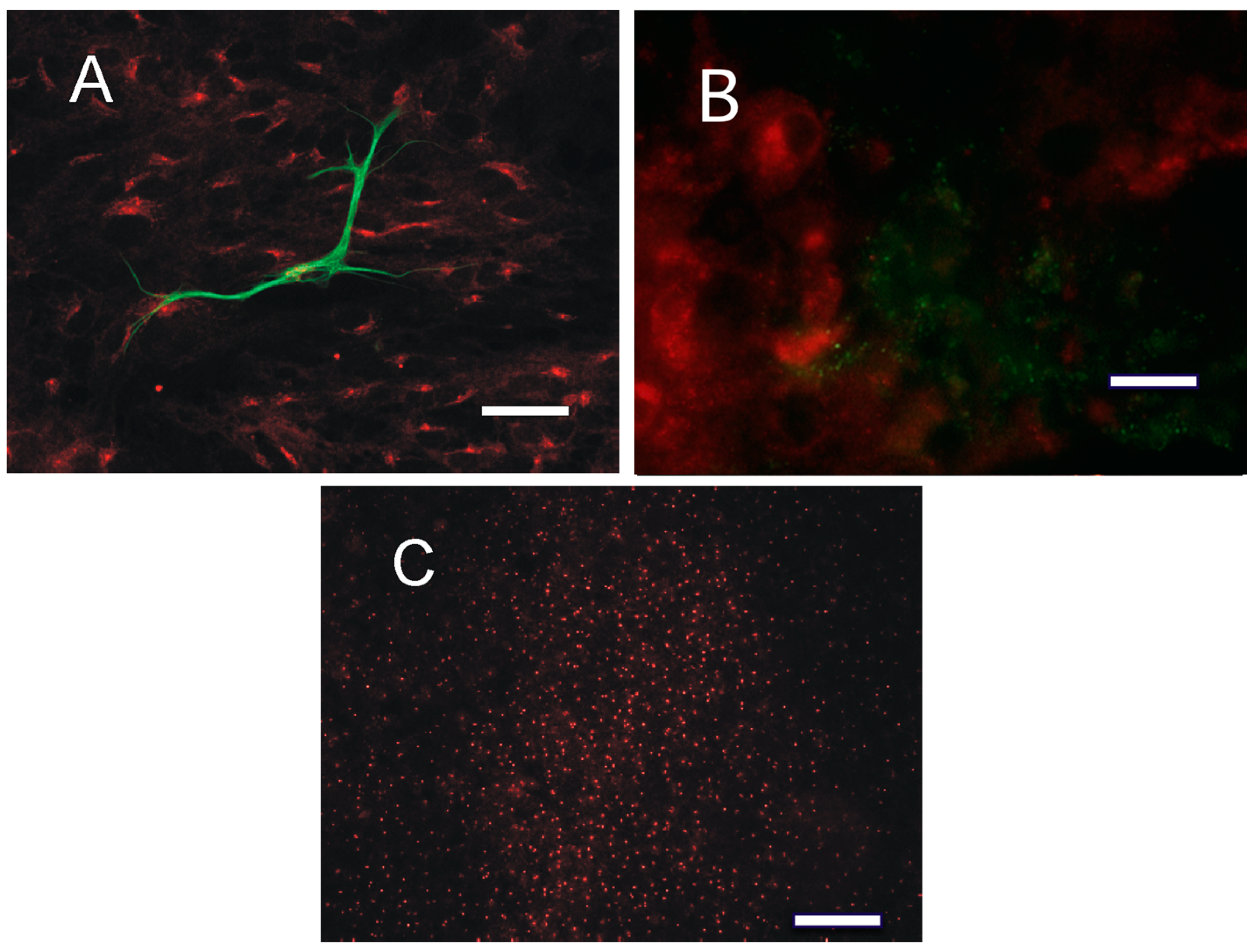
2.2. PKA Distribution in Glioma Cell Cultures
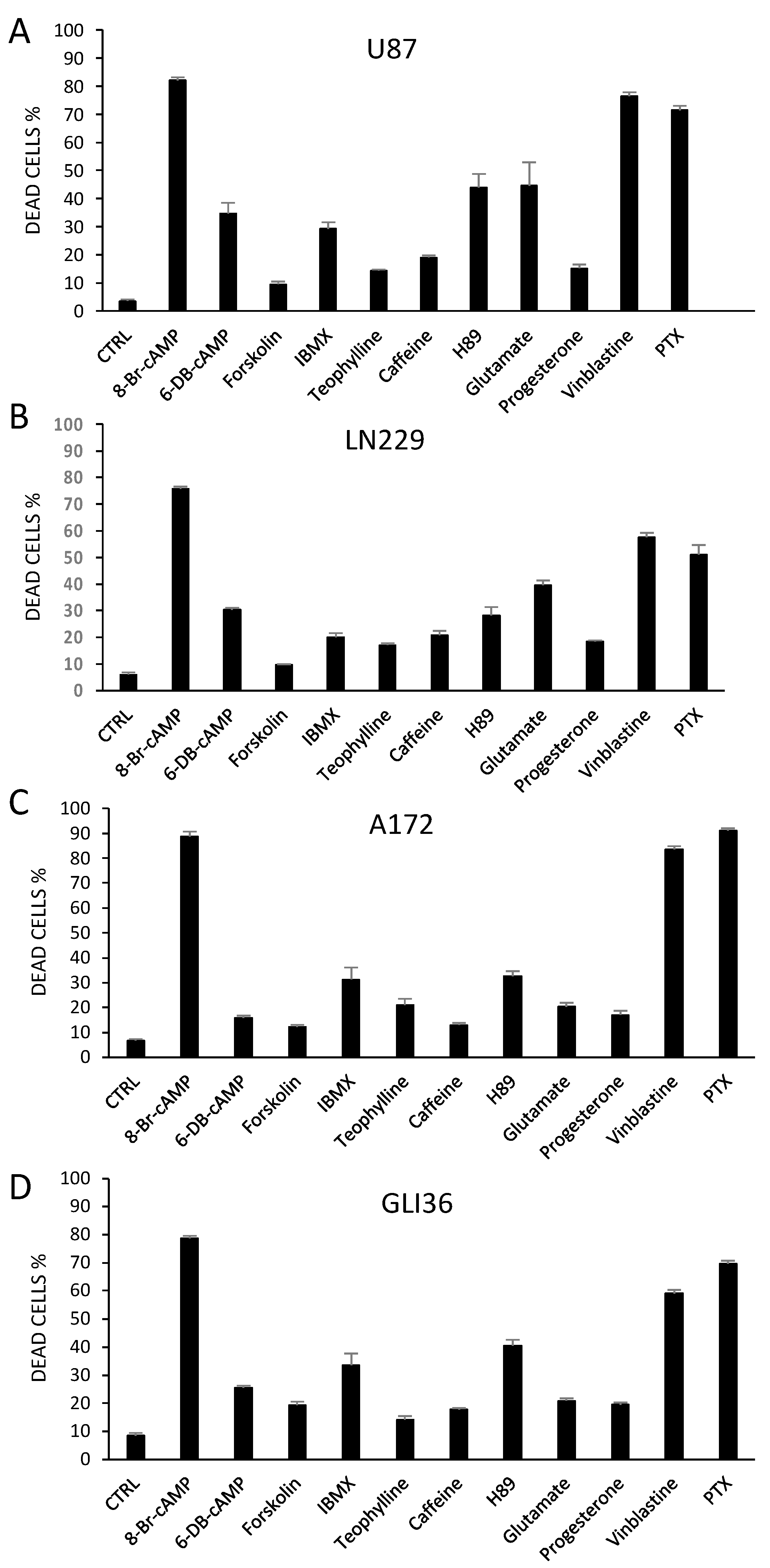
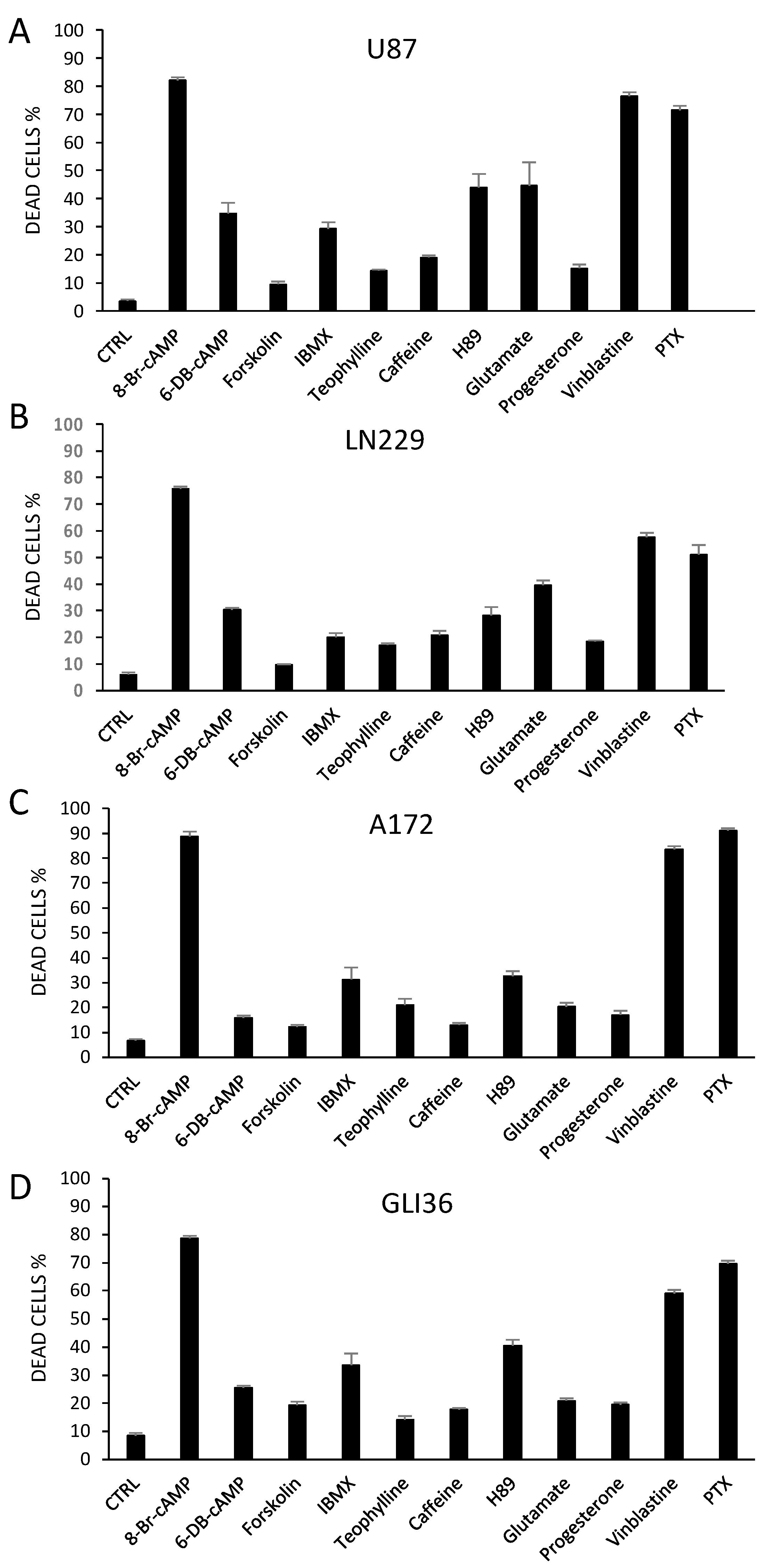
2.3. Altering PKA Activity Results in Cell Death
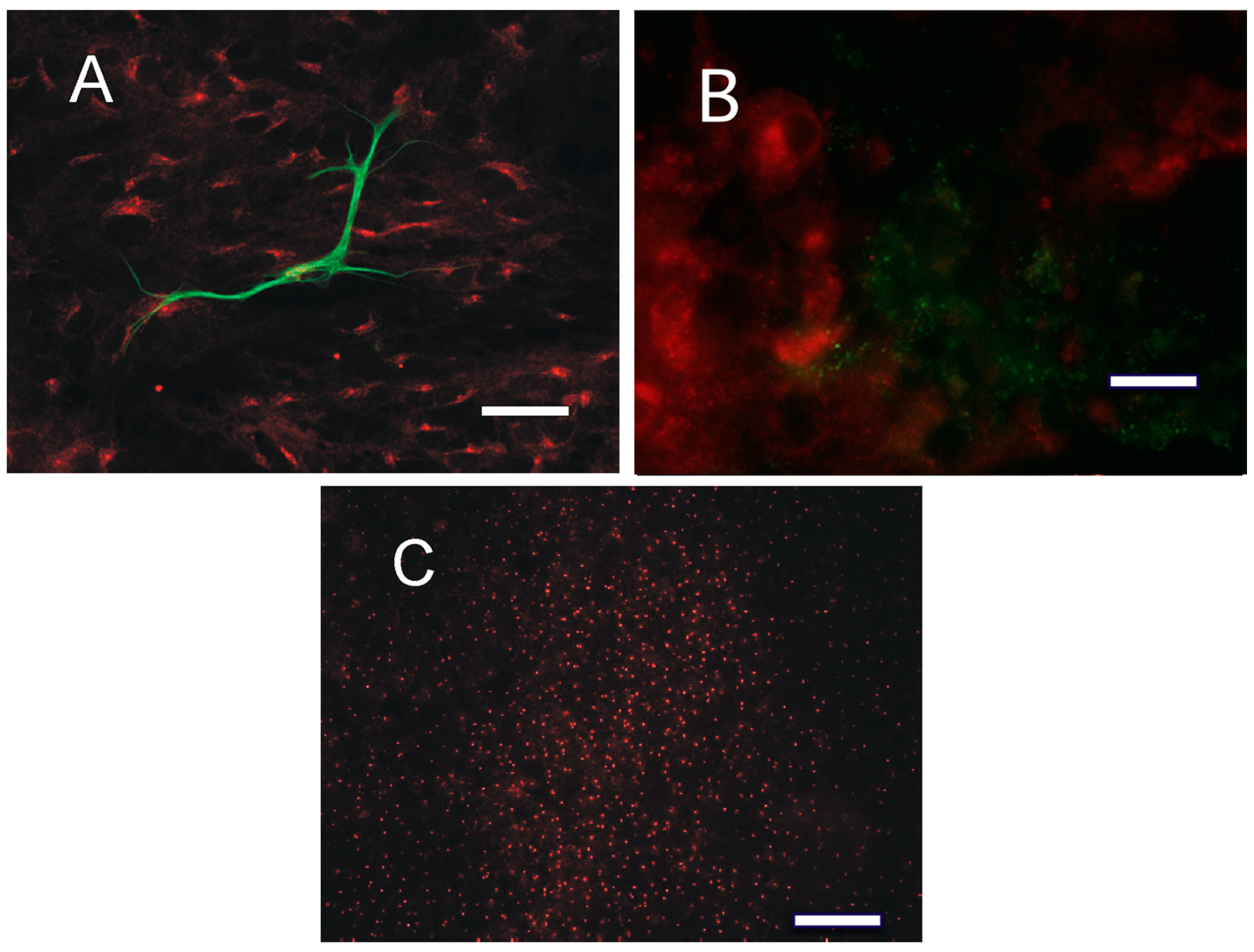
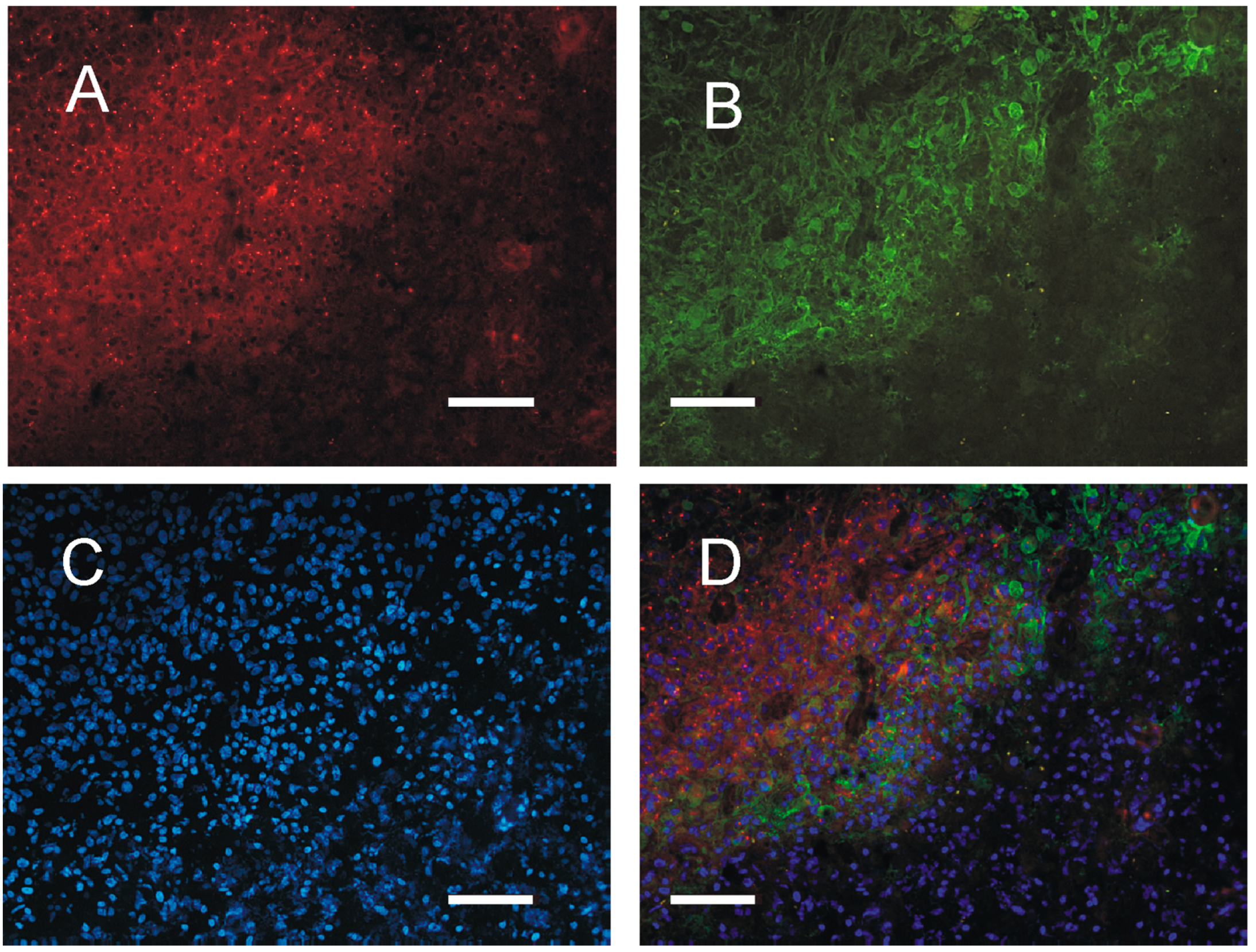
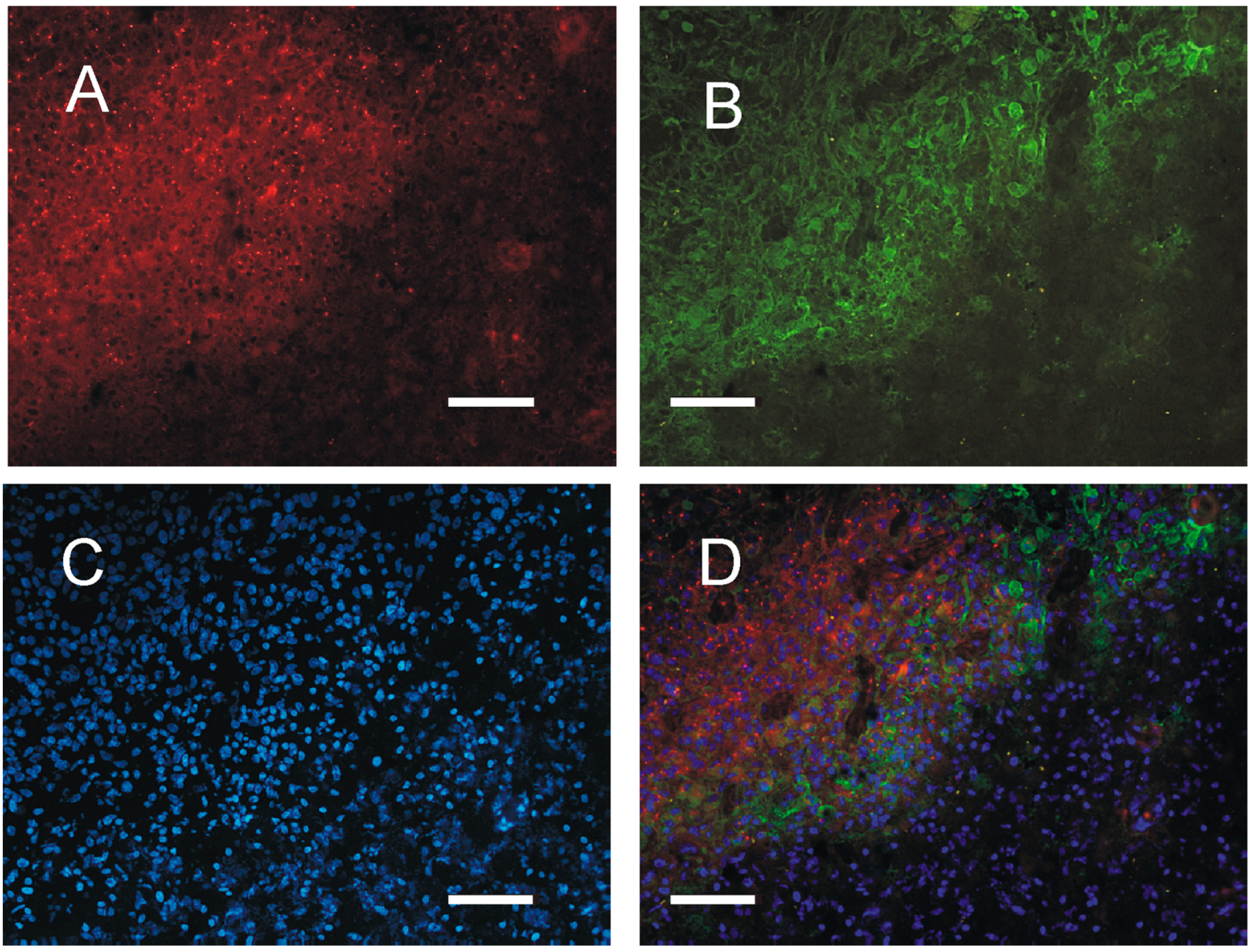
2.4. Distribution of PKA in the Tumor Tissue
3. Discussion
4. Materials and Methods
4.1. Analysis of Gene Expression
4.2. Patients
4.3. Chemicals
4.4. Cell Cultures and Pharmacological Treatments
4.5. Cell Death Count
4.6. Western Blots
4.7. Immunocytochemistry and Immunohistochemistry
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| cAMP | cyclic AMP |
| PKA | Protein kinase A, cAMP-dependent protein kinase |
| DMEM | Dulbecco Modified Eagle’s Medium |
| GFAP | Glial fibrillary acidic protein |
| IBMX | Isobutylmethylxantine |
| GBM | Glioblastoma multiforme |
| R | Regulatory subunits of cAMP dependent protein kinases |
References
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. Task force on gliomas. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef]
- Kleppe, R.; Krakstad, C.; Selheim, F.; Kopperud, R.; Døskeland, S.O. The cAMP-dependent protein kinase pathway as therapeutic target: Possibilities and pitfalls. Curr. Top. Med. Chem. 2011, 11, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S. Protein kinases controlling the onset of mitosis. Cell. Mol. Life Sci. 2006, 63, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Chiaradonna, F.; Balestrieri, C.; Gaglio, D.; Vanoni, M. RAS and PKA pathways in cancer: New insight from transcriptional analysis. Front. Biosci. 