Local Acetaldehyde—An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis


Abstract
:1. Introduction
2. Acetaldehyde
2.1. Genotoxicity and Mutagenicity
2.2. Carcinogenicity in Experimental Animals
2.3. Carcinogenicity in Humans
2.3.1. ALDH2-Deficiency as a Unique Human Cancer Model
2.3.2. Quantitative Assessment of the Carcinogenicity of Acetaldehyde in Humans
2.3.3. Implications of the ALDH2 Cancer Model for Regulatory Authorities
3. Microbial Metabolism—A Major Source of Local Acetaldehyde in the Upper GI Tract
3.1. Salivary Acetaldehyde—Instant and Long-Term Exposure
3.2. Oral and Esophageal Microbiome
3.2.1. Oral and Esophageal Microbiome in Health and Disease
3.2.2. Candida Yeasts—A Major Contributor to Local Alcohol Metabolism
3.2.3. Biofilm Lifestyle—The Preferred Mode of Microbial Growth in the GI Tract
3.2.4. The Effect of Alcohol and Smoking on the Microbiome and Microbiome Ethanol Metabolism
3.3. Gastric Microbiome
4. Host Metabolism of Ethanol and Acetaldehyde in the Upper GI Tract
4.1. Oral Mucosa and Salivary Glands
4.2. Esophagus and Stomach
5. Exogenous Sources for Local Acetaldehyde Exposure
6. Preventive Actions—Detection of Risk Groups
7. Minimization of Acetaldehyde Exposure
7.1. Population Level
7.2. Individual Level
8. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose-response meta-analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Baloch, Z.; He, T.T.; Xia, X. Alcohol consumption and gastric cancer risk: A meta-analysis. Med. Sci. Monit. 2017, 23, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment. Mutagenicity of Alcohol (Ethanol) and Its Metabolite Acetaldehyde. Public Health England: UK, 2016. Available online: https://www.gov.uk/government/publications/mutagenicity-of-alcohol-ethanol-and-its-metabolite-acetaldehyde (accessed 22 November 2017).
- Phillips, B.J.; Jenkinson, P. Is ethanol genotoxic? A review of the published data. Mutagenesis 2001, 16, 91–101. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Personal Habits and Indoor Combustions. In A Review of Human Carcinogens; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Lyon, France, 2012; Volume 100E, pp. 373–499. [Google Scholar]
- Secretan, B.; Straif, K.; Baan, R.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part E: Tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 2009, 10, 1033–1034. [Google Scholar] [CrossRef]
- Lachenmeier, D.W.; Salaspuro, M. ALDH2-deficiency as genetic epidemiologic and biochemical model for the carcinogenicity of acetaldehyde. Regul. Toxicol. Pharmacol. 2017, 86, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M. Key role of local acetaldehyde in upper GI tract carcinogenesis. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, A.; Ohashi, S.; Hirohashi, K.; Amanuma, Y.; Matsuda, T.; Muto, M. Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int. J. Mol. Sci. 2017, 18, 1943. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.F.; et al. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Theruvathu, J.A.; Jaruga, P.; Nath, R.G.; Dizdaroglu, M.; Brooks, P.J. Polyamines stimulate the formation of mutagenic 1,N2-propanodeoxyguanosine adducts from acetaldehyde. Nucleic Acids Res. 2005, 33, 3513–3520. [Google Scholar] [CrossRef] [PubMed]
- Homann, N.; Jousimies-Somer, H.; Jokelainen, K.; Heine, R.; Salaspuro, M. High acetaldehyde levels in saliva after ethanol consumption: Methodological aspects and pathogenetic implications. Carcinogenesis 1997, 18, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Linderborg, K.; Salaspuro, M.; Vakevainen, S. A single sip of a strong alcoholic beverage causes exposure to carcinogenic concentrations of acetaldehyde in the oral cavity. Food Chem. Toxicol. 2011, 49, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Tabor, C.W.; Tabor, H. Polyamines. Annu Rev. Biochem. 1984, 53, 749–790. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Matsumoto, A.; Uchida, M.; Kanaly, R.A.; Misaki, K.; Shibutani, S.; Kawamoto, T.; Kitagawa, K.; Nakayama, K.I.; Tomokuni, K.; et al. Increased formation of hepatic N2-ethylidene-2′-deoxyguanosine DNA adducts in aldehyde dehydrogenase 2-knockout mice treated with ethanol. Carcinogenesis 2007, 28, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Nagayoshi, H.; Matsumoto, A.; Nishi, R.; Kawamoto, T.; Ichiba, M.; Matsuda, T. Increased formation of gastric N(2)-ethylidene-2′-deoxyguanosine DNA adducts in aldehyde dehydrogenase-2 knockout mice treated with ethanol. Mutat. Res. 2009, 673, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Oyama, T.; Matsuda, T.; Isse, T.; Yamaguchi, T.; Tanaka, M.; Tsuji, M.; Kawamoto, T. The effect of ethanol on the formation of N2-ethylidene-dG adducts in mice: Implications for alcohol-related carcinogenicity of the oral cavity and esophagus. Biomarkers 2012, 17, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, Y.; Muto, M.; Hori, K.; Nagayoshi, H.; Yokoyama, A.; Chiba, T.; Matsuda, T. Combination of ADH1B*2/ALDH2*2 polymorphisms alters acetaldehyde-derived DNA damage in the blood of japanese alcoholics. Cancer Sci. 2012, 103, 1651–1655. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; Meng, L.; Bliss, R.L.; Jensen, J.A.; Hatsukami, D.K.; Hecht, S.S. Kinetics of DNA adduct formation in the oral cavity after drinking alcohol. Cancer Epidemiol. Biomark. Prev. 2012, 21, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; Juanes, R.C.; Khariwala, S.; Baker, E.J.; Daunais, J.B.; Grant, K.A. Increased levels of the acetaldehyde-derived DNA adduct N2-ethyldeoxyguanosine in oral mucosa DNA from rhesus monkeys exposed to alcohol. Mutagenesis 2016, 31, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; Meng, L.; Bliss, R.L.; Jensen, J.A.; Hatsukami, D.K.; Hecht, S.S. Time course of DNA adduct formation in peripheral blood granulocytes and lymphocytes after drinking alcohol. Mutagenesis 2012, 27, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Gromadzinska, J.; Mistry, Y.; Cordell, R.; Juren, T.; Segerback, D.; Farmer, P.B. Detection of acetaldehyde derived N(2)-ethyl-2′-deoxyguanosine in human leukocyte DNA following alcohol consumption. Mutat. Res. 2012, 737, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Lindros, K.O. Human blood acetaldehyde levels: With improved methods, a clearer picture emerges. Alcohol. Clin. Exp. Res. 1983, 7, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Nuutinen, H.U.; Salaspuro, M.P.; Valle, M.; Lindros, K.O. Blood acetaldehyde concentration gradient between hepatic and antecubital venous blood in ethanol-intoxicated alcoholics and controls. Eur. J. Clin. Investig. 1984, 14, 306–311. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Re-evaluation of some organic chemicals, hydrazine and hydrogen peroxide. Acetaldehyde. IARC Monogr. Eval. Carcinog. Risks Hum. 1999, 71 Pt 2, 319–335. [Google Scholar]
- Woutersen, R.A.; Appelman, L.M.; Feron, V.J.; Van der Heijden, C.A. Inhalation toxicity of acetaldehyde in rats. II. Carcinogenicity study: Interim results after 15 months. Toxicology 1984, 31, 123–133. [Google Scholar] [CrossRef]
- Woutersen, R.A.; Appelman, L.M.; Van Garderen-Hoetmer, A.; Feron, V.J. Inhalation toxicity of acetaldehyde in rats. III. Carcinogenicity study. Toxicology 1986, 41, 213–231. [Google Scholar] [CrossRef]
- Feron, V.J.; Kruysse, A.; Woutersen, R.A. Respiratory tract tumours in hamsters exposed to acetaldehyde vapour alone or simultaneously to benzo(a)pyrene or diethylnitrosamine. Eur. J. Cancer Clin. Oncol. 1982, 18, 13–31. [Google Scholar] [CrossRef]
- Soffritti, M.; Belpoggi, F.; Lambertin, L.; Lauriola, M.; Padovani, M.; Maltoni, C. Results of long-term experimental studies on the carcinogenicity of formaldehyde and acetaldehyde in rats. Ann. N. Y. Acad. Sci. 2002, 982, 87–105. [Google Scholar] [CrossRef]
- Bittersohl, G. Epidemiological research on cancer risk by aldol and aliphatic aldehydes. Environ. Qual. Saf. 1975, 4, 235–238. [Google Scholar] [PubMed]
- Maejima, R.; Iijima, K.; Kaihovaara, P.; Hatta, W.; Koike, T.; Imatani, A.; Shimosegawa, T.; Salaspuro, M. Effects of aldh2 genotype, ppi treatment and l-cysteine on carcinogenic acetaldehyde in gastric juice and saliva after intragastric alcohol administration. PLoS ONE 2015, 10, e0120397. [Google Scholar] [CrossRef] [PubMed]
- Vakevainen, S.; Tillonen, J.; Agarwal, D.P.; Srivastava, N.; Salaspuro, M. High salivary acetaldehyde after a moderate dose of alcohol in aldh2-deficient subjects: Strong evidence for the local carcinogenic action of acetaldehyde. Alcohol. Clin. Exp. Res. 2000, 24, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Vakevainen, S.; Tillonen, J.; Salaspuro, M. 4-methylpyrazole decreases salivary acetaldehyde levels in ALDH2-deficient subjects but not in subjects with normal ALDH2. Alcohol. Clin. Exp. Res. 2001, 25, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Kamada, Y.; Imazeki, H.; Hayashi, E.; Murata, S.; Kinoshita, K.; Yokoyama, T.; Kitagawa, Y. Effects of ADH1B and ALDH2 genetic polymorphisms on alcohol elimination rates and salivary acetaldehyde levels in intoxicated japanese alcoholic men. Alcohol. Clin. Exp. Res. 2016, 40, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Tsutsumi, E.; Imazeki, H.; Suwa, Y.; Nakamura, C.; Mizukami, T.; Yokoyama, T. Salivary acetaldehyde concentration according to alcoholic beverage consumed and aldehyde dehydrogenase-2 genotype. Alcohol. Clin. Exp. Res. 2008, 32, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Boccia, S.; Hashibe, M.; Galli, P.; De Feo, E.; Asakage, T.; Hashimoto, T.; Hiraki, A.; Katoh, T.; Nomura, T.; Yokoyama, A.; et al. Aldehyde dehydrogenase 2 and head and neck cancer: A meta-analysis implementing a mendelian randomization approach. Cancer Epidemiol. Biomark. Prev. 2009, 18, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Oze, I.; Hosono, S.; Ito, H.; Watanabe, M.; Ishioka, K.; Ito, S.; Tajika, M.; Yatabe, Y.; Niwa, Y.; et al. The aldehyde dehydrogenase 2 (ALDH2) Glu504Lys polymorphism interacts with alcohol drinking in the risk of stomach cancer. Carcinogenesis 2013, 34, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.T.; Wong, T.Y.; Ou, C.Y.; Fang, S.Y.; Chen, K.C.; Hsiao, J.R.; Huang, C.C.; Lee, W.T.; Lo, H.I.; Huang, J.S.; et al. The interplay between alcohol consumption, oral hygiene, ALDH2 and ADH1B in the risk of head and neck cancer. Int. J. Cancer 2014, 135, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Yokoyama, A.; Yokoyama, T.; Huang, Y.C.; Wu, S.Y.; Shao, Y.; Niu, J.; Wang, J.; Liu, Y.; Zhou, X.Q.; et al. Relationship between genetic polymorphisms of ALDH2 and ADH1B and esophageal cancer risk: A meta-analysis. World J. Gastroenterol. 2010, 16, 4210–4220. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Muramatsu, T.; Ohmori, T.; Yokoyama, T.; Okuyama, K.; Takahashi, H.; Hasegawa, Y.; Higuchi, S.; Maruyama, K.; Shirakura, K.; et al. Alcohol-related cancers and aldehyde dehydrogenase-2 in japanese alcoholics. Carcinogenesis 1998, 19, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Ohmori, T.; Muramatsu, T.; Higuchi, S.; Yokoyama, T.; Matsushita, S.; Matsumoto, M.; Maruyama, K.; Hayashida, M.; Ishii, H. Cancer screening of upper aerodigestive tract in japanese alcoholics with reference to drinking and smoking habits and aldehyde dehydrogenase-2 genotype. Int. J. Cancer 1996, 68, 313–316. [Google Scholar] [CrossRef]
