Single-Molecule Tethered Particle Motion: Stepwise Analyses of Site-Specific DNA Recombination


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. TPM as a Probe for Polymer–Ligand Interactions
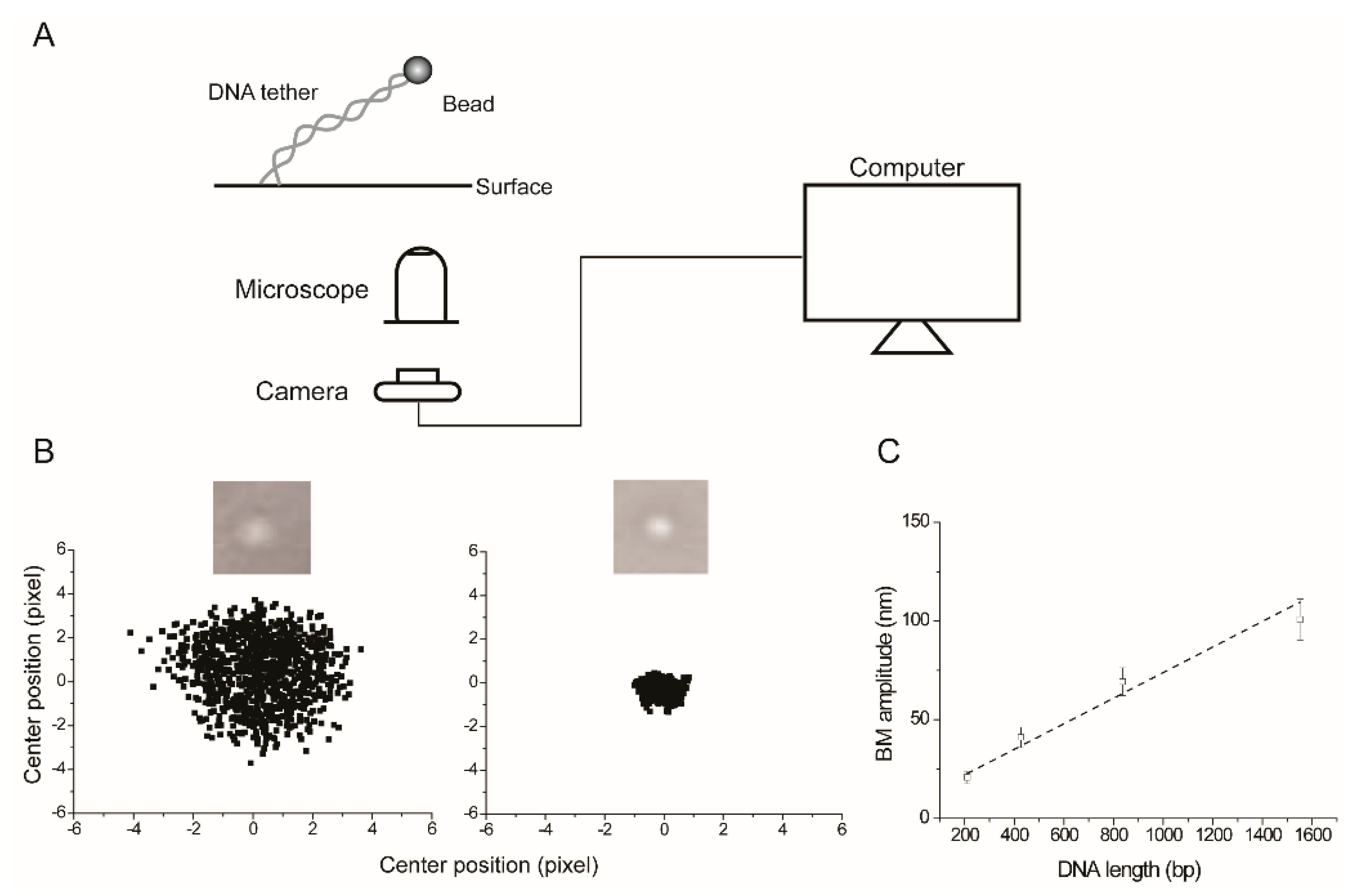