2008, 13, 5257–5278. [Google Scholar] [CrossRef] [PubMed]
- Canton, D.A.; Scott, J.D. Anchoring proteins encounter mitotic kinases. Cell Cycle 2013, 12, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.S.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia 2005, 7, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Odreman, F.; Vindigni, M.; Gonzales, M.L.; Niccolini, B.; Candiano, G.; Zanotti, B.; Skrap, M.; Pizzolitto, S.; Stanta, G.; Vindigni, A. Proteomic studies on low- and high-grade human brain astrocytomas. J. Proteome Res. 2005, 4, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ilouz, R.; Lev-Ram, V.; Bushong, E.A.; Stiles, T.L.; Friedmann-Morvinski, D.; Douglas, C.; Goldberg, G.; Ellisman, M.H.; Taylor, S.S. Isoform-specific subcellular localization and function of protein kinase A identified by mosaic imaging of mouse brain. eLife 2017, 12, E17681. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, K.; Mehta, S.; Ramamurthy, S.; Ronnett, G.V.; Zhou, F.Q.; Zhang, J. AKAP-mediated feedback control of cAMP gradients in developing hippocampal neurons. Nat. Chem. Biol. 2017, 13, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Localization of Triton-insoluble cAMP-dependent kinase type RIbeta in rat and mouse brain. J. Neurocytol. 