Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics

{kind=link}
{kind=link}
Abstract
:1. Introduction: The Alarming Context of Pancreatic Cancer
2. PDAC Therapeutics: Challenges and Opportunities
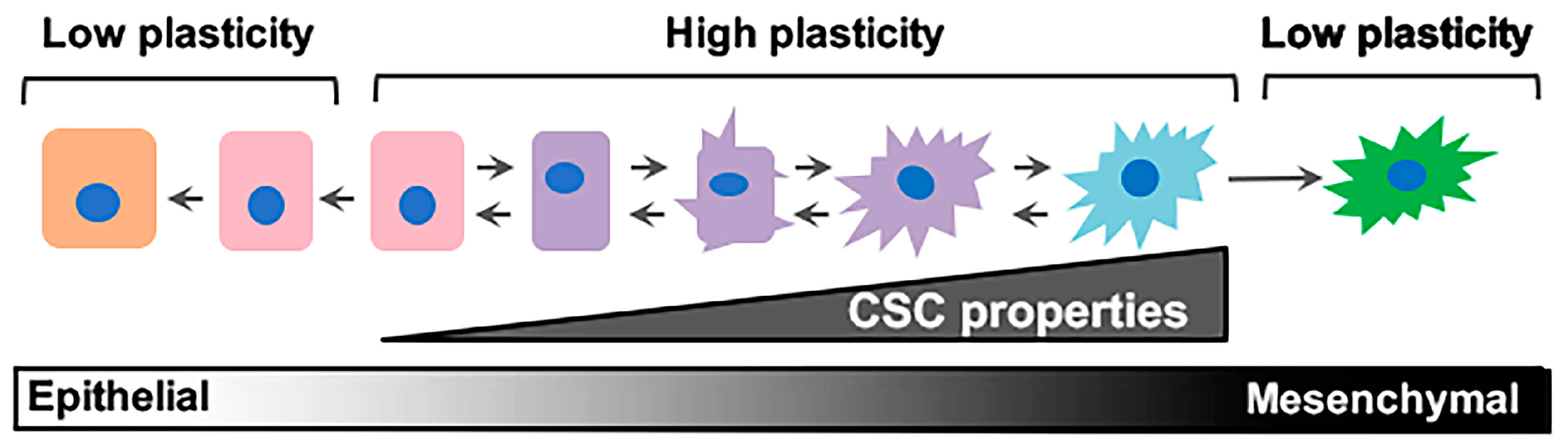
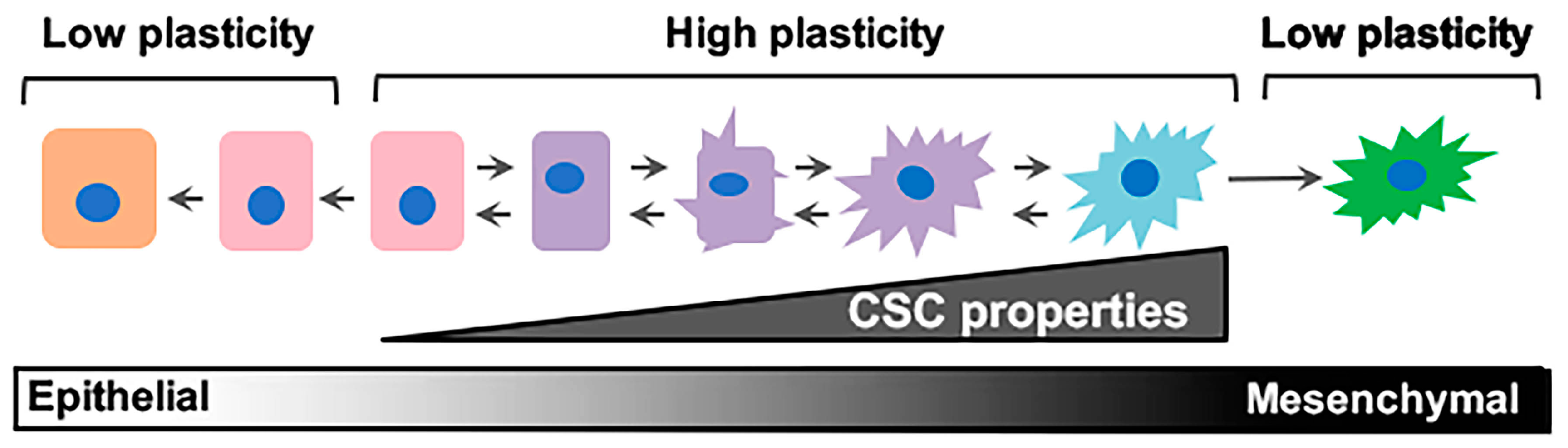
3. The Elusive Problem Child: Epithelial–Mesenchymal/Cancer Stem Cell (E–M/CSC) Plasticity