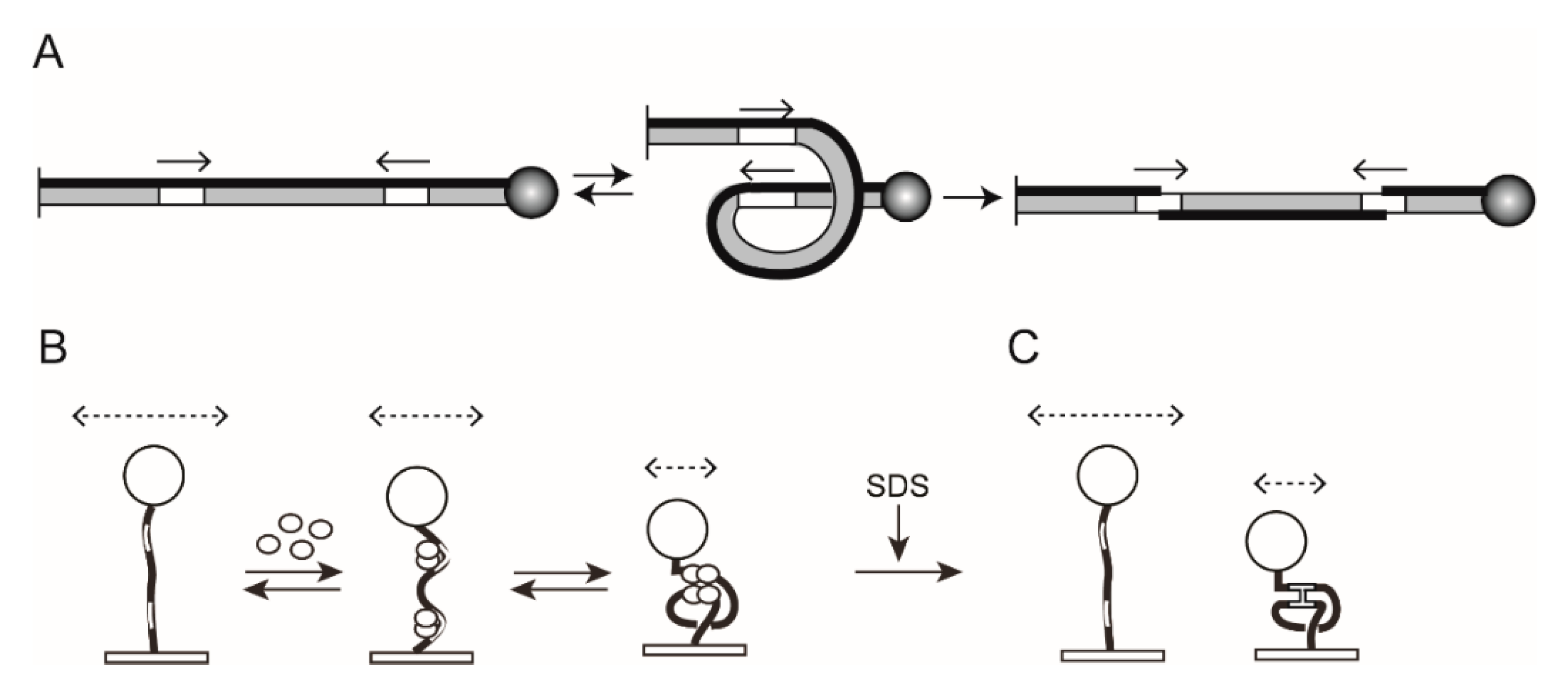
2.1. The Experimental Set Up for TPM
2.2. Biological Applications of TPM
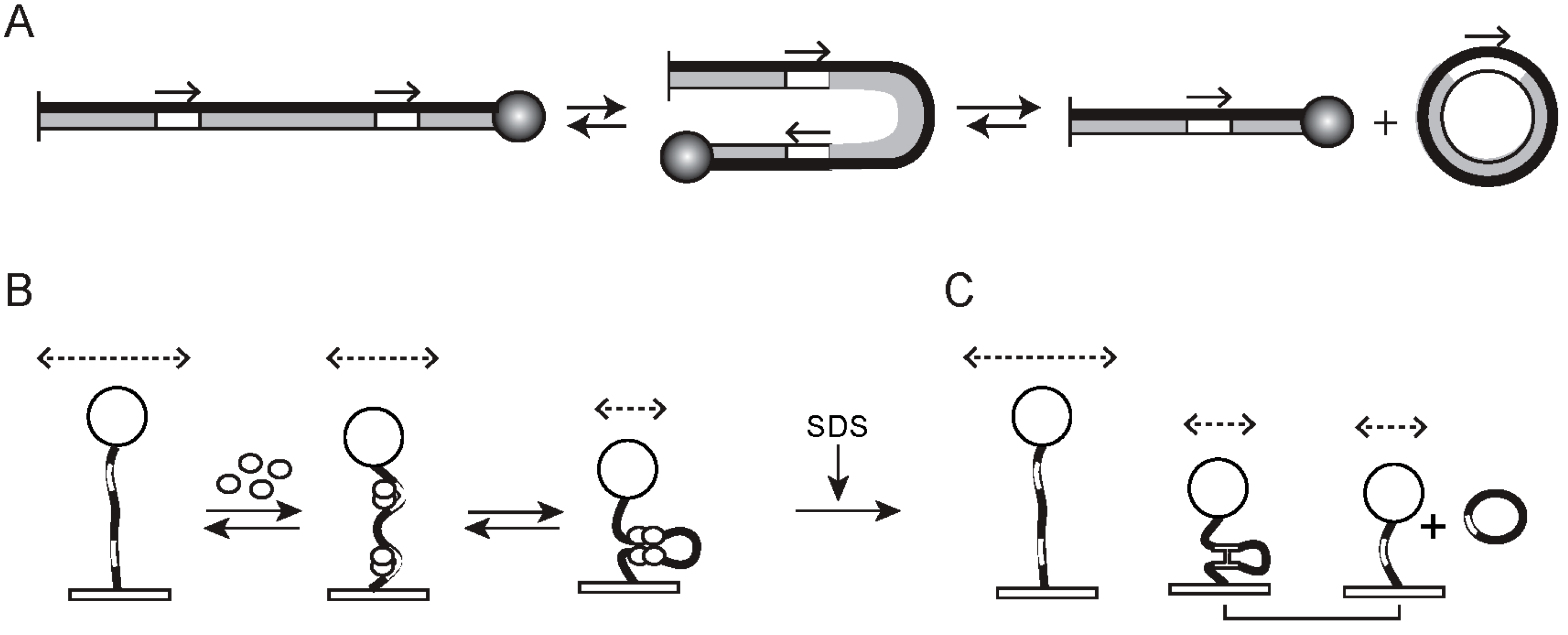
2.3. Technical and Analytical Improvements of Basic TPM