2001, 30, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Clustered distribution of cAMP-dependent protein kinase regulatory isoform RIalpha during the development of the rat brain. J. Comp. Neurol. 2002, 451, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Regional variations in the localization of insoluble kinase A regulatory isoforms during rodent brain development. J. Chem. Neuroanat. 2004, 27, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Cavaggioni, A.; Redaelli, M.; Malatesta, M.; Zancanaro, C.; Caretta, A. Selective Distribution of Protein Kinase A Regulatory Subunit RIIAlpha in Rodent Gliomas. Neuro-Oncology 2008, 10, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Hinton, D.R.; Zidovetzki, R.; Hofman, F.M. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab. Investig. 1998, 78, 165–174. [Google Scholar] [PubMed]
- Mucignat-Caretta, C.; Denaro, L.; Redaelli, M.; D’Avella, D.; Caretta, A. Protein kinase A regulatory subunit distribution in medulloblastoma. BMC Cancer 2010, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Caretta, A.; Mucignat-Caretta, C. Protein kinase a in Cancer. Cancers 2011, 3, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Zorzan, M.; Giordan, E.; Redaelli, M.; Caretta, A.; Mucignat-Caretta, C. Molecular targets in glioblastoma. Future Oncol. 2015, 11, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Ohgaki, H.; Kleihues, P.; Collins, V.P. Molecular pathogenesis of astrocytic tumours. J. Neurooncol. 2004, 70, 137–160. [Google Scholar] [CrossRef] [PubMed]
