Single Cell Isolation Using Optical Tweezers


Abstract
1. Introduction
1.1. Single Cell Isolation Methods
1.2. Single Cell Manipulation and Isolation Using Optical Tweezers
2. Materials and Methods
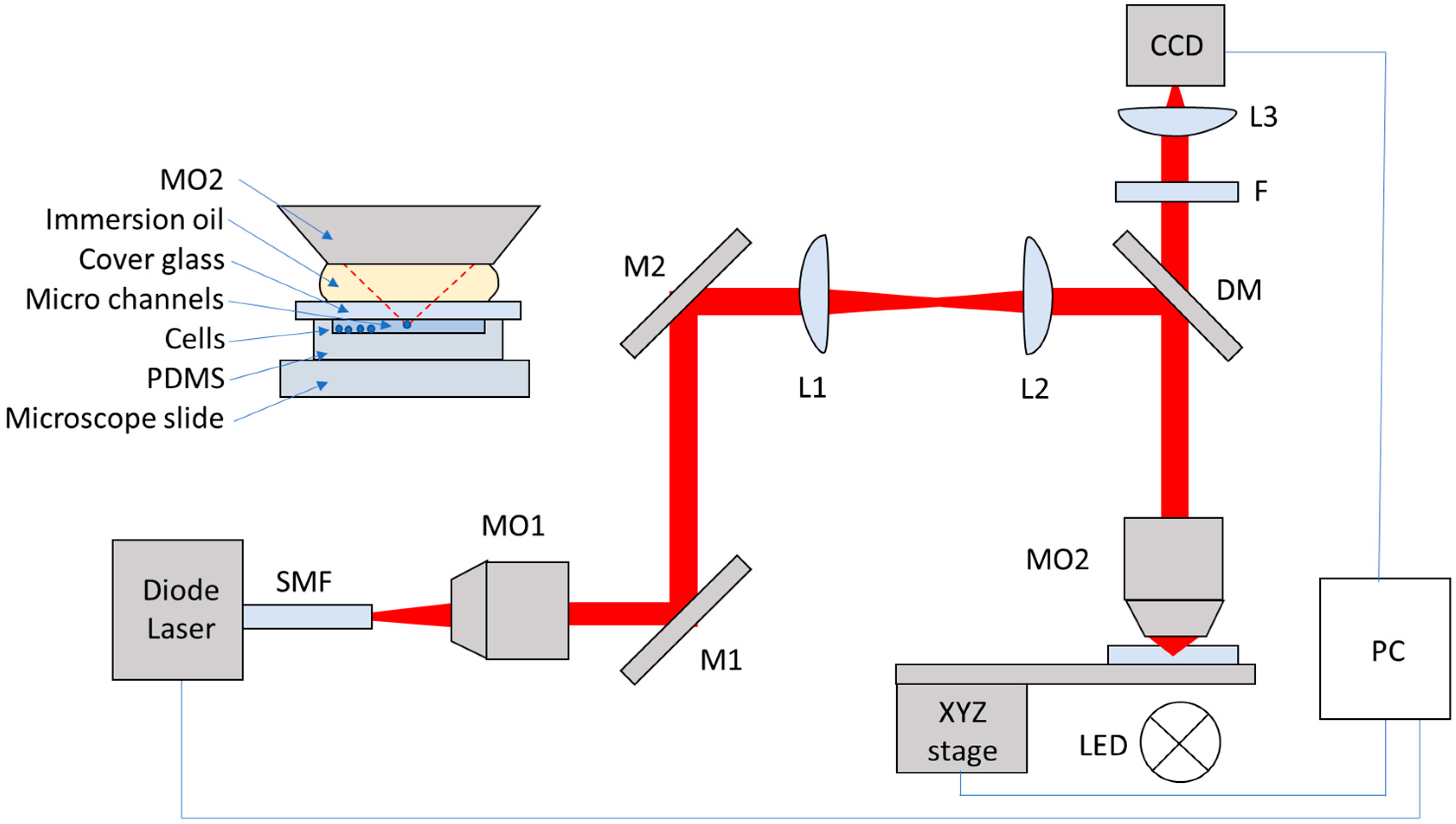
2.1. Optical Tweezers System
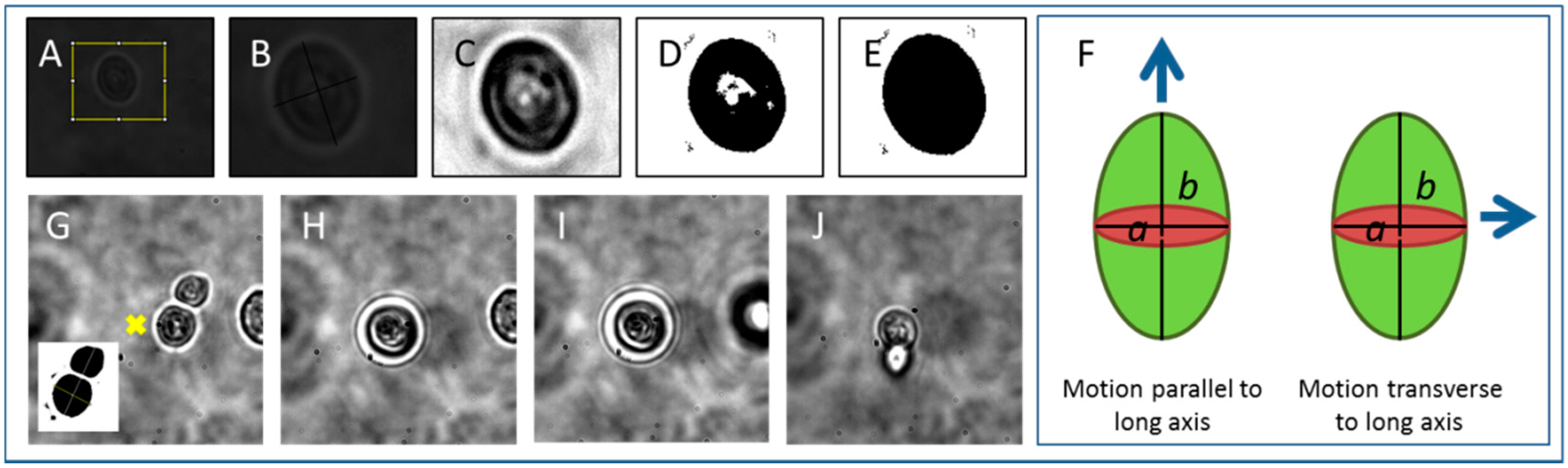
2.2. Cell Tweezing, Imaging and Tracking
2.3. Device Design and Fabrication
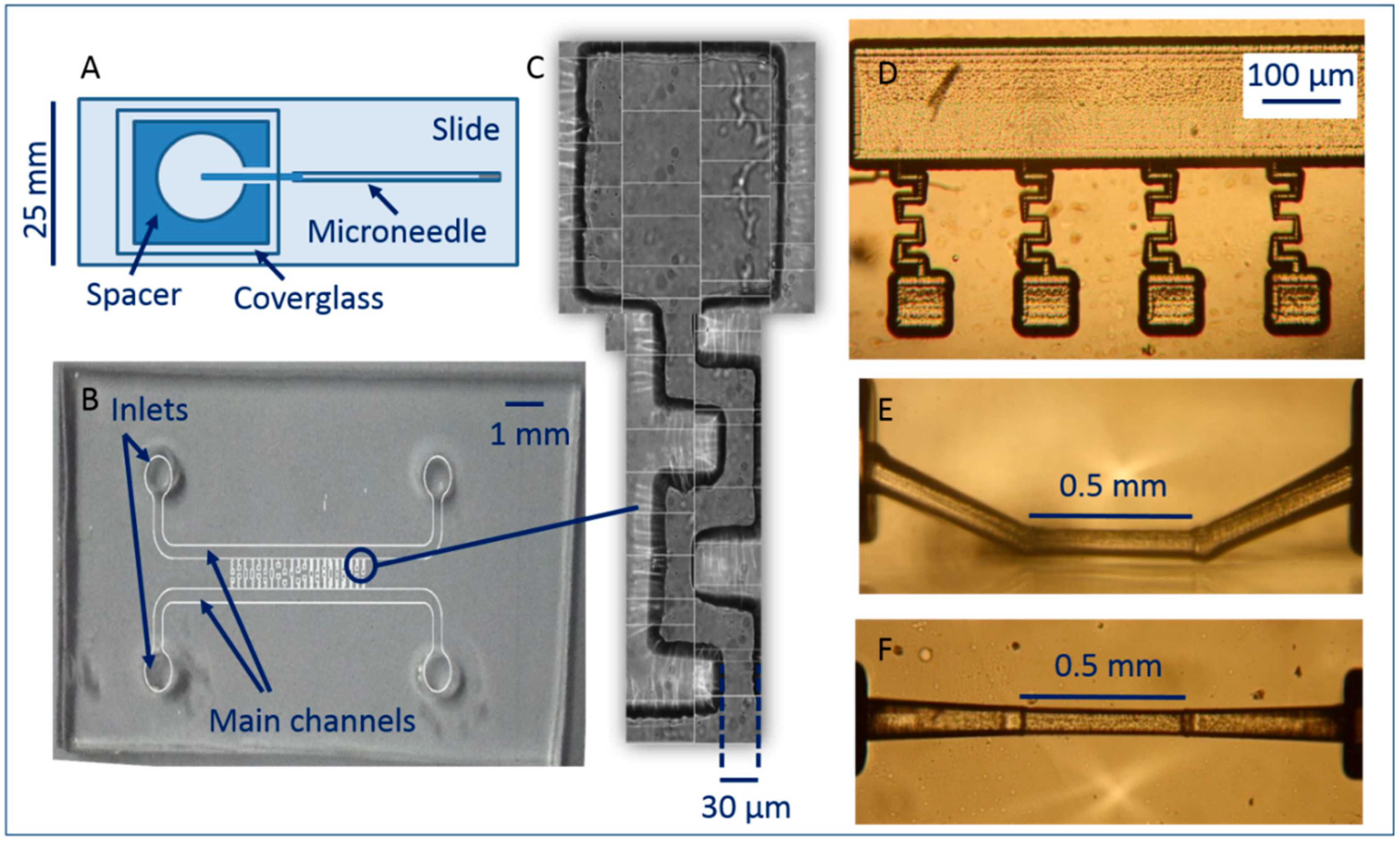
2.3.1. Hollow Glass Microneedle Channel (Micropipette)
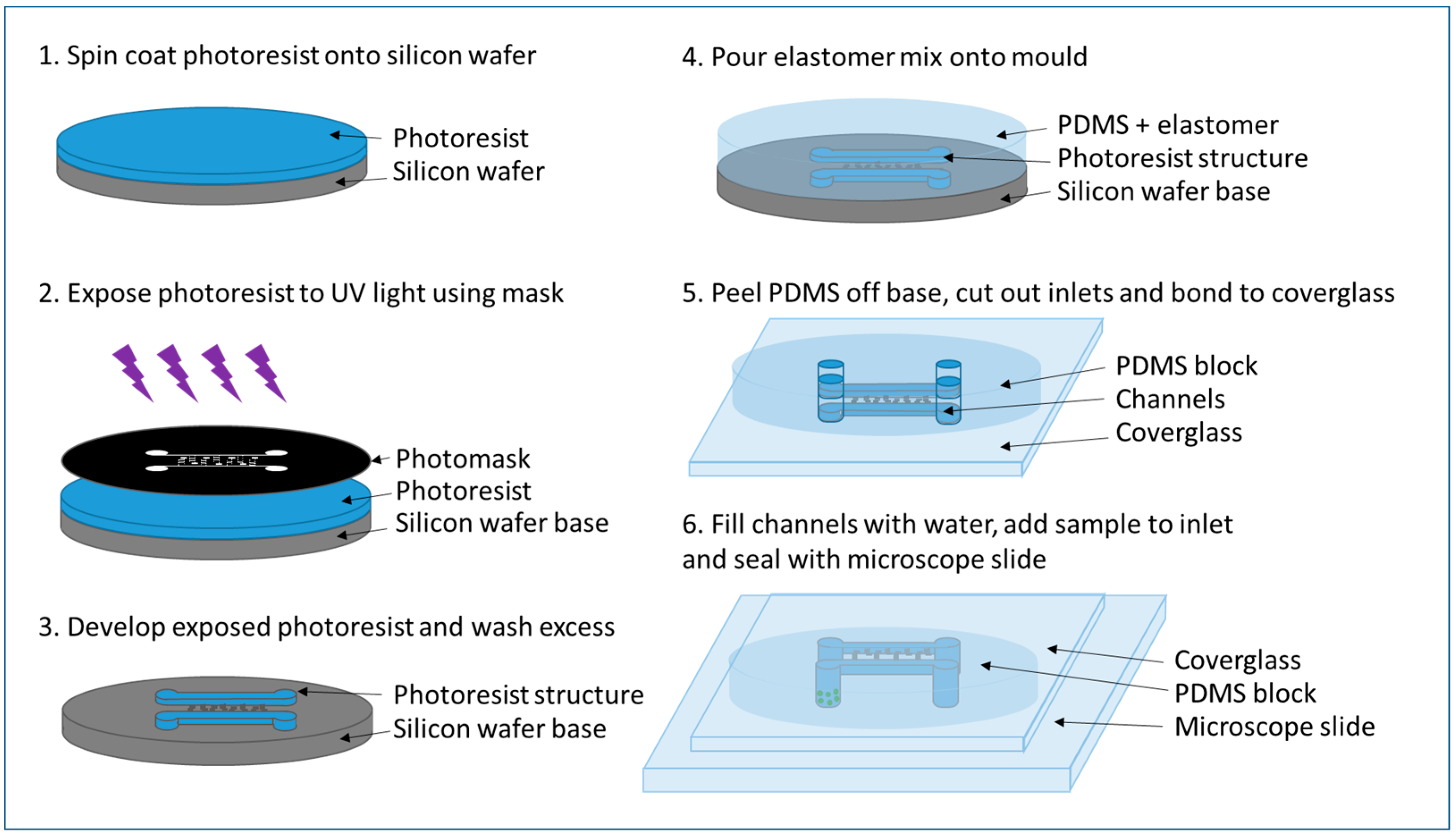
2.3.2. PDMS Chip