2.4. TPM Analysis of Site-Specific Recombination
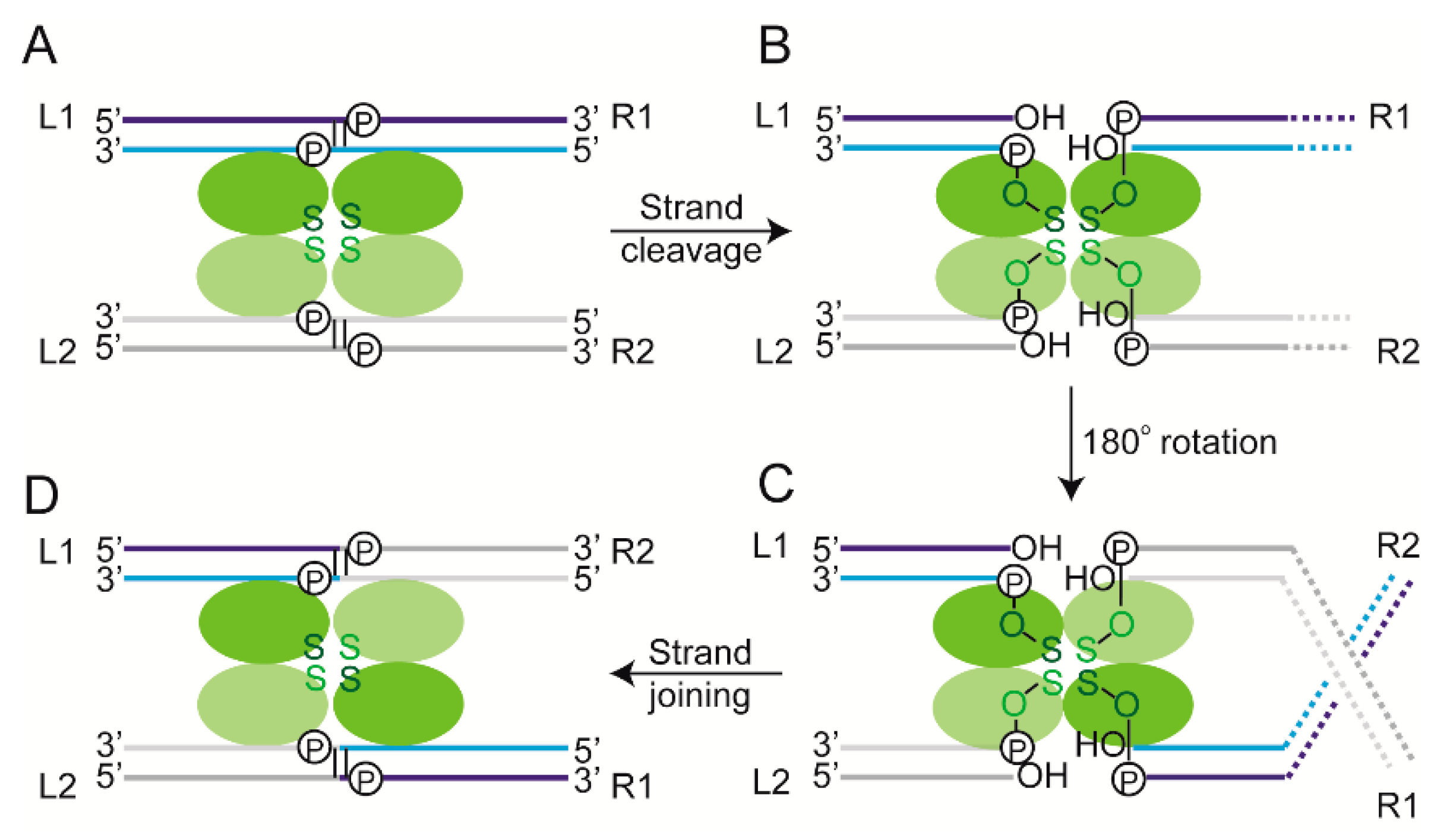
2.5. Recombination Mediated by Conservative Site-Specific Recombinases
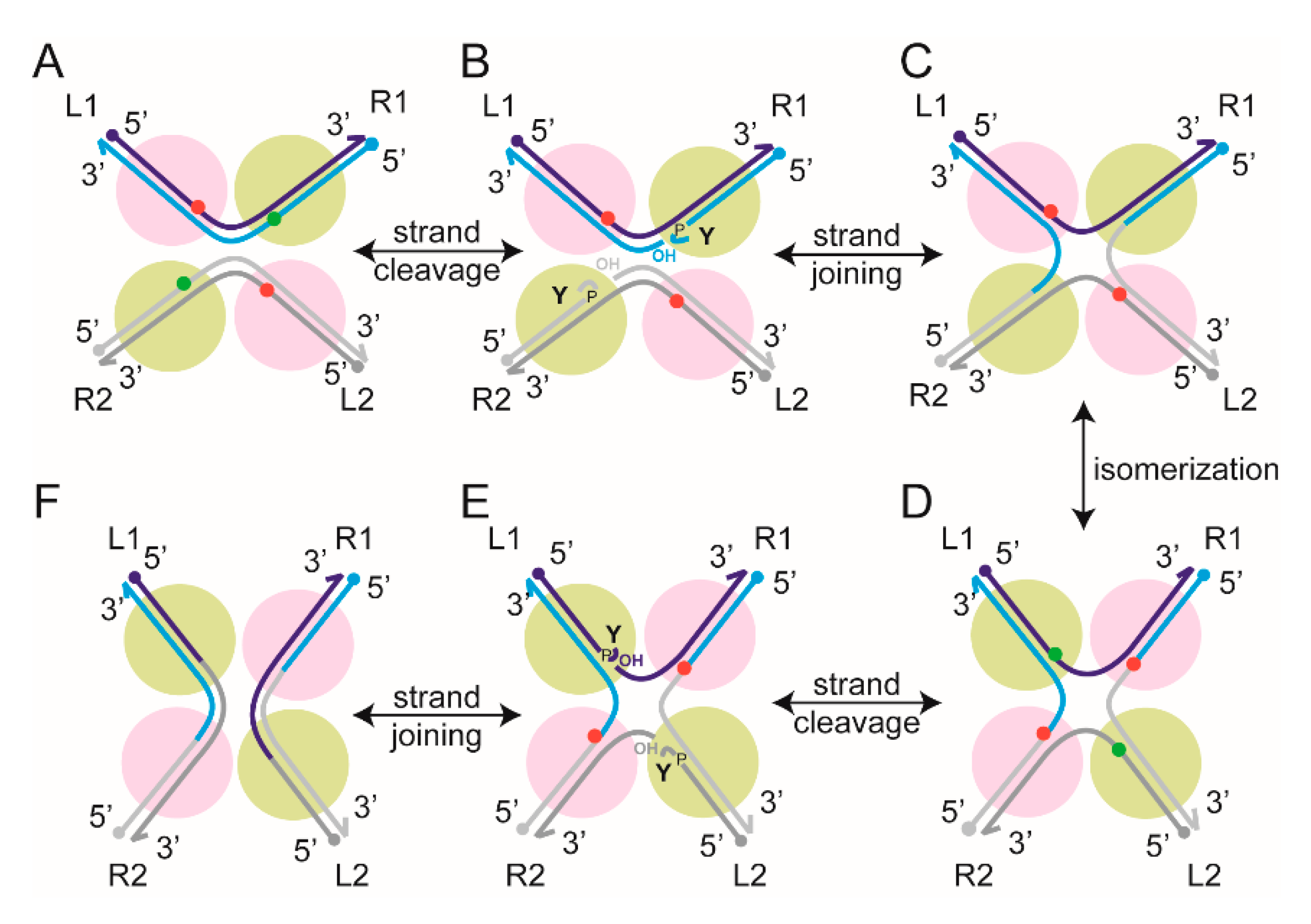