- Doskeland, S.O.; Maronde, E.; Gjertsen, B.T. The genetic subtypes of cAMP-dependent protein kinase–functionally different or redundant? Biochim. Biophys. Acta 1993, 1178, 249–258. [Google Scholar] [CrossRef]
- Frattola, L.; Canal, N.; Ferrarese, C.; Tonini, C.; Tonon, G.; Villani, R.; Trabucchi, M. Multiple forms of protein kinase from normal human brain and glioblastoma. Cancer Res. 1983, 43, 1321–1324. [Google Scholar] [PubMed]
- Schwartz, J.P.; Costa, E. Protein kinase translocation following beta-adrenergic receptor activation in C6 glioma cells. J. Biol. Chem. 1980, 255, 2943–2948. [Google Scholar] [PubMed]
- Hussain, M.; Tang, F.; Liu, J.; Zhang, J.; Javeed, A. Dichotomous role of protein kinase A type I (PKAI) in the tumor microenvironment: A potential target for ‘two-in-one’ cancer chemoimmunotherapeutics. Cancer Lett. 2015, 369, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, C.; Alberghina, L.; Vanoni, M.; Chiaradonna, F. Data recovery and integration from public databases uncovers transformation-specific transcriptional downregulation of cAMP-PKA pathway-encoding genes. BMC Bioinform. 2009, 10 (Suppl. S12), S1. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Cabrera, M.; Vega, L.; Hidalgo, J.; Velasco, A. Golgi structural stability and biogenesis depend on associated PKA activity. J. Cell Sci. 2006, 119, 3764–3775. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Liau, B.B.; Wallace, L.C.; Morton, A.R.; Xie, Q.; Dixit, D.; Factor, D.C.; Kim, L.J.Y.; Morrow, J.J.; Wu, Q.; et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 2017, 5. [Google Scholar] [CrossRef]
- Feng, H.; Li, Y.; Yin, Y.; Zhang, W.; Hou, Y.; Zhang, L.; Li, L.; Xie, B.; Gao, W.Q.; Sarkaria, J.N.; et al. Protein kinase A-dependent phosphorylation of Dock180 at serine residue 1250 is important for glioma growth and invasion stimulated by platelet derived-growth factor receptor α. Neuro-Oncology 2015, 17, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.J.; Ball, M.; Andrade, M.F.; McDermid, A.; Stanimirovic, D.B. Insulin-like growth factor binding protein-4 (IGFBP-4) is a novel anti-angiogenic and anti-tumorigenic mediator secreted by dibutyryl cyclic AMP (dB-cAMP)-differentiated glioblastoma cells. Glia 2006, 53, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Wadsten, P.; Su, S.; Rawlinson, N.; Hofman, F.M.; Hill, C.K.; Schönthal, A.H. The type IV phosphodiesterase inhibitor rolipram induces expression of the cell cycle inhibitors p21(Cip1) and p27(Kip1), resulting in growth inhibition, increased differentiation, and subsequent apoptosis of malignant A-172 glioma cells. Cancer Biol. Ther. 2002, 1, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yin, W.; Wang, X.; Zhu, W.; Huang, Y.; Yan, G. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 13438–13443. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Onuma, T.; Birukawa, N.; Abe, M.; Ito, E.; Chen, Z.; Urano, A. Change of morphology and cytoskeletal protein gene expression during dibutyryl cAMP-induced differentiation in C6 glioma cells. Cell. Mol. Neurobiol. 2008, 28, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.N.; Cole, W.C.; Yan, X.D.; Nahreini, P.; Kumar, B.; Hanson, A.; Prasad, J.E. Defects in cAMP-pathway may initiate carcinogenesis in dividing nerve cells: A review. Apoptosis 2003, 8, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Helmbrecht, K.; Rensing, L. Different constitutive heat shock protein 70 expression during proliferation and differentiation of rat C6 glioma cells. Neurochem. Res. 1999, 24, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Choi, M.R.; Song, D.K.; Huh, S.O.; Kim, Y.H.; Suh, H.W. Activation of adenylate cyclase results in down-regulation of c-jun mRNA expression in rat C6 glioma cells. Neurosci. Lett. 1999, 276, 53–56. [Google Scholar] [CrossRef]
- Kuzhikandathil, E.V.; Molloy, G.R. Proximal promoter of the rat brain creatine kinase gene lacks a consensus CRE element but is essential for the cAMP-mediated increased transcription in glioblastoma cells. J. Neurosci. Res. 1999, 56, 371–385. [Google Scholar] [CrossRef]