2.3.3. Ultrafast Laser Inscription and Selective Chemical Etching of Cell Isolation Chambers
3. Results and Discussion
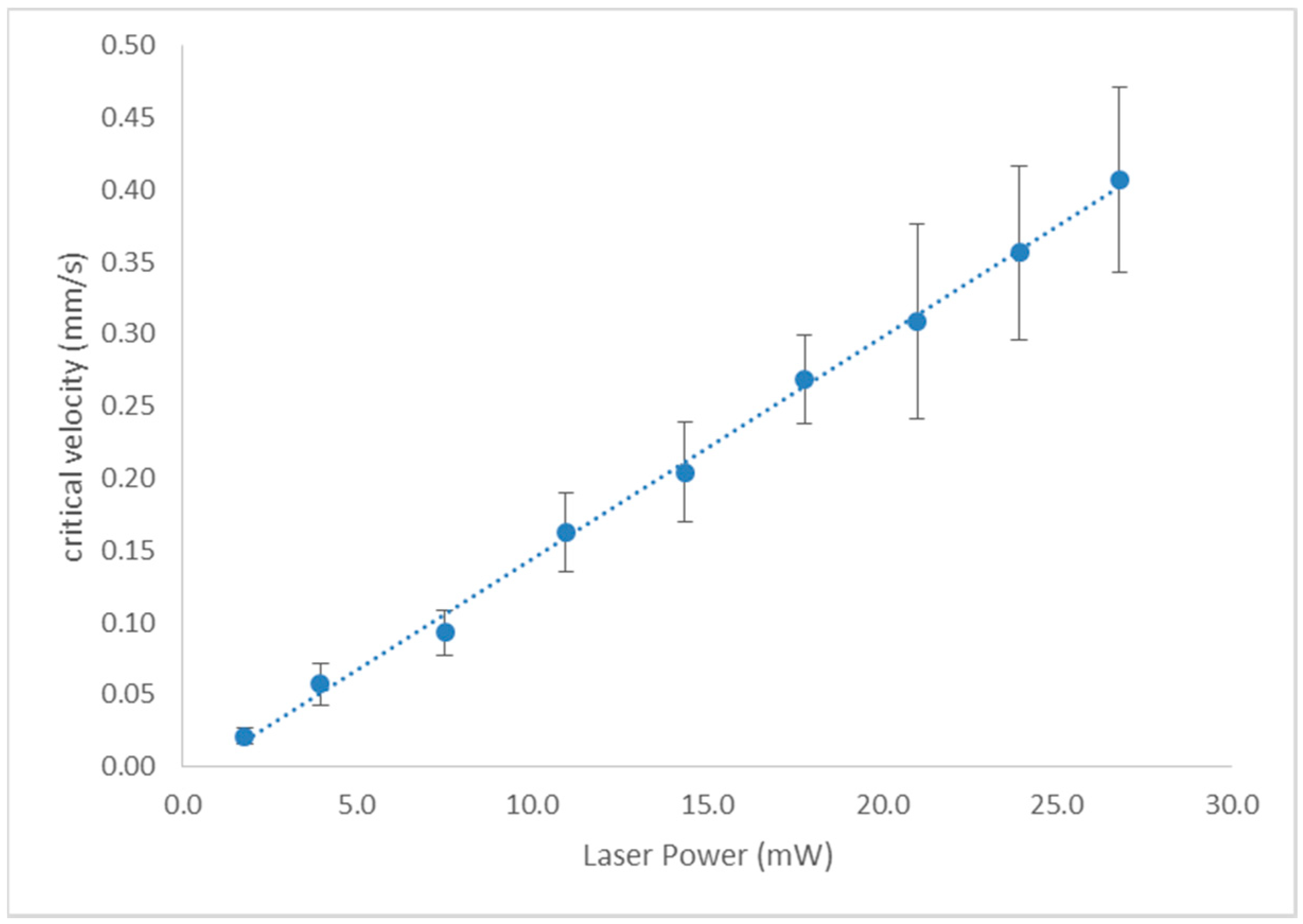
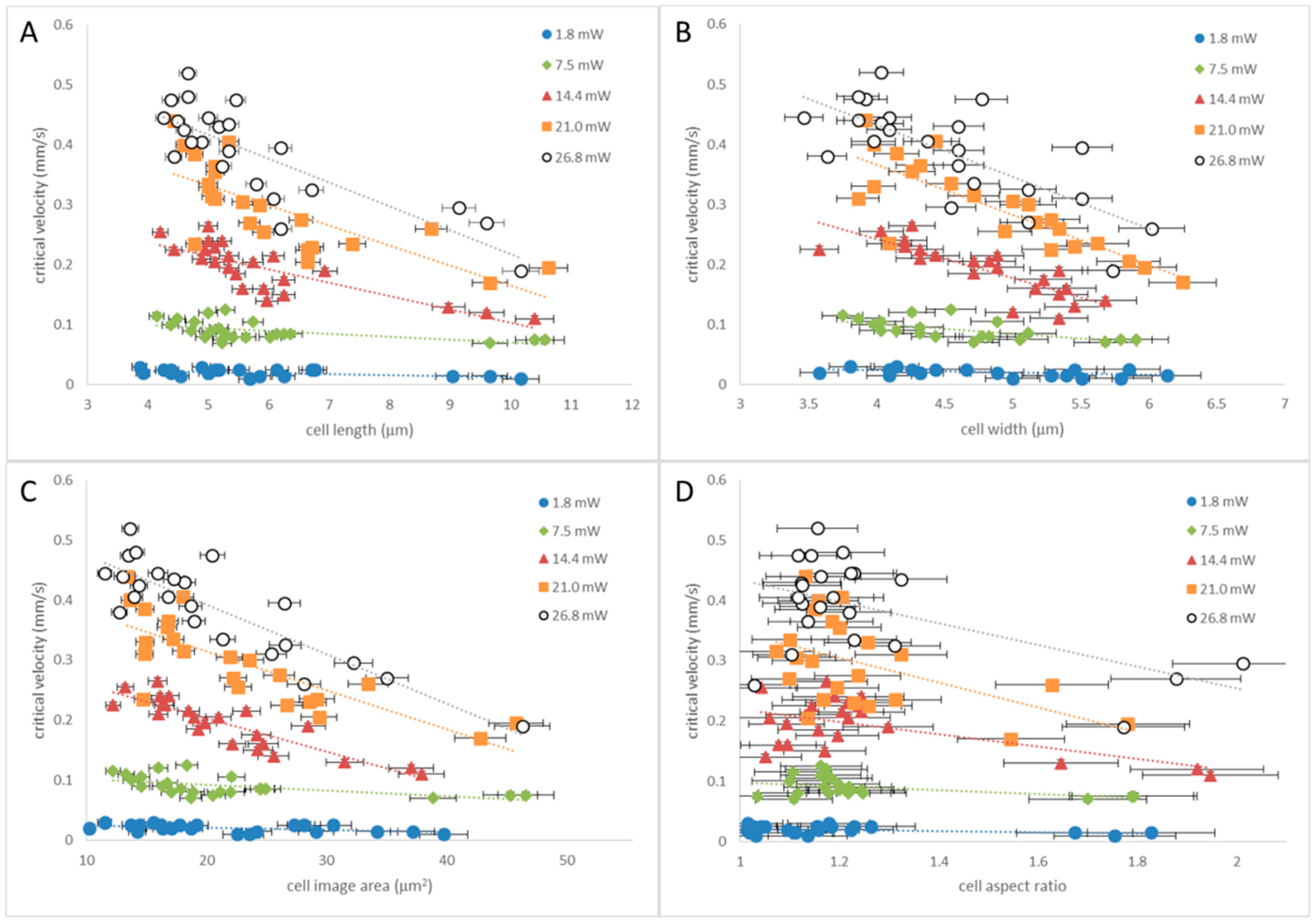
3.1. Tweezing Characterization
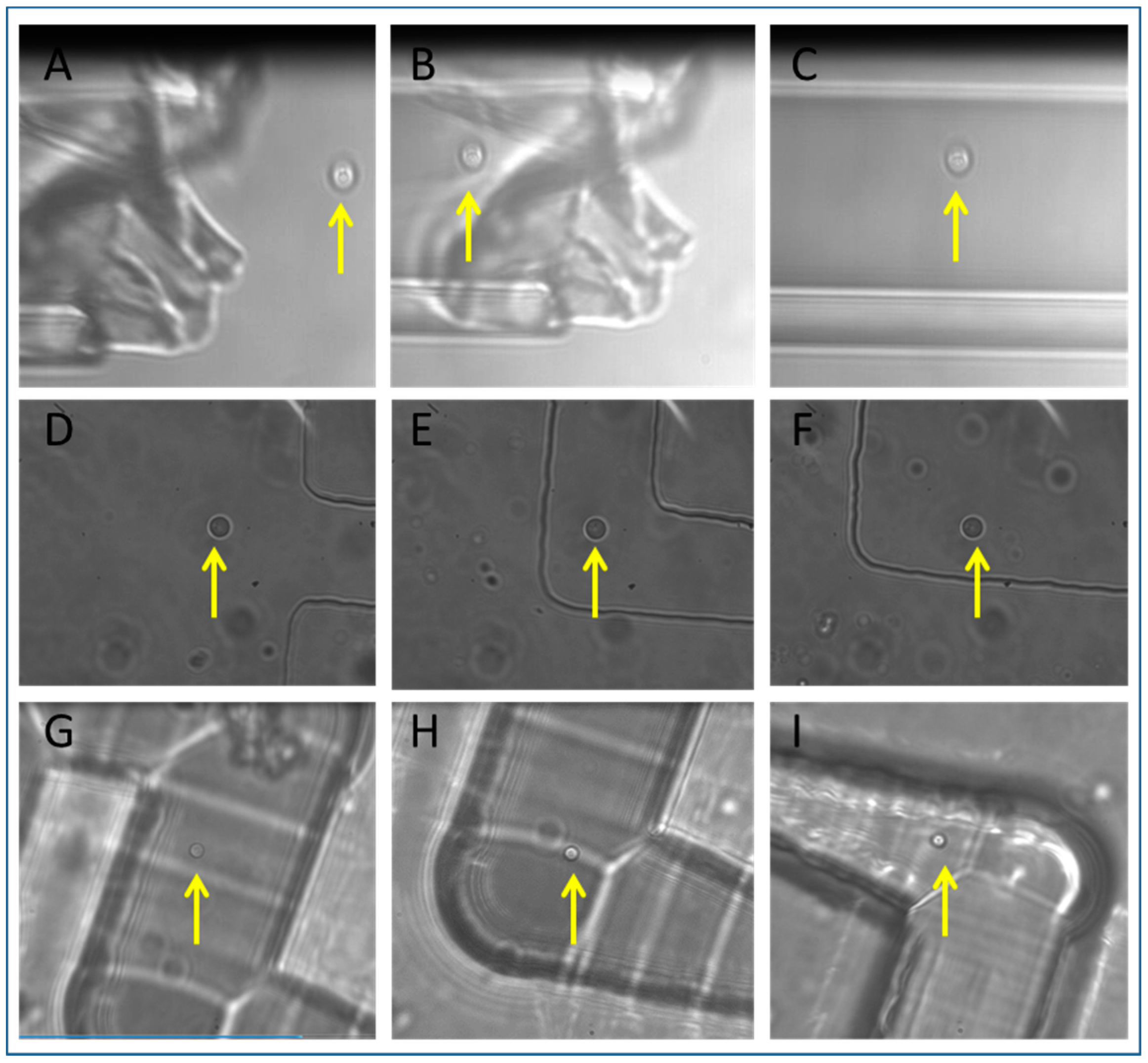
3.2. Tweezing in Isolation Devices
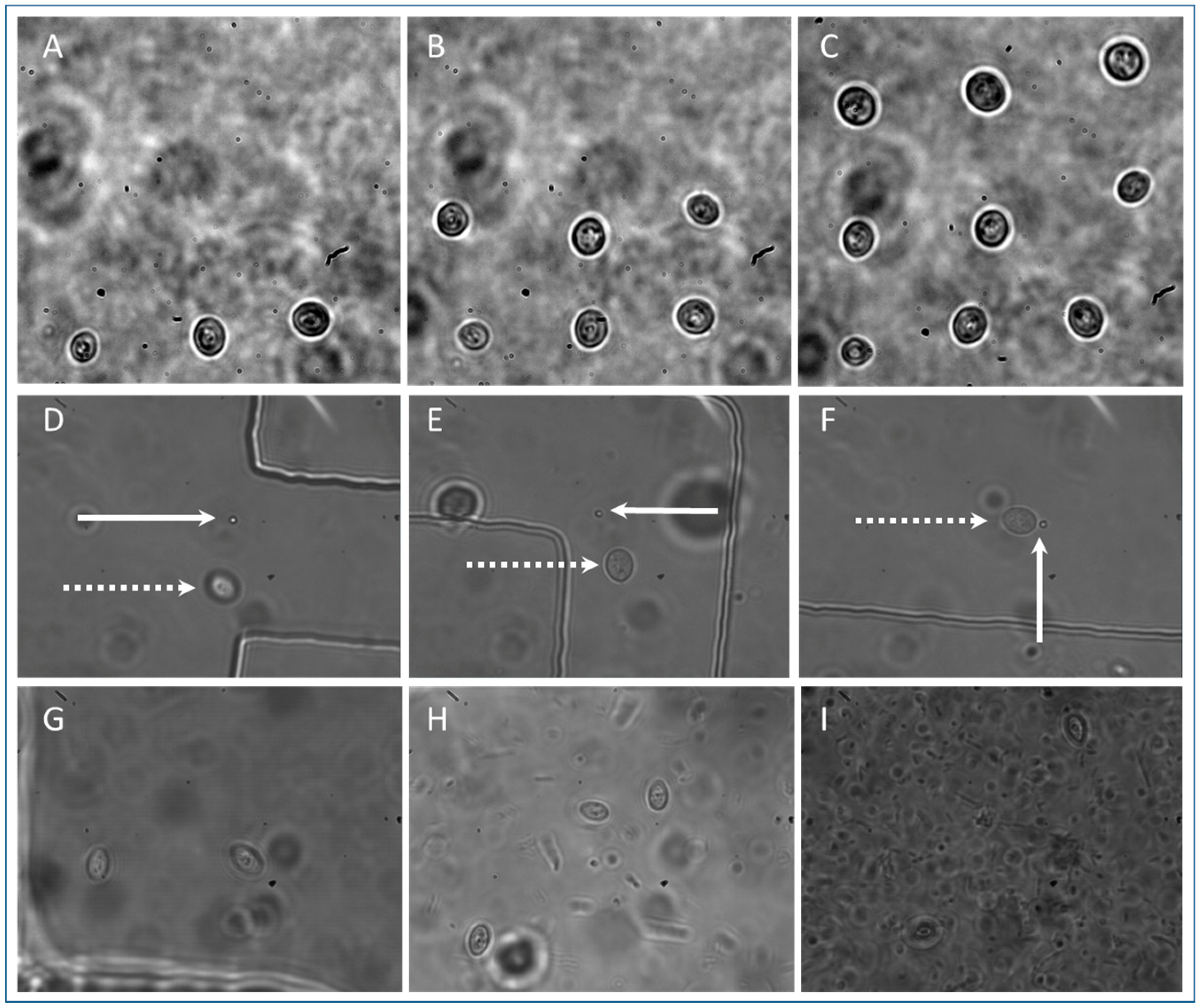
3.3. S. cerevisiae Isolation
3.4. Isolation of Other Cell Types
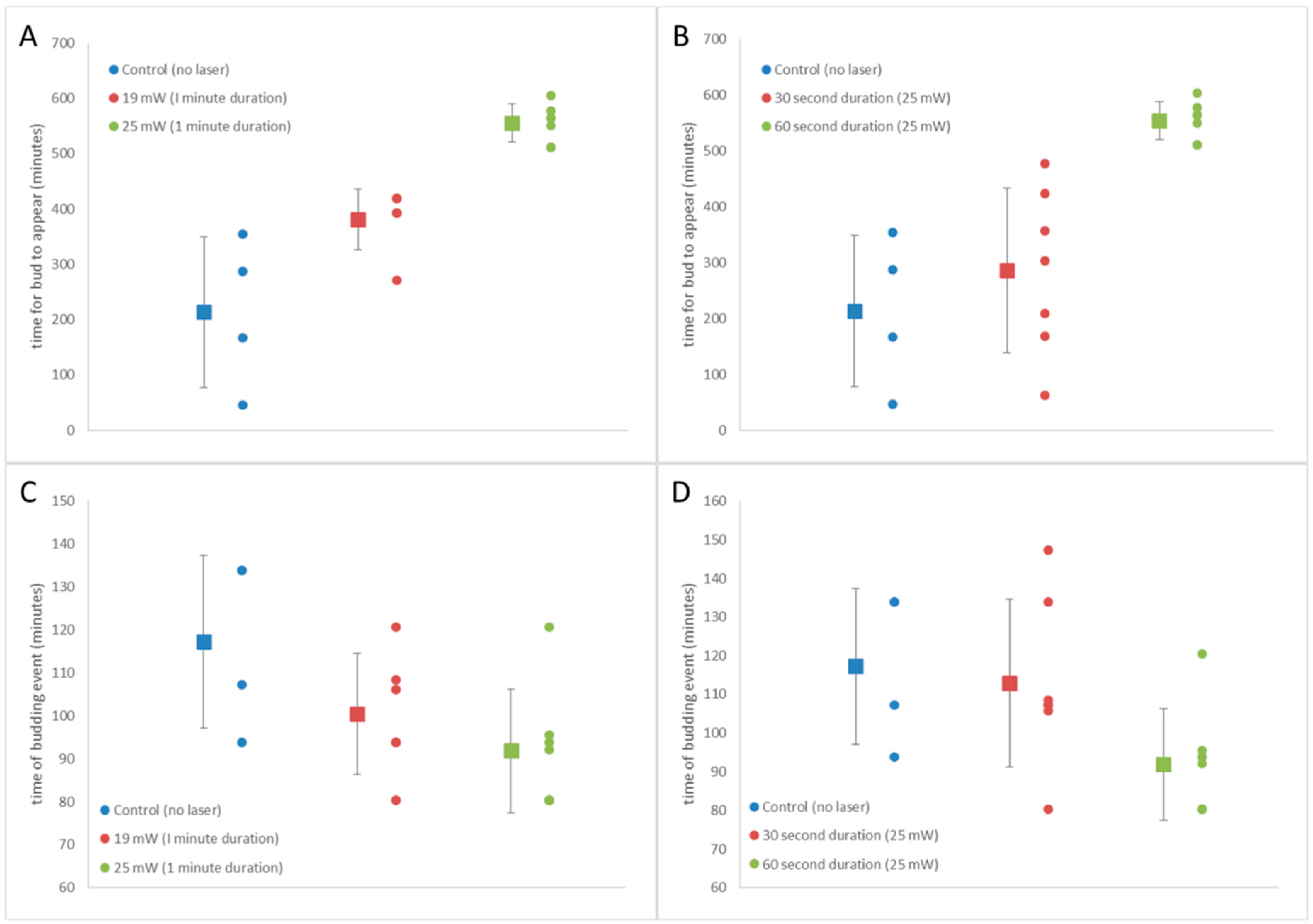
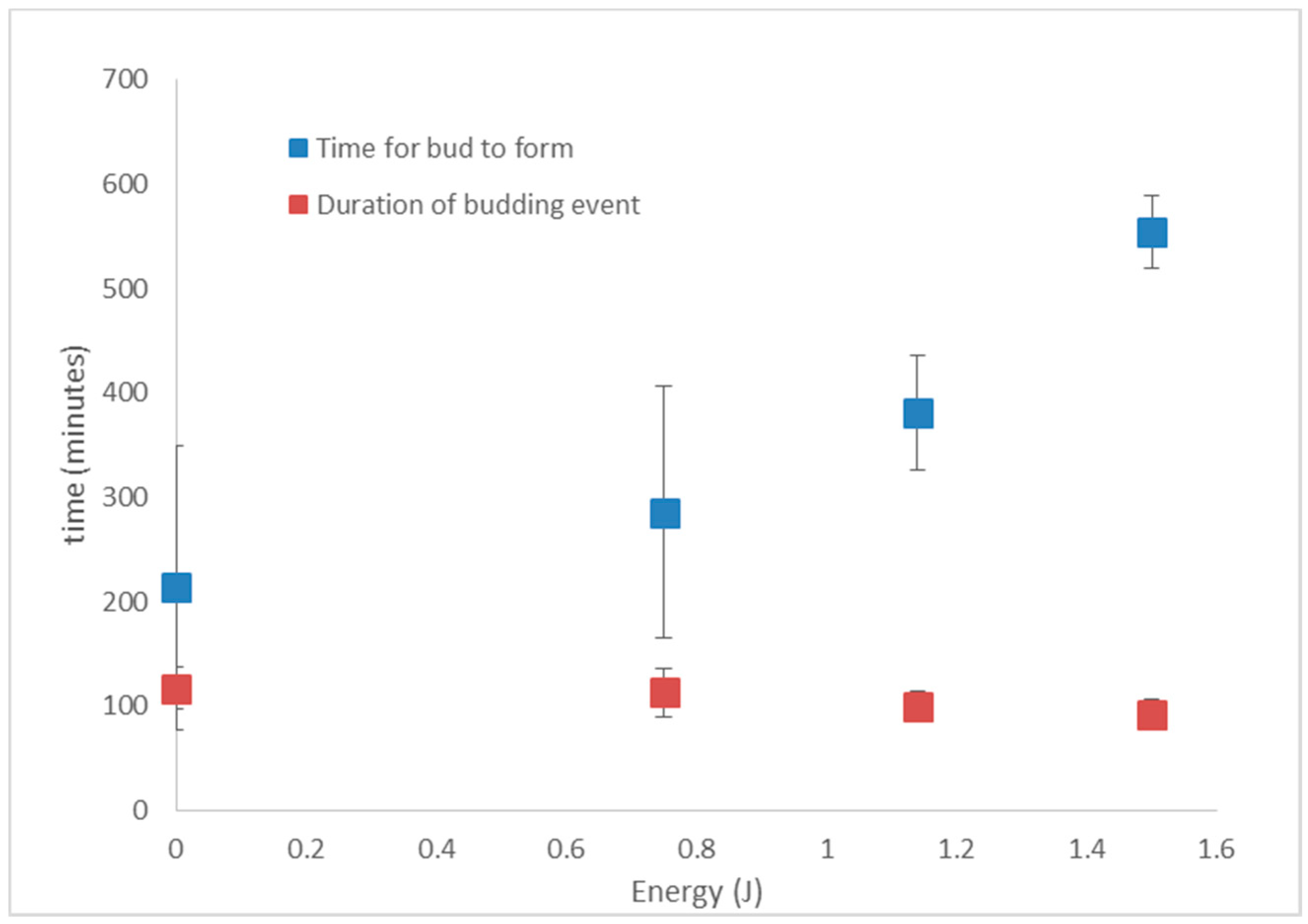
3.5. S. cerevisiae Doubling Time