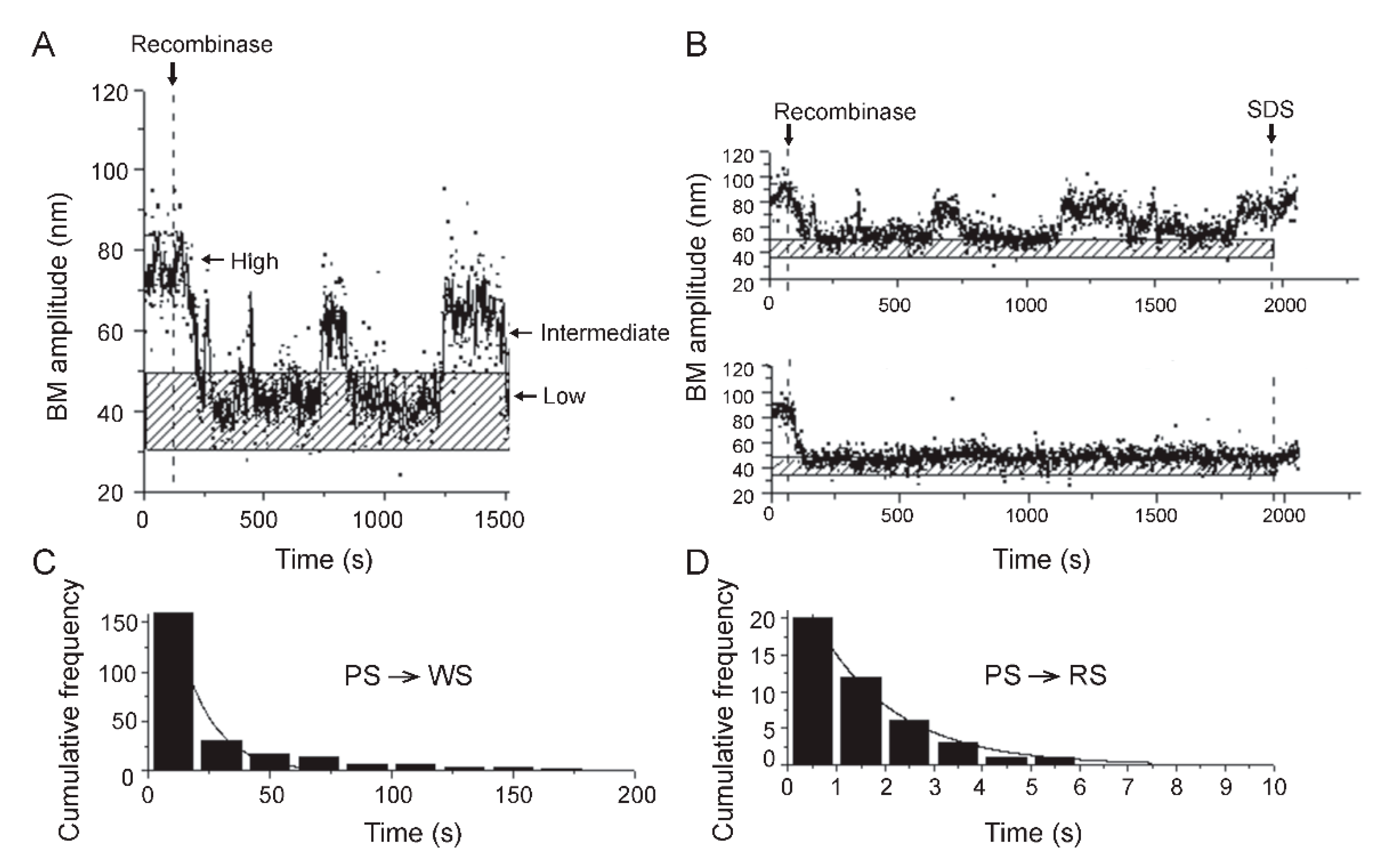
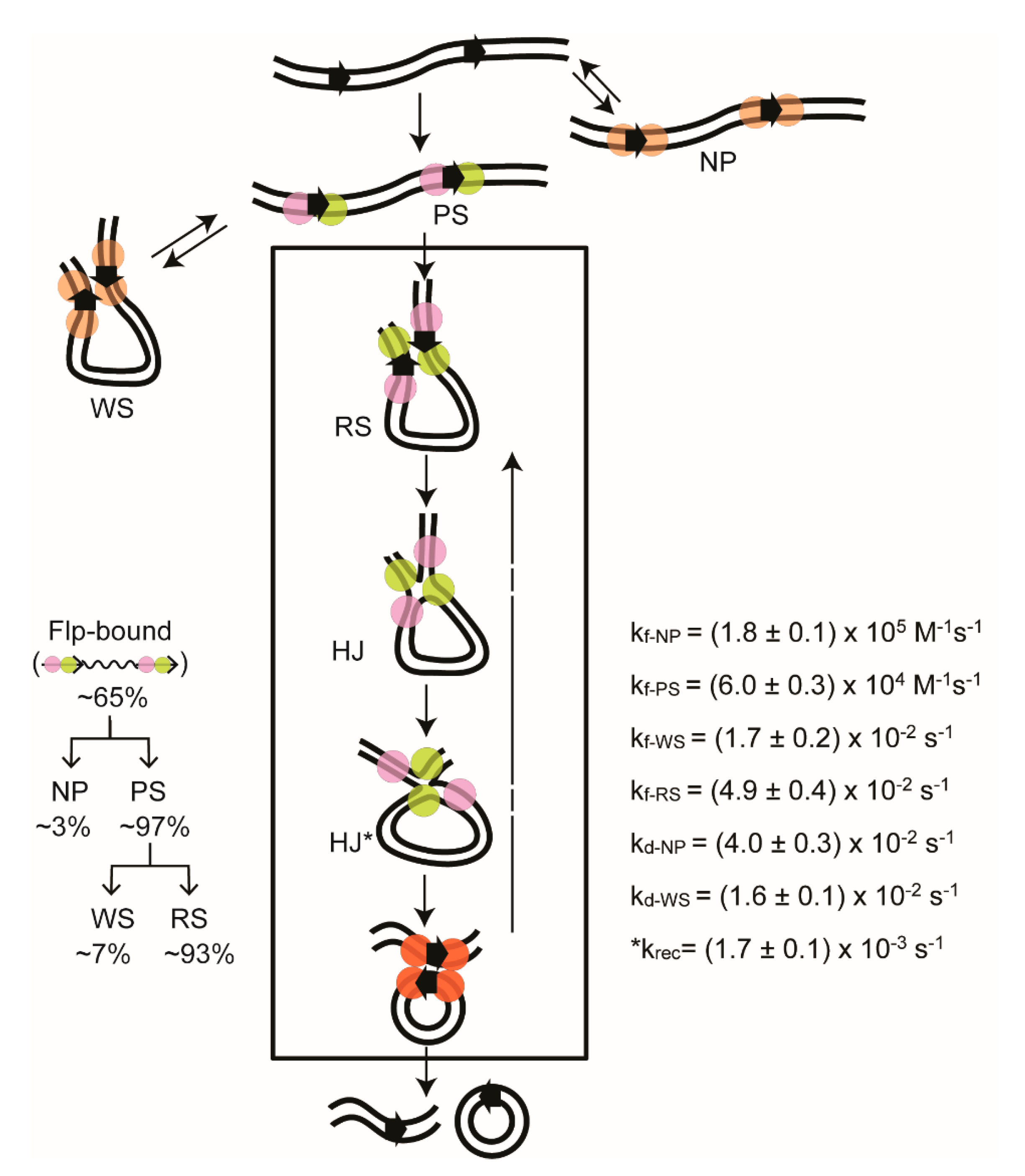
2.6. Steps of Site-Specific Recombination Reported by TPM
2.7. Thermodynamic and Kinetic Features of Individual Steps of Site-Specific Recombination