4. Intrinsic and Extrinsic Drivers of E-M/CSC Plasticity
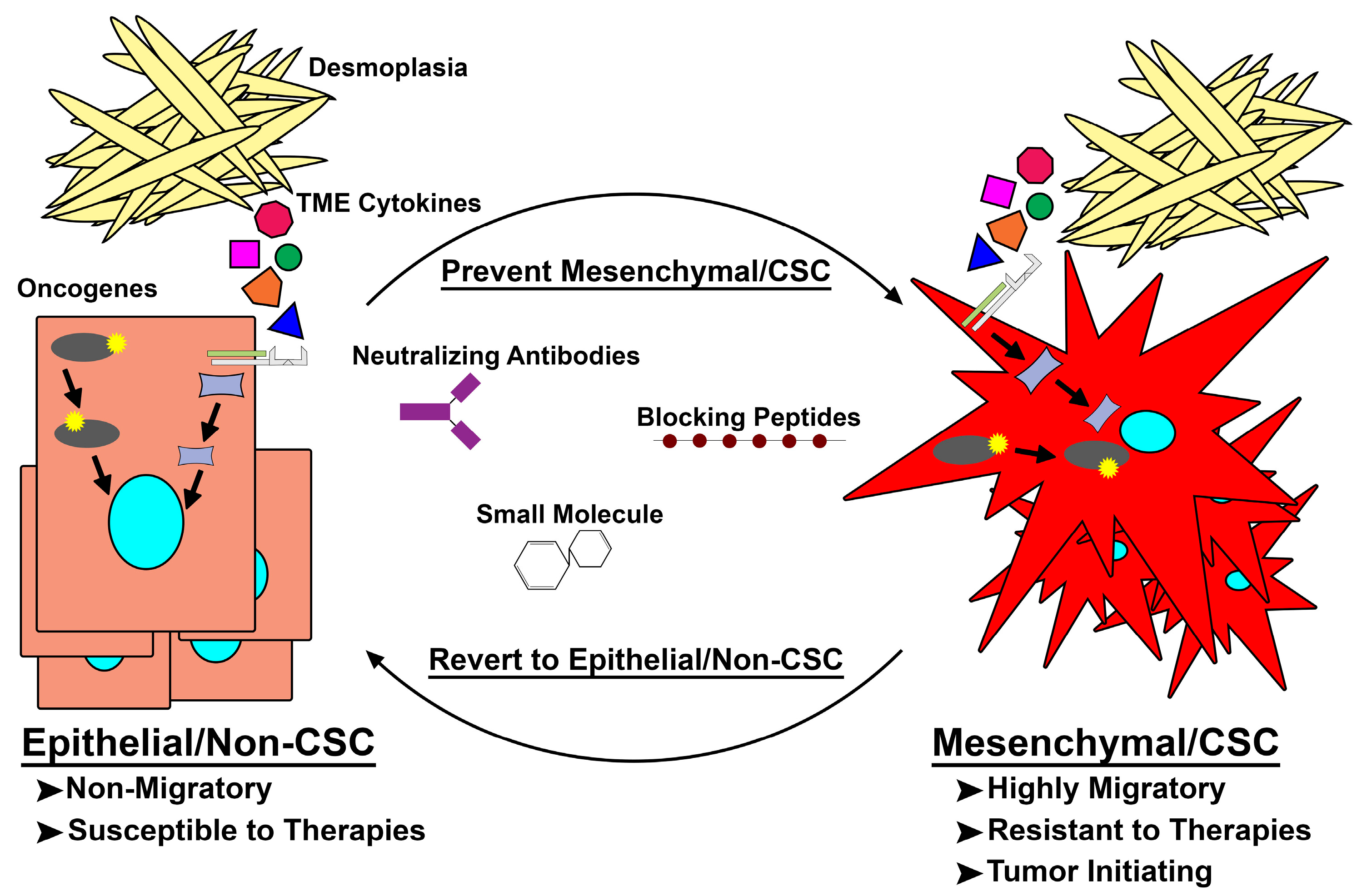
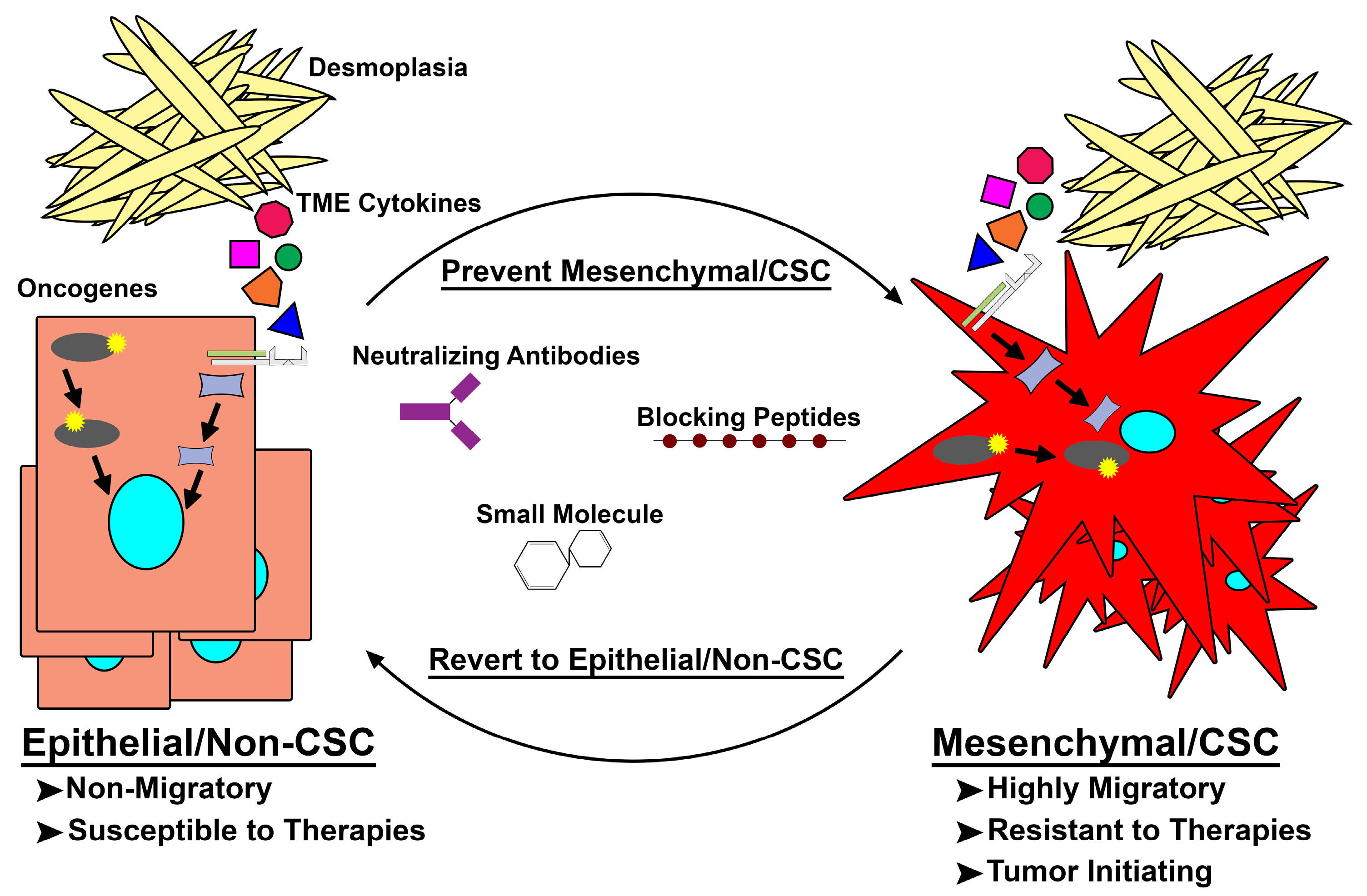
5. Latest in Mes/CSC Therapeutics and the Future of Targeting Mes/CSC Plasticity
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Muniraj, T.; Jamidar, P.A.; Aslanian, H.R. Pancreatic cancer: A comprehensive review and update. Dis. Mon. 2013, 59, 368–402. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, L.; Chen, Y.; Lian, G.; Qian, C.; Chen, S.; Li, J.; Huang, K. Pancreatic cancer epidemiology, detection, and management. Gastroenterol. Res. Pract. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Seer Cancer Stat facts: Pancreas Cancer. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 12 October 2017).