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DiCarlo, D.; Lee, L.P. Dynamic single cell analysis for quantitative biology. Anal. Chem. 2006, 78, 7918–7925. [Google Scholar]
- Mohan, R.; Sanpitakseree, C.; Desai, A.V.; Sevgen, S.E.; Schroeder, C.M.; Kenis, P.A.J. A microfluidic approach to study the effect of bacterial interactions on antimicrobial susceptibility in polymicrobial cultures. RSC Adv. 2015, 5, 35211. [Google Scholar] [CrossRef]
- Gierahn, T.M.; Wadsworth, M.H.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395. [Google Scholar] [CrossRef] [PubMed]
- L’Haridon, S.; Markx, G.H.; Ingham, C.J.; Paterson, L.; Duthoit, F.; Le Blay, G. New approaches for bringing the uncultured into culture. In Marine Microbiome, An Untapped Source of Biodiversity and Biotechnological Potential; Stal, L.J., Cretoiu, M.S., Eds.; Springer: Basel, Switzerland, 2016; pp. 401–434. [Google Scholar]
- Barber, M.A. Use of the single cell method in obtaining pure cultures of anaerobes. J. Exp. Med. 1920, 32, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015, 16, 16897–16919. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, K.I. The isolation and cultivation of single organisms. In Methods in Microbiology; Norris, J.R., Ribbons, D.W., Eds.; Academic Press Inc.: New York, NY, USA, 1969; Volume 1. [Google Scholar]
- Frohlich, J.; Konig, H. New techniques for isolation of single prokaryotic cells. FEMS Microbiol. Rev. 2000, 24, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser capture microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; McMillan, I.; Norris, M.H.; Hoang, T.T. Single prokaryotic cell isolation and total transcript amplification protocol for transcriptomic analysis. Nat. Protoc. 2015, 10, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Hulett, H.R.; Bonner, W.A.; Barrett, J.; Herzenberg, L.A. Cell sorting: Automated separation of mammalian cells as a function of intracellular fluorescence. Science 1969, 166, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Middlebrook, A.; Pennebaker, K.; Chang, C.; Shum, E.; Fan, C.; Weaver, S. A Complete Workflow from Single Cell Isolation to mRNA Sequencing Analysis, Complete Workflow from Single Cell Isolation to mRNA Sequencing Analysis. 2016. Available online: http://www.bd.com/genomics (accessed on 20 August 2018).