- Yokoyama, A.; Yokoyama, T.; Omori, T.; Matsushita, S.; Mizukami, T.; Takahashi, H.; Higuchi, S.; Maruyama, K.; Ishii, H.; Hibi, T. Helicobacter pylori, chronic atrophic gastritis, inactive aldehyde dehydrogenase-2, macrocytosis and multiple upper aerodigestive tract cancers and the risk for gastric cancer in alcoholic japanese men. J. Gastroenterol. Hepatol. 2007, 22, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.R.; Wu, G.S.; Pakstis, A.J.; Tong, L.; Oota, H.; Kidd, K.K.; Zhang, Y.P. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2487lys. Gene 2009, 435, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J.; Enoch, M.A.; Goldman, D.; Li, T.K.; Yokoyama, A. The alcohol flushing response: An unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009, 6, e50. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; Akram, A.; Kondapally Seshasai, S.R.; Cole, D.; Watts, G.F.; Hovingh, G.K.; Kastelein, J.J.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Pooling and expanding registries of familial hypercholesterolaemia to assess gaps in care and improve disease management and outcomes: Rationale and design of the global eas familial hypercholesterolaemia studies collaboration. Atheroscler. Suppl. 2016, 22, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Helminen, A.; Vakevainen, S.; Salaspuro, M. ALDH2 genotype has no effect on salivary acetaldehyde without the presence of ethanol in the systemic circulation. PLoS ONE 2013, 8, e74418. [Google Scholar] [CrossRef] [PubMed]
- Tramacere, I.; Negri, E.; Bagnardi, V.; Garavello, W.; Rota, M.; Scotti, L.; Islami, F.; Corrao, G.; Boffetta, P.; La Vecchia, C. A meta-analysis of alcohol drinking and oral and pharyngeal cancers. Part 1: Overall results and dose-risk relation. Oral Oncol. 2010, 46, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, V.; Salaspuro, M. Synergistic effect of alcohol drinking and smoking on in vivo acetaldehyde concentration in saliva. Int. J. Cancer 2004, 111, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, J.; Straif, K.; Agudo, A.; Ahrens, W.; Bezerra Dos Santos, A.; Boccia, S.; Cadoni, G.; Canova, C.; Castellsague, X.; Chen, C.; et al. Low frequency of cigarette smoking and the risk of head and neck cancer in the inhance consortium pooled analysis. Int. J. Epidemiol. 2016, 45, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Uebelacker, M.; Lachenmeier, D.W. Quantitative determination of acetaldehyde in foods using automated digestion with simulated gastric fluid followed by headspace gas chromatography. J. Autom. Methods Manag. Chem. 2011, 2011, 907317. [Google Scholar] [CrossRef] [PubMed]
- Nuutinen, H.; Lindros, K.O.; Salaspuro, M. Determinants of blood acetaldehyde level during ethanol oxidation in chronic alcoholics. Alcohol. Clin. Exp. Res. 1983, 7, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.J.; Peng, T.K.; Yin, S.J. Expression and activities of class iv alcohol dehydrogenase and class III aldehyde dehydrogenase in human mouth. Alcohol 1996, 13, 257–262. [Google Scholar] [CrossRef]
- Pavlova, S.I.; Jin, L.; Gasparovich, S.R.; Tao, L. Multiple alcohol dehydrogenases but no functional acetaldehyde dehydrogenase causing excessive acetaldehyde production from ethanol by oral streptococci. Microbiology 2013, 159, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.T.; Novak-Frazer, L.; Collins, R.; Dawsey, S.P.; Dawsey, S.M.; Abnet, C.C.; White, R.E.; Freedman, N.D.; Mwachiro, M.; Bowyer, P.; et al. Alcohol and acetaldehyde in african fermented milk mursik—A possible etiologic factor for high incidence of esophageal cancer in western kenya. Cancer Epidemiol. Biomark. Prev. 2013, 22, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Sohnius, E.M. The role of acetaldehyde outside ethanol metabolism in the carcinogenicity of alcoholic beverages: Evidence from a large chemical survey. Food Chem. Toxicol. 2008, 46, 2903–2911. [Google Scholar] [CrossRef] [PubMed]
- Paiano, V.; Bianchi, G.; Davoli, E.; Negri, E.; Fanelli, R.; Fattore, E. Risk assessment for the italian population of acetaldehyde in alcoholic and non-alcoholic beverages. Food Chem. 2014, 154, 26–31. [Google Scholar] [CrossRef] [PubMed]
- The Joint FAO/WHO Expert Committee on Food Additives (JECFA). Saturated aliphatic acyclic linear primary alcohols, aldehydes, and acids. Safety evaluation of certain food additives and contaminants. WHO Food Addit. 1998, 40, 148–188. [Google Scholar]
- Okamura, H.; Abe, H.; Hasegawa-Baba, Y.; Saito, K.; Sekiya, F.; Hayashi, S.M.; Mirokuji, Y.; Maruyama, S.; Ono, A.; Nakajima, M.; et al. The japan flavour and fragrance materials association‘s (JFFMA) safety assessment of acetal food flavouring substances uniquely used in japan. Food Addit. Contam. Part A 2015, 32, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Safety evaluation of certain food additives and contaminants. International programme on chemical safety. WHO Food Addit. 2000, 44. Available online: http://www.inchem.org/documents/jecfa/jecmono/v44jec08.htm (accessed on 22 November 2017).
- Lachenmeier, D.W.; Monakhova, Y.B. Short-term salivary acetaldehyde increase due to direct exposure to alcoholic beverages as an additional cancer risk factor beyond ethanol metabolism. J. Exp. Clin. Cancer Res. 2011, 30, 3. [Google Scholar] [CrossRef] [PubMed]
- Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Risk Characterisation Methods. COC/G 06—Version 1.0; Public Health England: UK, 2012. Available online: https://www.gov.uk/government/publications/cancer-risk-characterisation-methods (accessed on 22 November 2017).