2.8. Insights on Tyrosine Site-Specific Recombination Gained from TPM Studies
2.9. Reversibility of the Chemical Steps of Recombination and Directionality of the Reaction
2.10. Characterization of the Active Site Pentad Mutants of Cre and Flp by TPM
2.11. Other Studies on Tyrosine Recombination Using TPM and TPM Related Methods
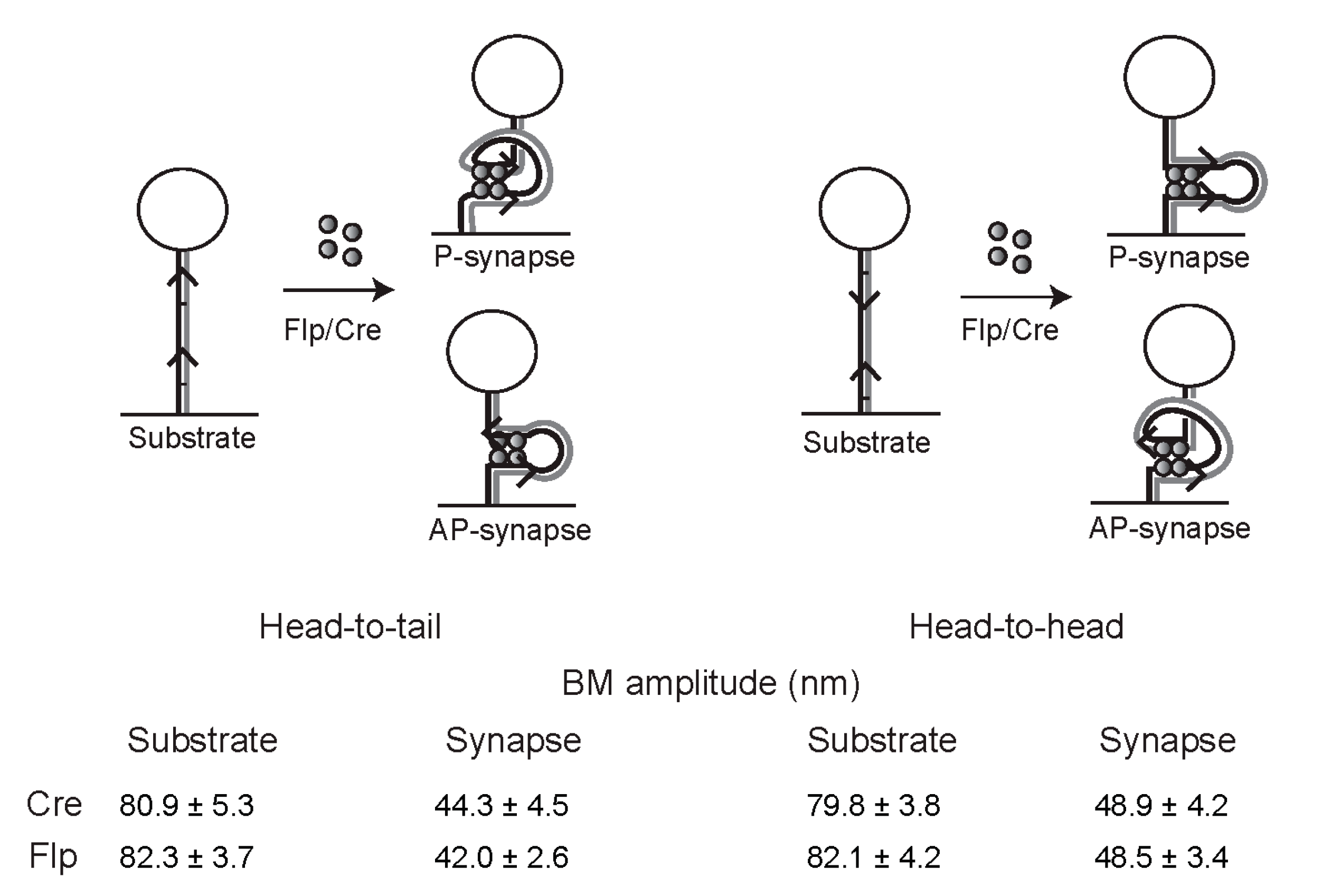
2.12. Geometry of Target Sites within the Tyrosine Family Recombination Synapse