- Aichler, M.; Seiler, C.; Tost, M.; Siveke, J.; Mazur, P.K.; Da Silva-Buttkus, P.; Bartsch, D.K.; Langer, P.; Chiblak, S.; Durr, A.; et al. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: A comparative study in transgenic mice and human tissues. J. Pathol. 2012, 226, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schonhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic KRAS is completely dependent on wild-type PI 3-kinase p110α. Genes Dev. 2014, 28, 2621–2635. [Google Scholar] [CrossRef] [PubMed]
- Di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells and pancreatic cancer cells: An unholy alliance. Cancer Res. 2008, 68, 7707–7710. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Renz, B.; Reichert, M.; Rustgi, A.; Wang, T.C. Cellular plasticity and heterogeneity in pancreatic regeneration and malignancy. Cancer Cell Microenviron. 2016, 3. [Google Scholar] [CrossRef]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Marsh Rde, W.; Talamonti, M.S.; Katz, M.H.; Herman, J.M. Pancreatic cancer and FOLFIRINOX: A new era and new questions. Cancer Med. 2015, 4, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Ko, A.H. Erlotinib in the treatment of advanced pancreatic cancer. Biologics 2008, 2, 83–95. [Google Scholar] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the national cancer institute of canada clinical trials group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Kim, G. nab-Paclitaxel for the treatment of pancreatic cancer. Cancer Manag. Res. 2017, 9, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-Paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.; El Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. Updated survival from a randomized phase III trial (MPACT) of nab-paclitaxel plus gemcitabine versus gemcitabine alone for patients (pts) with metastatic adenocarcinoma of the pancreas. J. Clin. Oncol. 2014, 32, 178. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting ras membrane association: Back to the future for anti-ras drug discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Athuluri-Divakar, S.K.; Vasquez-Del Carpio, R.; Dutta, K.; Baker, S.J.; Cosenza, S.C.; Basu, I.; Gupta, Y.K.; Reddy, M.V.; Ueno, L.; Hart, J.R.; et al. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell 2016, 165, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.; et al. Small molecule inhibition of the KRAS-pdedelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in RAS and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Dy, G.K.; Erlichman, C.; Reid, J.M.; Sloan, J.A.; Pitot, H.C.; Alberts, S.R.; Goldberg, R.M.; Hanson, L.J.; Atherton, P.J.; et al. A phase I trial of ISIS 2503, an antisense inhibitor of H-ras, in combination with gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2003, 9, 115–123. [Google Scholar] [PubMed]
- Alberts, S.R.; Schroeder, M.; Erlichman, C.; Steen, P.D.; Foster, N.R.; Moore, D.F., Jr.; Rowland, K.M., Jr.; Nair, S.; Tschetter, L.K.; Fitch, T.R. Gemcitabine and ISIS-2503 for patients with locally advanced or metastatic pancreatic adenocarcinoma: A north central cancer treatment group phase II trial. J. Clin. Oncol. 2004, 22, 4944–4950. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Der, C.J.; Wang-Gillam, A.; Cox, A.D. Targeting RAS-mutant cancers: Is ERK the key? Trends Cancer 2015, 1, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Stuhlmiller, T.J.; Angus, S.P.; Zawistowski, J.S.; Graves, L.M. Molecular pathways: Adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin. Cancer Res. 2014, 20, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Sen, B.; Saigal, B.; Parikh, N.; Gallick, G.; Johnson, F.M. Sustained SRC inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009, 69, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Masamune, A.; Shimosegawa, T. Alteration of pancreatic cancer cell functions by tumor-stromal cell interaction. Front. Physiol. 2013, 4, 318. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grunert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, S.; Muschel, R.J.; Brunner, T.B. The stromal compartments in pancreatic cancer: Are there any therapeutic targets? Cancer Lett. 2014, 343, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Neoptolemos, J.P. Understanding metastasis in pancreatic cancer: A call for new clinical approaches. Cell 2012, 148, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, L.A.; Blanco, M.; Castosa, R.; Concha, A.; Valladares, M.; Calvo, L.; Figueroa, A. Clinical implications of epithelial cell plasticity in cancer progression. Cancer Lett. 2015, 366, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Poon, R.T.; Yuen, A.P.; Ling, M.T.; Kwok, W.K.; Wang, X.H.; Wong, Y.C.; Guan, X.Y.; Man, K.; Chau, K.L.; et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin. Cancer Res. 2006, 12, 5369–5376. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Morel, A.P.; Hinkal, G.; Bastid, J.; Puisieux, A. Twisting an embryonic transcription factor into an oncoprotein. Oncogene 2010, 29, 3173–3184. [Google Scholar] [CrossRef] [PubMed]
- Frixen, U.H.; Behrens, J.; Sachs, M.; Eberle, G.; Voss, B.; Warda, A.; Lochner, D.; Birchmeier, W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 1991, 113, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Barriere, G.; Riouallon, A.; Renaudie, J.; Tartary, M.; Rigaud, M. Mesenchymal and stemness circulating tumor cells in early breast cancer diagnosis. BMC Cancer 2012, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Tosoni, D.; Confalonieri, S.; Mazzarol, G.; Vecchi, M.; Ronzoni, S.; Bernard, L.; Viale, G.; Pelicci, P.G.; Di Fiore, P.P. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010, 140, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Kripke, M.L. Metastasis results from preexisting variant cells within a malignant-tumor. Science 1977, 197, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 2259–2265. [Google Scholar]
- Nowell, P.C. Mechanisms of tumor progression. Cancer Res. 1986, 2203–2207. [Google Scholar]
- Southam, C.M.; Brunschwig, A. Quantitative studies of autotransplantation of human cancer. Cancer 1961, 14, 971–978. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator ZEB1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Ince, T.A.; Richardson, A.L.; Bell, G.W.; Saitoh, M.; Godar, S.; Karnoub, A.E.; Iglehart, J.D.; Weinberg, R.A. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007, 12, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Cipriano, R.; Bryson, B.L.; Gilmore, H.L.; Jackson, M.W. Tumor microenvironmental signaling elicits epithelial-mesenchymal plasticity through cooperation with transforming genetic events. Neoplasia 2013, 15, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Potent EMT and CSC phenotypes are induced by oncostatin-M in pancreatic cancer. Mol. Cancer Res. 2017, 15, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, R.; Graham, J.; Miskimen, K.L.; Bryson, B.L.; Bruntz, R.C.; Scott, S.A.; Brown, H.A.; Stark, G.R.; Jackson, M.W. FAM83B mediates EGFR- and RAS-driven oncogenic transformation. J. Clin. Investig. 2012, 122, 3197–3210. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.Q.; Yan, T.T.; Liu, W.; Zhu, X.Q.; Tian, X.L.; Fu, X.L.; Hua, R.; Zhang, J.F.; Huo, Y.M.; Liu, D.J.; et al. High expression of FAM83B predicts poor prognosis in patients with pancreatic ductal adenocarcinoma and correlates with cell cycle and cell proliferation. J. Cancer 2017, 8, 3154–3165. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, R.; Miskimen, K.L.; Bryson, B.L.; Foy, C.R.; Bartel, C.A.; Jackson, M.W. Conserved oncogenic behavior of the FAM83 family regulates MAPK signaling in human cancer. Mol. Cancer Res. 2014, 12, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Meier, R.; Furuta, S.; Lenburg, M.E.; Kenny, P.A.; Xu, R.; Bissell, M.J. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J. Clin. Investig. 2012, 122, 3211–3220. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, J.; Liu, Z.; Liang, Q.; Zhang, N.; Jin, Y. FAM83A is amplified and promotes cancer stem cell-like traits and chemoresistance in pancreatic cancer. Oncogenesis 2017, 6, e300. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ma, C.; Xu, Q.; Cheng, L.; Xiao, L.; Xu, D.; Gao, Y.; Wang, J.; Song, H. A rapid nested polymerase chain reaction method to detect circulating cancer cells in breast cancer patients using multiple marker genes. Oncol. Lett. 2014, 7, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liao, G.Q.; He, P.; Zhu, H.; Liu, P.H.; Qu, Y.M.; Song, X.M.; Xu, Q.W.; Gao, Q.; Zhang, Y.; et al. Detection of circulating cancer cells in lung cancer patients with a panel of marker genes. Biochem. Biophys. Res. Commun. 2008, 372, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Snijders, A.M.; Lee, S.Y.; Hang, B.; Hao, W.; Bissell, M.J.; Mao, J.H. FAM83 family oncogenes are broadly involved in human cancers: An integrative multi-omics approach. Mol. Oncol. 2017, 11, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, A.; Higuchi, H.; Takaishi, H.; Matsuzaki, Y.; Suzuki, S.; Izumiya, M.; Iizuka, H.; Sakai, G.; Hozawa, S.; Azuma, T.; et al. Side population of pancreatic cancer cells predominates in TGF-β-mediated epithelial to mesenchymal transition and invasion. Int. J. Cancer 2009, 124, 2771–2779. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Perales, S.; Alejandre, M.J.; Iglesias, J.; Palomino, R.J.; Martin, M.; Caba, O.; Prados, J.C.; Aranega, A.; Delgado, J.R.; et al. Serum cytokine profile in patients with pancreatic cancer. Pancreas 2014, 43, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Poling, J.; Gajawada, P.; Richter, M.; Lorchner, H.; Polyakova, V.; Kostin, S.; Shin, J.; Boettger, T.; Walther, T.; Rees, W.; et al. Therapeutic targeting of the oncostatin M receptor-β prevents inflammatory heart failure. Basic Res. Cardiol. 2014, 109, 396. [Google Scholar] [CrossRef] [PubMed]
- Plater-Zyberk, C.; Buckton, J.; Thompson, S.; Spaull, J.; Zanders, E.; Papworth, J.; Life, P.F. Amelioration of arthritis in two murine models using antibodies to oncostatin M. Arthritis Rheum. 2001, 44, 2697–2702. [Google Scholar] [CrossRef]
- Choy, E.H.; Bendit, M.; McAleer, D.; Liu, F.; Feeney, M.; Brett, S.; Zamuner, S.; Campanile, A.; Toso, J. Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti-oncostatin M monoclonal antibody in rheumatoid arthritis: Results from phase II randomized, placebo-controlled trials. Arthritis Res. Ther. 2013, 15, R132. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1α signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 2017, 32, 392. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Ali, S.; Ahmad, A.; Azmi, A.S.; Li, Y.; Banerjee, S.; Kong, D.; Sethi, S.; Aboukameel, A.; Padhye, S.B.; et al. Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and MIR-21, which can be attenuated by CDF treatment. PLoS ONE 2012, 7, e50165. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, O.; Shimizu, K.; Semba, S.; Chiba, S.; Ku, Y.; Yokozaki, H.; Hori, Y. Hypoxia induces tumor aggressiveness and the expansion of CD133-positive cells in a hypoxia-inducible factor-1α-dependent manner in pancreatic cancer cells. Pathobiology 2011, 78, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.P.; Eibl, G.; Reber, H.A.; Hines, O.J. Influence of hypoxia and neoangiogenesis on the growth of pancreatic cancer. Mol. Cancer 2003, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Wang, M.J.; Lee, D.; Liang, C.C.; Lin, S. Hypoxia-inducible factor-1α regulates matrix metalloproteinase-1 activity in human bone marrow-derived mesenchymal stem cells. FEBS Lett. 2008, 582, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; Hiasa, Y.; Kumagi, T.; Yamanishi, H.; Azemoto, N.; Kobata, T.; Matsuura, B.; Abe, M.; Onji, M. Increased B cell-activating factor promotes tumor invasion and metastasis in human pancreatic cancer. PLoS ONE 2013, 8, e71367. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCS and CSCS in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Li, Q.; Liu, S.; Ning, N.; Zhang, X.; Xu, Y.; Chang, A.E.; Wicha, M.S. Concise review: Targeting cancer stem cells using immunologic approaches. Stem Cells 2015, 33, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Sachlos, E.; Risueno, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Kolev, V.N.; Wright, Q.G.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 2015, 75, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Liu, L.; Kallifatidis, G.; Baumann, B.; Mattern, J.; Gladkich, J.; Wirth, T.; Schemmer, P.; Buchler, M.W.; Zoller, M.; et al. Synergistic activity of sorafenib and sulforaphane abolishes pancreatic cancer stem cell characteristics. Cancer Res. 2010, 70, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Okada, M.; Shibuya, K.; Seino, M.; Sato, A.; Takeda, H.; Seino, S.; Yoshioka, T.; Kitanaka, C. JNK suppression of chemotherapeutic agents-induced ros confers chemoresistance on pancreatic cancer stem cells. Oncotarget 2015, 6, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.Y.; Sharma, S.; Zhou, Y.Q.; Yao, H.P.; Hu, X.; Zhang, R.; Wang, M.H. Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells. Mol. Cancer Ther. 2014, 13, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Ding, Q.; Miyazaki, Y.; Kuwahata, T.; Tsukasa, K.; Takao, S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci. Rep. 2013, 3, 3230. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Nall, D.; Tang, S.N.; Meeker, D.; Passarini, J.; Sharma, J.; Srivastava, R.K. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and krasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PLoS ONE 2011, 6, e16530. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011, 307, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.N.; Liang, Y.; Zhou, L.J.; Chen, S.P.; Chen, G.; Zhang, T.P.; Kang, T.; Zhao, Y.P. Combination of salinomycin and gemcitabine eliminates pancreatic cancer cells. Cancer Lett. 2011, 313, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Fan, P.; Bauer, N.; Gladkich, J.; Ryschich, E.; Bazhin, A.V.; Giese, N.A.; Strobel, O.; Hackert, T.; et al. Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget 2015, 6, 9999–10015. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Azmi, A.S.; Ali, S.; Zaiem, F.; Sarkar, F.H. Metformin may function as anti-cancer agent via targeting cancer stem cells: The potential biological significance of tumor-associated miRNAs in breast and pancreatic cancers. Ann. Transl. Med. 2014, 2, 59. [Google Scholar] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating csc function mediated by deregulating mirnas in pancreatic cancer cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Ritchie, R.L.; Marya, A.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl. Oncol. 2013, 6, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Schmitt, T.M.; Hulbert, A.; Brockenbrough, J.S.; Nguyen, H.; Cuevas, C.; Dotson, A.M.; Tan, X.; Hotes, J.L.; Greenberg, P.D.; et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 2015, 28, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: A phase I trial of safety and immune activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [PubMed]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of algenpantucel-l immunotherapy to standard adjuvant therapy for pancreatic cancer: A phase 2 study. J. Gastrointest. Surg. 2013, 17, 94–101. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics. Cancers 2018, 10, 14. https://doi.org/10.3390/cancers10010014
Smigiel JM, Parameswaran N, Jackson MW. Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics. Cancers. 2018; 10(1):14. https://doi.org/10.3390/cancers10010014
Chicago/Turabian StyleSmigiel, Jacob M., Neetha Parameswaran, and Mark W. Jackson. 2018. "Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics" Cancers 10, no. 1: 14. https://doi.org/10.3390/cancers10010014
APA StyleSmigiel, J. M., Parameswaran, N., & Jackson, M. W. (2018). Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics. Cancers, 10(1), 14. https://doi.org/10.3390/cancers10010014