- Benford, D.J. The use of dose-response data in a margin of exposure approach to carcinogenic risk assessment for genotoxic chemicals in food. Mutagenesis 2016, 31, 329–331. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Opinion of the scientific committee on a request from EFSA related to a harmonised approach for risk assessment of substances which are both genotoxic and carcinogenic. EFSA J. 2005, 3, 282. [Google Scholar] [CrossRef]
- Kurkivuori, J.; Salaspuro, V.; Kaihovaara, P.; Kari, K.; Rautemaa, R.; Gronroos, L.; Meurman, J.H.; Salaspuro, M. Acetaldehyde production from ethanol by oral streptococci. Oral Oncol. 2007, 43, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Muto, M.; Hitomi, Y.; Ohtsu, A.; Shimada, H.; Kashiwase, Y.; Sasaki, H.; Yoshida, S.; Esumi, H. Acetaldehyde production by non-pathogenic Neisseria in human oral microflora: Implications for carcinogenesis in upper aerodigestive tract. Int. J. Cancer 2000, 88, 342–350. [Google Scholar] [CrossRef]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Light alcohol drinking and cancer: A meta-analysis. Ann. Oncol. 2013, 24, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Kaihovaara, P.; Rudnai, P.; Znaor, A.; Lissowska, J.; Swiatkowska, B.; Mates, D.; Pandics, T.; Salaspuro, M. Acetaldehyde level in spirits from central european countries. Eur. J. Cancer Prev 2011, 20, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Launoy, G.; Milan, C.; Day, N.E.; Faivre, J.; Pienkowski, P.; Gignoux, M. Oesophageal cancer in france: Potential importance of hot alcoholic drinks. Int. J. Cancer 1997, 71, 917–923. [Google Scholar] [CrossRef]
- Linderborg, K.; Joly, J.P.; Visapaa, J.P.; Salaspuro, M. Potential mechanism for calvados-related oesophageal cancer. Food Chem. Toxicol. 2008, 46, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Bik, E.M.; Long, C.D.; Armitage, G.C.; Loomer, P.; Emerson, J.; Mongodin, E.F.; Nelson, K.E.; Gill, S.R.; Fraser-Liggett, C.M.; Relman, D.A. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010, 4, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Chen, C.Y.; Hayes, R.B. Oral microbiome and oral and gastrointestinal cancer risk. Cancer Causes Control 2012, 23, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Norder Grusell, E.; Dahlen, G.; Ruth, M.; Ny, L.; Quiding-Jarbrink, M.; Bergquist, H.; Bove, M. Bacterial flora of the human oral cavity, and the upper and lower esophagus. Dis. Esophagus 2013, 26, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Abnet, C.C.; Kamangar, F.; Islami, F.; Nasrollahzadeh, D.; Brennan, P.; Aghcheli, K.; Merat, S.; Pourshams, A.; Marjani, H.A.; Ebadati, A.; et al. Tooth loss and lack of regular oral hygiene are associated with higher risk of esophageal squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3062–3068. [Google Scholar] [CrossRef] [PubMed]
- Hashim, D.; Sartori, S.; Brennan, P.; Curado, M.P.; Wunsch-Filho, V.; Divaris, K.; Olshan, A.F.; Zevallos, J.P.; Winn, D.M.; Franceschi, S.; et al. The role of oral hygiene in head and neck cancer: Results from international head and neck cancer epidemiology (inhance) consortium. Ann. Oncol. 2016, 27, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Sepehr, A.; Kamangar, F.; Fahimi, S.; Saidi, F.; Abnet, C.C.; Dawsey, S.M. Poor oral health as a risk factor for esophageal squamous dysplasia in northeastern iran. Anticancer Res. 2005, 25, 543–546. [Google Scholar] [PubMed]
- Marttila, E.; Bowyer, P.; Sanglard, D.; Uittamo, J.; Kaihovaara, P.; Salaspuro, M.; Richardson, M.; Rautemaa, R. Fermentative 2-carbon metabolism produces carcinogenic levels of acetaldehyde in candida albicans. Mol. Oral Microbiol. 2013, 28, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Homann, N.; Tillonen, J.; Meurman, J.H.; Rintamaki, H.; Lindqvist, C.; Rautio, M.; Jousimies-Somer, H.; Salaspuro, M. Increased salivary acetaldehyde levels in heavy drinkers and smokers: A microbiological approach to oral cavity cancer. Carcinogenesis 2000, 21, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, Q.; Hua, H.; Chen, F. Changes in the salivary microbiota of oral leukoplakia and oral cancer. Oral Oncol. 2016, 56, e6–e8. [Google Scholar] [CrossRef] [PubMed]
- Moritani, K.; Takeshita, T.; Shibata, Y.; Ninomiya, T.; Kiyohara, Y.; Yamashita, Y. Acetaldehyde production by major oral microbes. Oral Dis. 2015, 21, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Tillonen, J.; Homann, N.; Rautio, M.; Jousimies-Somer, H.; Salaspuro, M. Role of yeasts in the salivary acetaldehyde production from ethanol among risk groups for ethanol-associated oral cavity cancer. Alcohol. Clin. Exp. Res. 1999, 23, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.T.; Uittamo, J.; Salaspuro, M.; Rautemaa, R. Acetaldehyde production from ethanol and glucose by non-Candida albicans yeasts in vitro. Oral Oncol. 2009, 45, e245–e248. [Google Scholar] [CrossRef] [PubMed]