- Davey, H.M.; Winson, M.K. Using flow cytometry to quantify microbial heterogeneity. Curr. Issues Mol. Biol. 2003, 5, 9–15. [Google Scholar] [PubMed]
- Connell, J.L.; Wessel, A.K.; Parsek, M.R.; Ellington, A.D.; Whiteley, M.; Shear, J.B. Probing prokaryotic social behaviors with bacterial “lobster traps”. mBio 2010, 1, e00202–e00210. [Google Scholar] [CrossRef] [PubMed]
- Probst, C.; Grünberger, A.; Wiechert, W.; Kohlheyer, D. Polydimethylsiloxane (PDMS) sub-micron traps for single-cell analysis of bacteria. Micromachines 2013, 4, 357–369. [Google Scholar] [CrossRef]
- Lu, H.; Caen, O.; Vrignon, J.; Zonta, E.; El Harrak, Z.; Nizard, P.; Baret, J.-C.; Taly, V. High throughput single cell counting in droplet-based microfluidics. Sci. Rep. 2017, 7, 1366. [Google Scholar] [CrossRef] [PubMed]
- Mazutis, L.; Gilbert, J.; Ung, W.L.; Weitz, D.A.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef] [PubMed]
- Applegate, R.W.; Squier, J.; Vestad, T.; Oakey, J.; Marr, D.W.; Bado, P.; Dugan, M.A.; Said, A.A. Microfluidic sorting system based on optical waveguide integration and diode laser bar trapping. Lab Chip 2006, 6, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, O.; Grenvall, C.; Nordin, M.; Evander, M.; Lauell, T. Acoustic actuated fluorescence activated sorting of microparticles. Lab Chip 2014, 14, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Evander, M.; Johansson, L.; Lilliehorn, T.; Piskur, J.; Lindvall, M.; Johansson, S.; Almqvist, M.; Laurell, T.; Nilsson, J. Noninvasive acoustic cell trapping in a microfluidic perfusion system for online bioassays. Anal. Chem. 2007, 79, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Voldman, J. Electrical forces for microscale cell manipulation. Annu. Rev. Biomed. Eng. 2006, 8, 425–454. [Google Scholar] [CrossRef] [PubMed]
- Zborowski, M.; Chalmers, J.J. Magnetic cell sorting. In Immunochemical Protocols; Burns, R., Ed.; Humana Press: New York, NY, USA, 2005; Volume 259. [Google Scholar]
- Wyatt Shields, C.; Reyes, C.D.; Lopez, G.P. Microfluidic cell sorting: A review of the advances in the separation of cells from debulking to rare cell isolation. Lab Chip 2015, 15, 1230–1249. [Google Scholar] [CrossRef] [PubMed]
- Ashkin, A.; Dziedzic, J.M.; Yamane, T. Optical trapping and manipulation of single cells using infrared laser beams. Nature 1987, 330, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, D.K.; Sonek, G.J.; Berns, M.W.; Chapman, C.F.; Tromberg, B.F. Evidence for localized cell heating induced by infrared optical tweezers. Biophys. J. 1995, 68, 2137–2144. [Google Scholar] [CrossRef]
- Liang, H.; Vu, K.T.; Krishnan, P.; Trang, T.C.; Shin, D.; Kimel, S.; Berns, M.W. Wavelength dependence of cell cloning efficiency after optical trapping. Biophys. J. 1996, 70, 1529–1533. [Google Scholar] [CrossRef]
- Liu, Y.; Sonek, G.J.; Berns, M.W.; Tromberg, B.J. Physiological monitoring of optical trapped cells: Assessing the effects of confinement by 1064-nm laser tweezers using microfluorometry. Biophys. J. 1996, 71, 2158–2167. [Google Scholar] [CrossRef]
- Neuman, K.C.; Chadd, E.H.; Liou, G.F.; Bergman, K.; Block, S.M. Characterization of photodamage to Escherichia coli in optical traps. Biophys. J. 1999, 77, 2856–2863. [Google Scholar] [CrossRef]
- Leitz, G.; Fallman, E.; Tuck, S.; Axner, O. Stress response in Caenorhabditis elegans caused by optical tweezers: wavelength, power, and time dependence. Biophys. J. 2002, 82, 2224–2231. [Google Scholar] [CrossRef]
- Ayano, S.; Wakamoto, Y.; Yamashita, S.; Yasuda, K. Quantitative measurement of damage caused by 1064-nm wavelength optical trapping of Escherichia coli cells using on-chip single cell cultivation system. Biochem. Biophys. Res. Commun. 2006, 350, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Haro-González, P.; Ramsay, W.T.; Maestro, L.M.; del Rosal, B.; Santacruz-Gomez, K.; Iglesias-de la Cruz, M.; Sanz-Rodríguez, F.; Choo, J.Y.; Rodriguez Sevilla, P.; Bettinelli, M.; et al. Quantum dot-based thermal spectroscopy and imaging of optically trapped microspheres and single cells. Small 2013, 9, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.C.; Skirtach, A.G.; Gauthier, R.C.; Grover, C.P. Automated single-cell sorting system based on optical trapping. J. Biomed. Opt. 2001, 6, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Wakamoto, Y.; Umehara, S.; Matsumura, K.; Inoue, I.; Yasuda, K. Development of non-destructive, non-contact single-cell based differential cell assay using on-chip microcultivation and optical tweezers. Sens. Actuators B 2003, 96, 693–700. [Google Scholar] [CrossRef]
- Umehara, S.; Inoue, I.; Wakamoto, Y.; Yasuda, K. Origin of individuality of two daughter cells during the division process examined by the simultaneous measurement of growth and swimming property using an on-chip single-cell cultivation system. Biophys. J. 2007, 93, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Enger, J.; Goksör, M.; Ramser, K.; Hagberg, P.; Hanstorp, D. Optical tweezers applied to a microfluidic system. Lab Chip 2004, 4, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Enger, J.; Nordlander, B.; Erjavec, N.; Ramser, K.; Goksör, M.; Hohmann, S.; Nyström, T.; Hanstorp, D. A microfluidic system in combination with optical tweezers for analyzing rapid and reversible cytological alterations in single cells upon environmental changes. Lab Chip 2007, 7, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Sott, K.; Lundqvist, F.; Sveningsson, M.; Scrimgeour, J.; Hanstrop, D.; Goksor, M.; Graneli, A. A microfluidic device for reversible environmental changes around single cells using optical tweezers for cell selection and positioning. Lab Chip 2010, 10, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Kasukurti, A.; Potcoava, M.; Desai, S.A.; Eggleton, C.; Marr, D.W.M. Single cell isolation using a DVD optical pickup. Opt. Express 2011, 19, 10377. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, S.; Kong, M.; Wang, Z.; Costa, K.D.; Li, R.A.; Sun, D. Enhanced cell sorting and manipulation with combined optical tweezer and microfluidic chip technologies. Lab Chip 2011, 11, 3656. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gou, X.; Chen, S.; Yan, X.; Sun, D. Cell manipulation tool with combined microwell array and optical tweezers for cell isolation and deposition. J. Micromech. Microeng. 2013, 23, 075006. [Google Scholar] [CrossRef]