- You, F.; Osawa, Y.; Hayashi, S.; Nakashima, S. Immediate early gene IEX-1 induces astrocytic differentiation of U87-MG human glioma cells. J. Cell Biochem. 2007, 100, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Anciaux, K.; Van Dommelen, K.; Nicolai, S.; Van Mechelen, E.; Slegers, H. Cyclic AMP-mediated induction of the glial fibrillary acidic protein is independent of protein kinase A activation in rat C6 glioma. J. Neurosci. Res. 1997, 48, 324–333. [Google Scholar] [CrossRef]
- Oh-hashi, K.; Hirata, Y.; Koga, H.; Kiuchi, K. GRP78-binding protein regulates cAMP-induced glial fibrillary acidic protein expression in rat C6 glioblastoma cells. FEBS Lett. 2006, 580, 3943–3947. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Qiu, J.; Li, Q.; Shi, Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends Cancer 2017, 3, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Payner, T.; Leaver, H.A.; Knapp, B.; Whittle, I.R.; Trifan, O.C.; Miller, S.; Rizzo, M.T. Microsomal prostaglandin E synthase-1 regulates human glioma cell growth via prostaglandin E(2)-dependent activation of type II protein kinase A. Mol. Cancer Ther. 2006, 5, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Kashima, T.; Vinters, H.V.; Campagnoni, A.T. Unexpected expression of intermediate filament protein genes in human oligodendroglioma cell lines. J. Neuropathol. Exp. Neurol. 1995, 54, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, M.; Mucignat-Caretta, C.; Cavaggioni, A.; Caretta, A.; D’Avella, D.; Denaro, L.; Cavirani, S.; Donofrio, G. Bovine herpesvirus 4 based vector as a potential oncolytic-virus for treatment of glioma. Virol. J. 2010, 7, 298. [Google Scholar] [CrossRef] [PubMed]