- Uittamo, J.; Siikala, E.; Kaihovaara, P.; Salaspuro, M.; Rautemaa, R. Chronic candidosis and oral cancer in APECED-patients: Production of carcinogenic acetaldehyde from glucose and ethanol by Candida albicans. Int. J. Cancer 2009, 124, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Bakri, M.M.; Cannon, R.D.; Holmes, A.R.; Rich, A.M. Detection of Candida albicans adh1 and adh2 mRNAs in human archival oral biopsy samples. J. Oral Pathol. Med. 2014, 43, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Alnuaimi, A.D.; Ramdzan, A.N.; Wiesenfeld, D.; O‘Brien-Simpson, N.M.; Kolev, S.D.; Reynolds, E.C.; McCullough, M.J. Candida virulence and ethanol-derived acetaldehyde production in oral cancer and non-cancer subjects. Oral Dis. 2016, 22, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Delsing, C.E.; Bleeker-Rovers, C.P.; van de Veerdonk, F.L.; Tol, J.; van der Meer, J.W.; Kullberg, B.J.; Netea, M.G. Association of esophageal candidiasis and squamous cell carcinoma. Med. Mycol. Case Rep. 2012, 1, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Rautemaa, R.; Hietanen, J.; Niissalo, S.; Pirinen, S.; Perheentupa, J. Oral and oesophageal squamous cell carcinoma—A complication or component of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED, APS-I). Oral Oncol. 2007, 43, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Fox, E.P.; Nett, J.E.; Sorrells, T.R.; Mitrovich, Q.M.; Hernday, A.D.; Tuch, B.B.; Andes, D.R.; Johnson, A.D. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 2012, 148, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.T.; Novak-Frazer, L.; Rautemaa, W.; Rajendran, R.; Sorsa, T.; Ramage, G.; Bowyer, P.; Rautemaa, R. A novel antifungal is active against Candida albicans biofilms and inhibits mutagenic acetaldehyde production in vitro. PLoS ONE 2014, 9, e101859. [Google Scholar] [CrossRef] [PubMed]
- Janus, M.M.; Crielaard, W.; Volgenant, C.M.; van der Veen, M.H.; Brandt, B.W.; Krom, B.P. Candida albicans alters the bacterial microbiome of early in vitro oral biofilms. J. Oral Microbiol. 2017, 9, 1270613. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.; Matthews, C.R.; Joshi, V.; de Jager, M.; Aspiras, M. Tobacco smoking affects bacterial acquisition and colonization in oral biofilms. Infect. Immun. 2011, 79, 4730–4738. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Gleber-Netto, F.O.; Fernandes, G.R.; Amorim, M.; Barbosa, L.F.; Francisco, A.L.; de Andrade, A.G.; Setubal, J.C.; Kowalski, L.P.; Nunes, D.N.; et al. Alcohol and tobacco consumption affects bacterial richness in oral cavity mucosa biofilms. BMC Microbiol. 2014, 14, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homann, N.; Tillonen, J.; Rintamaki, H.; Salaspuro, M.; Lindqvist, C.; Meurman, J.H. Poor dental status increases acetaldehyde production from ethanol in saliva: A possible link to increased oral cancer risk among heavy drinkers. Oral Oncol. 2001, 37, 153–158. [Google Scholar] [CrossRef]
- Marttila, E.; Uittamo, J.; Rusanen, P.; Lindqvist, C.; Salaspuro, M.; Rautemaa, R. Acetaldehyde production and microbial colonization in oral squamous cell carcinoma and oral lichenoid disease. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [PubMed]
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 141. [Google Scholar] [CrossRef] [PubMed]
- Tsuruya, A.; Kuwahara, A.; Saito, Y.; Yamaguchi, H.; Tsubo, T.; Suga, S.; Inai, M.; Aoki, Y.; Takahashi, S.; Tsutsumi, E.; et al. Ecophysiological consequences of alcoholism on human gut microbiota: Implications for ethanol-related pathogenesis of colon cancer. Sci. Rep. 2016, 6, 27923. [Google Scholar] [CrossRef] [PubMed]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A.; Francois, F.; Perez-Perez, G.; Blaser, M.J.; Relman, D.A. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. USA 2006, 103, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the gut microbiome using 16s or shotgun metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Torres, J.; Hu, N.; Medrano-Guzman, R.; Herrera-Goepfert, R.; Humphrys, M.S.; Wang, L.; Wang, C.; Ding, T.; Ravel, J.; et al. Molecular characterization of the human stomach microbiota in gastric cancer patients. Front. Cell. Infect. Microbiol. 2017, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Stockbruegger, R.W.; Seeberg, S.; Hellner, L.; Jaup, B.H.; Dotevall, G. Intragastric bacteria and nitrite after short-term treatment with different doses of antimuscarinic drugs. Scand. J. Gastroenterol. 1984, 19, 14–23. [Google Scholar] [PubMed]