- Probst, C.; Grunberger, A.; Wiechert, W.; Kohlheyer, D. Microfluidic growth chambers with optical tweezers for full spatial single-cell control and analysis of evolving microbes. J. Microbiol. Methods 2013, 95, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.H.; Sonek, G.J.; Berns, M.W. Parametric study of the forces on microspheres help by optical tweezers. Appl. Opt. 1994, 33, 1735. [Google Scholar] [CrossRef] [PubMed]
- Happel, J.; Brenner, H. Low Reynolds Number Hydrodynamics with Special Applications to Particulate Media; Springer: Basel, Switzerland, 1965. [Google Scholar]
- Jones, P.H.; Maragò, O.M.; Volpe, G. Optical Tweezers: Principles and Applications; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Borghese, F.; Denti, P.; Saija, R.; Iatı, M.A.; Marago, O.M. Radiation torque and force on optically trapped linear nanostructures. Phys. Rev. Lett. 2008, 100, 163903. [Google Scholar] [CrossRef] [PubMed]
- Hensley, Z.D.; Papavassiliou, D.V. Drag coefficient correction for spherical and nonspherical particles suspended in square microducts. Ind. Eng. Chem. Res. 2014, 53, 10465–10474. [Google Scholar] [CrossRef]
- Srivastava, D.K.; Yadav, R.R.; Srivastava, N. Oseen’s correction to Stokes Drag on axially symmetric particle in micropolar fluid. Int. J. Adv. Appl. Math. Mech. 2016, 3, 41–78. [Google Scholar]
- Choudhury, D.; Ramsay, W.T.; Kiss, R.; Willoughby, N.A.; Paterson, L.; Kar, A.K. A 3D mammalian cell separator biochip. Lab Chip 2012, 12, 948. [Google Scholar] [CrossRef] [PubMed]
- Keloth, A. Three Dimensional Optofluidic Devices for Manipulation of Particles and Cells. Ph.D. Thesis, Heriot Watt University, Edinburgh, UK, 2017. [Google Scholar]
- Steinhaus, B.; Garcia, M.L.; Shen, A.Q.; Angenent, L.T. A portable anaerobic microbioreactor reveals optimum growth conditions for the methanogen Methanosaeta concilii. Appl. Environ. Microbiol. 2007, 73, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.; Leach, J.; Padgett, M.J.; Blackburn, P.; Isaacs, N.; Goksor, M.; Hanstorp, D.; Wright, A.; Girkin, J.; Cooper, J. Creating permanent 3D arrangements of isolated cells using holographic optical tweezers. Lap Chip 2005, 5, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Braff, D.; David Shis, D.; Collins, J.J. Synthetic biology platform technologies for antimicrobial applications. Adv. Drug Delivery Rev. 2016, 105, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Leitao, R.M.; Kellog, D.R. The duration of mitosis and daughter cell size are modulated by nutrients in budding yeast. J. Cell Biol. 2017, 216, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Pilát, Z.; Jonáš, A.; Ježek, J.; Zemánek, P. Effects of infrared optical trapping on saccharomyces cerevisiae in a microfluidic system. Sensors 2017, 17, 2640. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Correction Applied Power (mW) | (N) | (N) | (N) | (N) | (N) | (N) |
|---|---|---|---|---|---|---|
| 1.8 ± 0.1 | 1.04 ± 0.46 × 10−12 | 1.88 ± 0.83 × 10−12 | 1.08 ± 0.48 × 10−12 | 1.94 ± 0.85 × 10−12 | 1.92 ± 0.84 × 10−12 | 1.09 ± 0.48 × 10−12 |
| 4.0 ± 0.1 | 2.87 ± 1.08 × 10−12 | 4.98 ± 1.87 × 10−12 | 2.98 ± 1.12 × 10−12 | 5.15 ± 1.93 × 10−12 | 5.13 ± 1.92 × 10−12 | 3.04 ± 1.14 × 10−12 |
| 7.5 ± 0.1 | 4.61 ± 1.32 × 10−12 | 7.87 ± 2.25 × 10−12 | 4.79 ± 1.37 × 10−12 | 8.13 ± 2.33 × 10−12 | 8.14 ± 2.33 × 10−12 | 4.93 ± 1.41 × 10−12 |
| 11.0 ± 0.1 | 8.12 ± 2.27 × 10−12 | 1.39 ± 0.39 × 10−11 | 8.43 ± 2.36 × 10−12 | 1.44 ± 0.40 × 10−11 | 1.44 ± 0.40 × 10−11 | 8.66 ± 2.43 × 10−12 |
| 14.4 ± 0.1 | 1.05 ± 0.31 × 10−11 | 1.81 ± 0.53 × 10−11 | 1.09 ± 0.32 × 10−11 | 1.87 ± 0.55 × 10−11 | 1.86 ± 0.55 × 10−11 | 1.11 ± 0.33 × 10−11 |
| 17.8 ± 0.1 | 1.35 ± 0.38 × 10−11 | 2.3 ± 0.58 × 10−11 | 1.4 ± 0.35 × 10−11 | 2.38 ± 0.60 × 10−11 | 2.38 ± 0.60 × 10−11 | 1.44 ± 0.36 × 10−11 |
| 21.0 ± 0.1 | 1.62 ± 0.60 × 10−11 | 2.74 ± 1.01 × 10−11 | 1.68 ± 0.62 × 10−11 | 2.83 ± 1.04 × 10−11 | 2.84 ± 1.05 × 10−11 | 1.74 ± 0.64 × 10−11 |
| 23.9 ± 0.1 | 1.77 ± 0.53 × 10−11 | 3.07 ± 0.92 × 10−11 | 1.84 ± 0.55 × 10−11 | 3.18 ± 0.96 × 10−11 | 3.17 ± 0.95 × 10−11 | 1.88 ± 0.57 × 10−11 |
| 26.8 ± 0.1 | 1.99 ± 0.58 × 10−11 | 3.41 ± 1.00 × 10−11 | 2.06 ± 0.61 × 10−11 | 3.52 ± 1.04 × 10−11 | 3.52 ± 1.04 × 10−11 | 2.12 ± 0.62 × 10−11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keloth, A.; Anderson, O.; Risbridger, D.; Paterson, L. Single Cell Isolation Using Optical Tweezers. Micromachines 2018, 9, 434. https://doi.org/10.3390/mi9090434
Keloth A, Anderson O, Risbridger D, Paterson L. Single Cell Isolation Using Optical Tweezers. Micromachines. 2018; 9(9):434. https://doi.org/10.3390/mi9090434
Chicago/Turabian StyleKeloth, Anusha, Owen Anderson, Donald Risbridger, and Lynn Paterson. 2018. "Single Cell Isolation Using Optical Tweezers" Micromachines 9, no. 9: 434. https://doi.org/10.3390/mi9090434
APA StyleKeloth, A., Anderson, O., Risbridger, D., & Paterson, L. (2018). Single Cell Isolation Using Optical Tweezers. Micromachines, 9(9), 434. https://doi.org/10.3390/mi9090434