- Seligman, A.M.; Shear, M.J. Experimental production of brain tumors in mice with methylcholanthrene. Am. J. Cancer 1939, 37, 364–395. [Google Scholar]
- Newcomb, E.W.; Zagzag, D. The murine GL261 glioma experimental model to assess novel brain tumor treatments. In CNS Cancer: Models, Markers, Prognostic Factors, Targets and Therapeutic Approaches; Meir, E.G., Ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 227–241. [Google Scholar]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mucignat-Caretta, C.; Denaro, L.; D’Avella, D.; Caretta, A. Protein Kinase A Distribution Differentiates Human Glioblastoma from Brain Tissue. Cancers 2018, 10, 2. https://doi.org/10.3390/cancers10010002
Mucignat-Caretta C, Denaro L, D’Avella D, Caretta A. Protein Kinase A Distribution Differentiates Human Glioblastoma from Brain Tissue. Cancers. 2018; 10(1):2. https://doi.org/10.3390/cancers10010002
Chicago/Turabian StyleMucignat-Caretta, Carla, Luca Denaro, Domenico D’Avella, and Antonio Caretta. 2018. "Protein Kinase A Distribution Differentiates Human Glioblastoma from Brain Tissue" Cancers 10, no. 1: 2. https://doi.org/10.3390/cancers10010002
APA StyleMucignat-Caretta, C., Denaro, L., D’Avella, D., & Caretta, A. (2018). Protein Kinase A Distribution Differentiates Human Glioblastoma from Brain Tissue. Cancers, 10(1), 2. https://doi.org/10.3390/cancers10010002