- Theisen, J.; Nehra, D.; Citron, D.; Johansson, J.; Hagen, J.A.; Crookes, P.F.; DeMeester, S.R.; Bremner, C.G.; DeMeester, T.R.; Peters, J.H. Suppression of gastric acid secretion in patients with gastroesophageal reflux disease results in gastric bacterial overgrowth and deconjugation of bile acids. J. Gastrointest. Surg. 2000, 4, 50–54. [Google Scholar] [CrossRef]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.C.; Rust, S.; Bode, C. The effect of cimetidine treatment on ethanol formation in the human stomach. Scand. J. Gastroenterol. 1984, 19, 853–856. [Google Scholar] [PubMed]
- Vakevainen, S.; Mentula, S.; Nuutinen, H.; Salmela, K.S.; Jousimies-Somer, H.; Farkkila, M.; Salaspuro, M. Ethanol-derived microbial production of carcinogenic acetaldehyde in achlorhydric atrophic gastritis. Scand. J. Gastroenterol. 2002, 37, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Vakevainen, S.; Tillonen, J.; Blom, M.; Jousimies-Somer, H.; Salaspuro, M. Acetaldehyde production and other ADH-related characteristics of aerobic bacteria isolated from hypochlorhydric human stomach. Alcohol. Clin. Exp. Res. 2001, 25, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Gail, M.H.; Shi, J.; Klepac-Ceraj, V.; Paster, B.J.; Dye, B.A.; Wang, G.Q.; Wei, W.Q.; Fan, J.H.; Qiao, Y.L.; et al. Association between upper digestive tract microbiota and cancer-predisposing states in the esophagus and stomach. Cancer Epidemiol. Biomark. Prev. 2014, 23, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Eom, C.S.; Jeon, C.Y.; Park, S.M. Acid suppressive drugs and gastric cancer: A meta-analysis of observational studies. World J. Gastroenterol. 2013, 19, 2560–2568. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kang, M.; Lee, J.S.; Inoue, M.; Sasazuki, S.; Tsugane, S. Fermented and non-fermented soy food consumption and gastric cancer in japanese and korean populations: A meta-analysis of observational studies. Cancer Sci. 2011, 102, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Praud, D.; Rota, M.; Pelucchi, C.; Bertuccio, P.; Rosso, T.; Galeone, C.; Zhang, Z.F.; Matsuo, K.; Ito, H.; Hu, J.; et al. Cigarette smoking and gastric cancer in the stomach cancer pooling (StoP) project. Eur. J. Cancer Prev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.S.; Kamangar, F.; Forman, D.; Islami, F. Pickled food and risk of gastric cancer—A systematic review and meta-analysis of english and chinese literature. Cancer Epidemiol. Biomark. Prev. 2012, 21, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Rokkas, T.; Rokka, A.; Portincasa, P. A systematic review and meta-analysis of the role of helicobacter pylori eradication in preventing gastric cancer. Ann. Gastroenterol. 2017, 30, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Sipponen, P.; Kekki, M.; Haapakoski, J.; Ihamaki, T.; Siurala, M. Gastric cancer risk in chronic atrophic gastritis: Statistical calculations of cross-sectional data. Int. J. Cancer 1985, 35, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Somi, M.H.; Mousavi, S.M.; Naghashi, S.; Faramarzi, E.; Jafarabadi, M.A.; Ghojazade, M.; Majdi, A.; Naseri Alavi, S.A. Is there any relationship between food habits in the last two decades and gastric cancer in North-western Iran? Asian Pac. J. Cancer Prev. 2015, 16, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, P.M.; Hendolin, P.; Kaihovaara, P.; Kronberg, L.; Meierjohann, A.; Millerhovf, A.; Paloheimo, L.; Sundelin, H.; Syrjanen, K.; Webb, D.L.; et al. Slow-release l-cysteine capsule prevents gastric mucosa exposure to carcinogenic acetaldehyde: Results of a randomised single-blinded, cross-over study of helicobacter-associated atrophic gastritis. Scand. J. Gastroenterol. 2017, 52, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Vakevainen, S.; Tillonen, J.; Salaspuro, M.; Jousimies-Somer, H.; Nuutinen, H.; Farkkila, M. Hypochlorhydria induced by a proton pump inhibitor leads to intragastric microbial production of acetaldehyde from ethanol. Aliment. Pharmacol. Ther. 2000, 14, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer; World Health Organization. Schistosomes, liver flukes and helicobacter pylori. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 177–241. [Google Scholar]
- Roine, R.P.; Salmela, K.S.; Hook-Nikanne, J.; Kosunen, T.U.; Salaspuro, M. Alcohol dehydrogenase mediated acetaldehyde production by Helicobacter pylori—A possible mechanism behind gastric injury. Life Sci. 1992, 51, 1333–1337. [Google Scholar] [CrossRef]
- Salmela, K.S.; Roine, R.P.; Koivisto, T.; Hook-Nikanne, J.; Kosunen, T.U.; Salaspuro, M. Characteristics of Helicobacter pylori alcohol dehydrogenase. Gastroenterology 1993, 105, 325–330. [Google Scholar] [CrossRef]
- Kaihovaara, P.; Salmela, K.S.; Roine, R.P.; Kosunen, T.U.; Salaspuro, M. Purification and characterization of Helicobacter pylori alcohol dehydrogenase. Alcohol. Clin. Exp. Res. 1994, 18, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.T.; Liao, C.S.; Yin, S.J. Human hepatic alcohol and aldehyde dehydrogenases: Genetic polymorphism and activities. Proc. Natl. Sci. Counc. Repub. China B 1997, 21, 106–111. [Google Scholar] [PubMed]
- Vondracek, M.; Xi, Z.; Larsson, P.; Baker, V.; Mace, K.; Pfeifer, A.; Tjalve, H.; Donato, M.T.; Gomez-Lechon, M.J.; Grafstrom, R.C. Cytochrome p450 expression and related metabolism in human buccal mucosa. Carcinogenesis 2001, 22, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Homann, N.; Stickel, F.; Konig, I.R.; Jacobs, A.; Junghanns, K.; Benesova, M.; Schuppan, D.; Himsel, S.; Zuber-Jerger, I.; Hellerbrand, C.; et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int. J. Cancer 2006, 118, 1998–2002. [Google Scholar] [CrossRef] [PubMed]
- Visapaa, J.P.; Gotte, K.; Benesova, M.; Li, J.; Homann, N.; Conradt, C.; Inoue, H.; Tisch, M.; Horrmann, K.; Vakevainen, S.; et al. Increased cancer risk in heavy drinkers with the alcohol dehydrogenase 1C*1 allele, possibly due to salivary acetaldehyde. Gut 2004, 53, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.J.; Chou, F.J.; Chao, S.F.; Tsai, S.F.; Liao, C.S.; Wang, S.L.; Wu, C.W.; Lee, S.C. Alcohol and aldehyde dehydrogenases in human esophagus: Comparison with the stomach enzyme activities. Alcohol. Clin. Exp. Res. 1993, 17, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Millonig, G.; Wang, Y.; Homann, N.; Bernhardt, F.; Qin, H.; Mueller, S.; Bartsch, H.; Seitz, H.K. Ethanol-mediated carcinogenesis in the human esophagus implicates CYP2E1 induction and the generation of carcinogenic DNA-lesions. Int. J. Cancer 2011, 128, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Egerer, G.; Simanowski, U.A.; Waldherr, R.; Eckey, R.; Agarwal, D.P.; Goedde, H.W.; von Wartburg, J.P. Human gastric alcohol dehydrogenase activity: Effect of age, sex, and alcoholism. Gut 1993, 34, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.J.; Liao, C.S.; Wu, C.W.; Li, T.T.; Chen, L.L.; Lai, C.L.; Tsao, T.Y. Human stomach alcohol and aldehyde dehydrogenases: Comparison of expression pattern and activities in alimentary tract. Gastroenterology 1997, 112, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Naito, Z.; Matsuda, N.; Onodera, H.; Sakurazawa, N.; Yamashita, N.; Kanazawa, Y.; Fujita, I.; Makino, H.; Uchida, E. Localization of cytochrome p4502e1 enzyme in normal and cancerous gastric mucosa and association with its genetic polymorphism in unoperated and remnant stomach. J. Nippon Med. Sch. 2011, 78, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Hayes, R.B.; Sartori, S.; Lee, Y.C.; Muscat, J.; Olshan, A.; Winn, D.M.; Castellsague, X.; Zhang, Z.F.; Morgenstern, H.; et al. Mouthwash use and cancer of the head and neck: A pooled analysis from the international head and neck cancer epidemiology consortium. Eur. J. Cancer Prev. 2016, 25, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Cancer prevention research—Then and now. Nat. Rev. Cancer 2009, 9, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wan, X.; Wang, Y.; Sun, Y.; Yang, G.; Wang, L. Time trends of esophageal and gastric cancer mortality in china, 1991–2009: An age-period-cohort analysis. Sci. Rep. 2017, 7, 6797. [Google Scholar] [CrossRef] [PubMed]
- Güzel-Seydim, Z.B.; Seydim, A.C.; Greene, A.K.; Bodine, A.B. Determination of organic acids and volatile flavor substances in kefir during fermentation. J. Food Compos. Anal. 2000, 13, 35–43. [Google Scholar] [CrossRef]
- Jeong, S.H.; Lee, S.H.; Jung, J.Y.; Choi, E.J.; Jeon, C.O. Microbial succession and metabolite changes during long-term storage of kimchi. J. Food Sci. 2013, 78, M763–M769. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Ren, J.S.; Taylor, P.R.; Kamangar, F. Pickled vegetables and the risk of oesophageal cancer: A meta-analysis. Br. J. Cancer 2009, 101, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Wakhisi, J.; Mining, S.; Mwangi, A.; Patel, R. Esophageal cancer, the topmost cancer at MTRH in the rift valley, Kenya, and its potential risk factors. ISRN Oncol. 2013, 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Wang, X.; Yu, I.T.; Huang, C.; Zhou, X.; Li, J.; Wang, D. Processed food consumption and risk of esophageal squamous cell carcinoma: A case-control study in a high risk area. Cancer Sci. 2012, 103, 2007–2011. [Google Scholar] [CrossRef] [PubMed]
- Augustin, J.; Augustin, E.; Cutrufelli, R.L.; Hagen, S.R.; Teitzel, C. Alcohol retention in food preparation. J. Am. Diet. Assoc. 1992, 92, 486–488. [Google Scholar] [PubMed]
- Haussmann, H.J. Use of hazard indices for a theoretical evaluation of cigarette smoke composition. Chem. Res. Toxicol. 2012, 25, 794–810. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Hoffmann, I.; El-Bayoumy, K. The less harmful cigarette: A controversial issue. A tribute to ernst l. Wynder. Chem. Res. Toxicol. 2001, 14, 767–790. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Monakhova, Y.B.; Hengen, J.; Kohl-Himmelseher, M.; Schussler, J.; Hahn, H.; Kuballa, T.; Lachenmeier, D.W. Electronic cigarettes: Overview of chemical composition and exposure estimation. Tob. Induc. Dis. 2014, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yuan, Z.; Lu, M.; Zhang, Y.; Jin, L.; Ye, W. Poor oral health is associated with an increased risk of esophageal squamous cell carcinoma—A population-based case-control study in China. Int. J. Cancer 2017, 140, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.T.; Leng, W.D.; Zhang, C.; Liu, J.; Cao, S.Y.; Huang, W. Meta-analysis on the association between toothbrushing and head and neck cancer. Oral Oncol. 2015, 51, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Agreus, L.; Kuipers, E.J.; Kupcinskas, L.; Malfertheiner, P.; Di Mario, F.; Leja, M.; Mahachai, V.; Yaron, N.; van Oijen, M.; Perez Perez, G.; et al. Rationale in diagnosis and screening of atrophic gastritis with stomach-specific plasma biomarkers. Scand. J. Gastroenterol. 2012, 47, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Tuyns, A.J.; Pequignot, G.; Jensen, O.M. Esophageal cancer in Ille-et-Vilaine in relation to levels of alcohol and tobacco consumption. Risks are multiplying. Bull. Cancer 1977, 64, 45–60. [Google Scholar] [PubMed]
- Salaspuro, V.; Hietala, J.; Kaihovaara, P.; Pihlajarinne, L.; Marvola, M.; Salaspuro, M. Removal of acetaldehyde from saliva by a slow-release buccal tablet of l-cysteine. Int. J. Cancer 2002, 97, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, V.J.; Hietala, J.M.; Marvola, M.L.; Salaspuro, M.P. Eliminating carcinogenic acetaldehyde by cysteine from saliva during smoking. Cancer Epidemiol. Biomark. Prev. 2006, 15, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Linderborg, K.; Marvola, T.; Marvola, M.; Salaspuro, M.; Farkkila, M.; Vakevainen, S. Reducing carcinogenic acetaldehyde exposure in the achlorhydric stomach with cysteine. Alcohol. Clin. Exp. Res. 2011, 35, 516–522. [Google Scholar] [CrossRef] [PubMed]
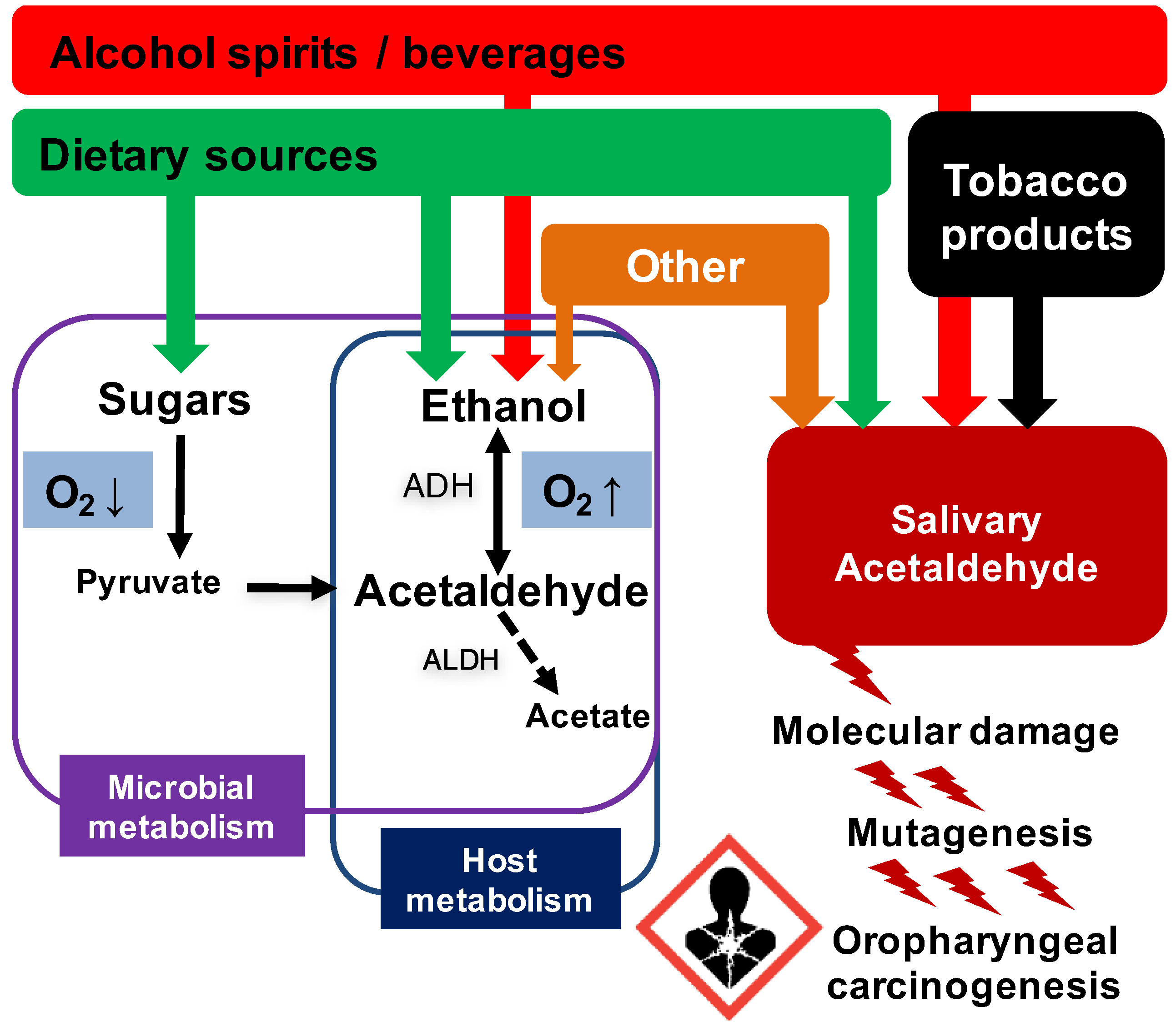

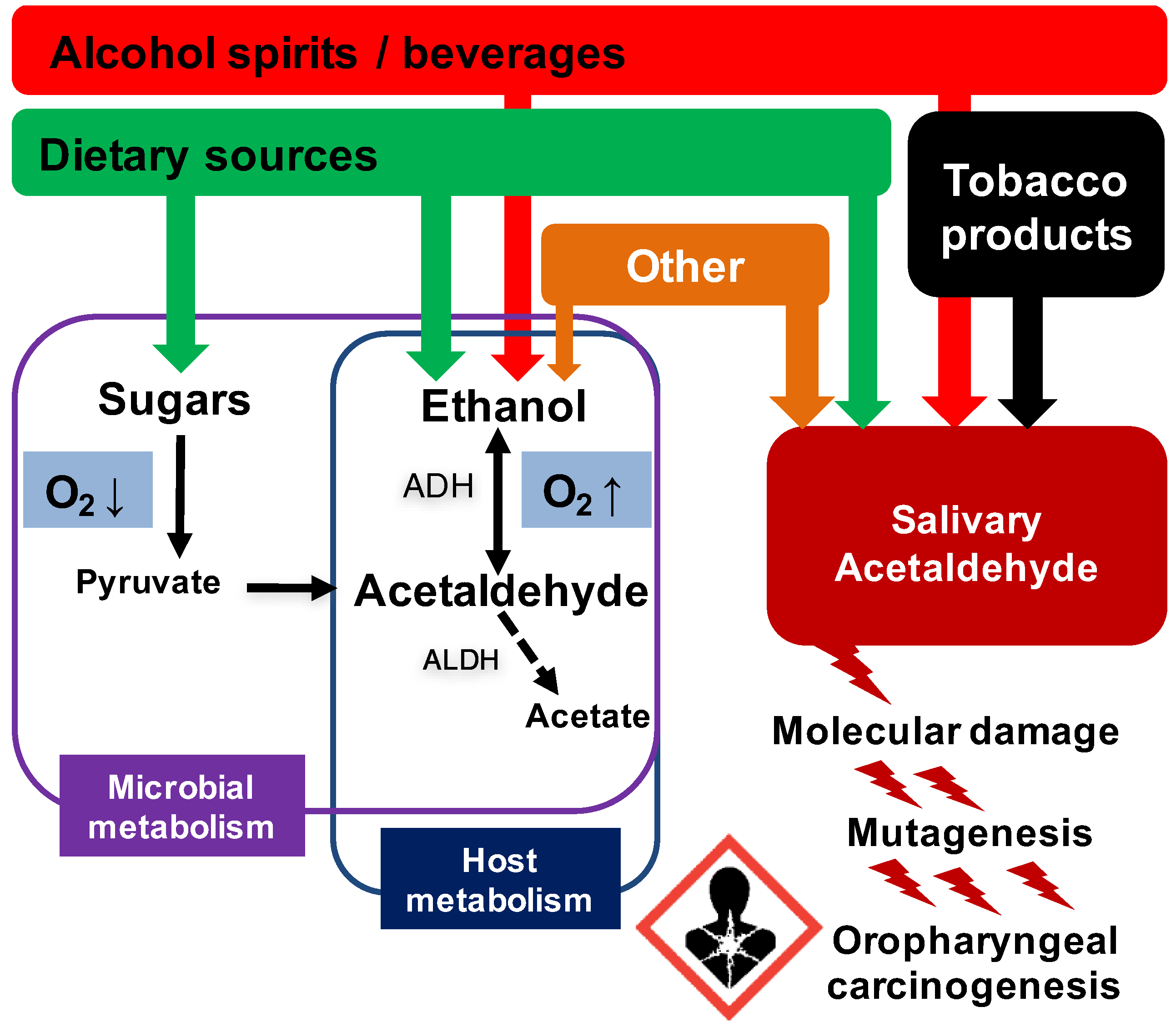{kind=link}
|
| Exposure Model | Salivary ACH Concentration (mg/L; µM) | Exposure Time (min) | ACH Exposure (mg/L × min)/day | OR/RR for Oropharyngeal Cancer |
|---|---|---|---|---|
| ALDH2-Deficiency Model | ||||
| - 3 doses (33 g ethanol)/day | 1.1; 25 1 | 283 2 | 311 1 | 1.68–2.61 3 |
| - 7 doses (77 g ethanol)/day | 1.1; 25 1 | 660 2 | 726 1 | 3.57–7.28 3 |
| 1 Dose 40% Alcohol, no ACH 4 | ||||
| - Instant | ||||
| 3 × first 5 min | 6.2; 150 | 15 | 93 | |
| 1 × next 5 min | 4.4; 100 | 5 | 22 | |
| instant total | 115 | 1.29 5 | ||
| - Long-term | ||||
| 20–80 min | 0.88; 20 | 60 | 53 | |
| - Total | ||||
| 0–80 min | 80 | 168 | ||
| Smoking 6 | ||||
| - 1 cigarette | 11.5; 261 | 5 | 58 | |
| - 3–5 cigarettes | 11.5; 261 | 15–25 | 173–288 | 2.017 |
| Yogurt | ||||
| - Safest-case scenario 8 | 2.4; 55 | 3 | 7 | ? |
| - Worst-case scenario 9 | 17.4; 395 | 3 × 5 | 261 | |
| Apple | ||||
| - Safest-case scenario 10 | 0.3; 7 | 3 | 0.9 | ? |
| - Worst-case scenario 11 | 2.4; 55 | 3 x 5 | 36 |
| Type of Cancer | Food Habit | OR | 95%CI | p-Value |
|---|---|---|---|---|
| Esophageal | Pickled vegetables 1 | 2.10 | 1.47–3.0 | <0.001 |
| Mursik milk 2 | 3.72 | 1.96–7.14 | <0.001 | |
| Gastric | Pickled vegetables 3 | 1.52 | 1.37–1.68 | <0.001 |
| Yogurt 4 | 16.26 | 2.1–125.7 | 0.008 | |
| Cheese 4 | 15.05 | 1.6–137.0 | 0.01 | |
| Moldy food 4 | 1.92 | 1.2–3.0 | 0.004 | |
| Pickling liquid 4 | ||||
| Brine | 4.76 | 2.2–10.4 | <0.001 | |
| Vinegar + brine | 3.34 | 1.6–7.0 | 0.002 |
Environmental risk factors:
Gene polymorphism-based risk conditions:
Disease-based risk conditions:
Iatrogenic risk conditions: |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieminen, M.T.; Salaspuro, M. Local Acetaldehyde—An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis. Cancers 2018, 10, 11. https://doi.org/10.3390/cancers10010011
Nieminen MT, Salaspuro M. Local Acetaldehyde—An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis. Cancers. 2018; 10(1):11. https://doi.org/10.3390/cancers10010011
Chicago/Turabian StyleNieminen, Mikko T., and Mikko Salaspuro. 2018. "Local Acetaldehyde—An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis" Cancers 10, no. 1: 11. https://doi.org/10.3390/cancers10010011
APA StyleNieminen, M. T., & Salaspuro, M. (2018). Local Acetaldehyde—An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis. Cancers, 10(1), 11. https://doi.org/10.3390/cancers10010011