2.13. TPM Analyses of a Serine Site-Specific Recombinase
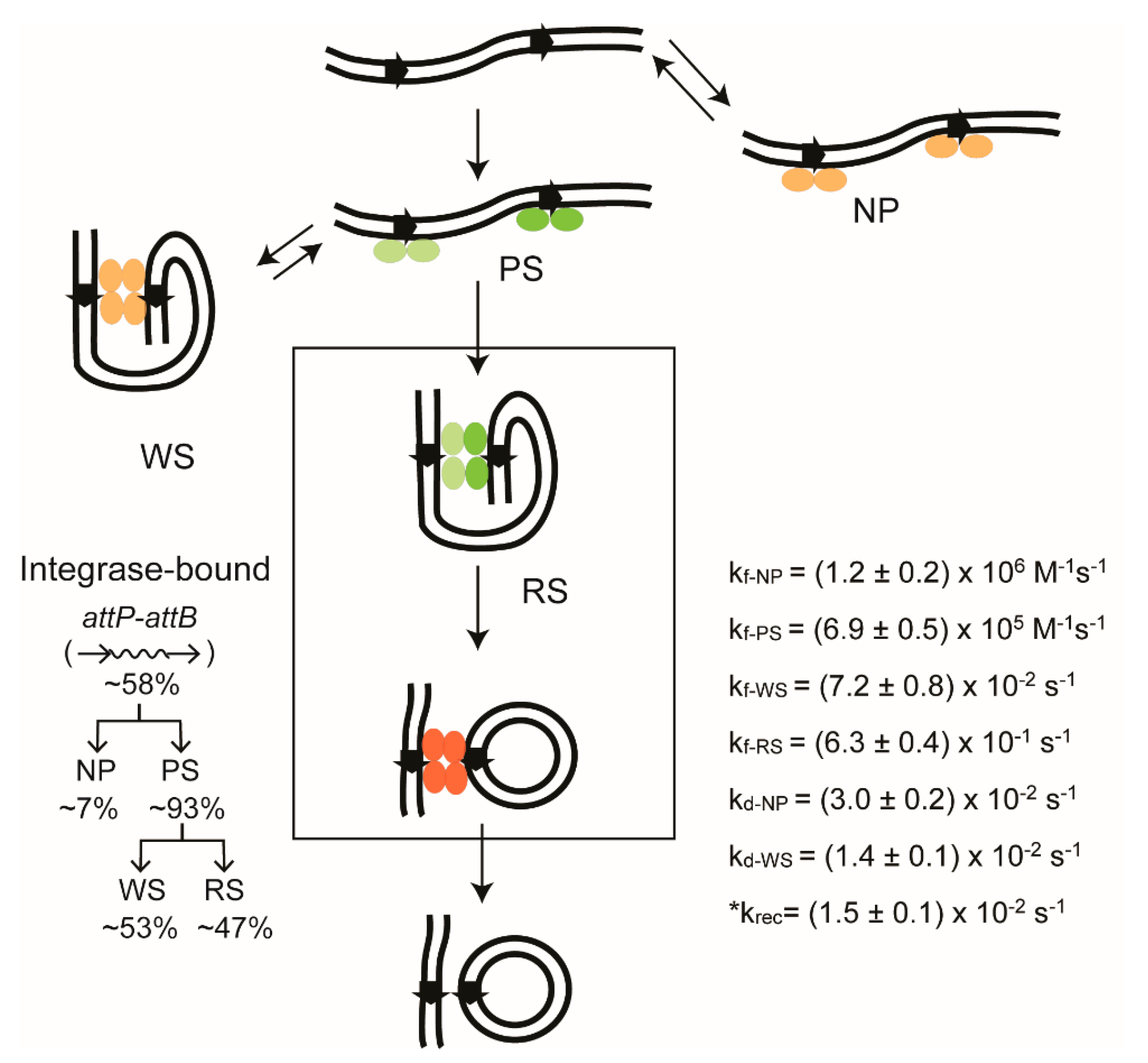
2.14. Stepwise Dissection of ϕC31 Integrase Mechanism
3. Summary and Perspective
Funding
Conflicts of Interest
References
- Finzi, L.; Dunlap, D.D. Single-molecule approaches to probe the structure, kinetics, and thermodynamics of nucleoprotein complexes that regulate transcription. J. Biol. Chem. 2010, 285, 18973–18978. [Google Scholar] [CrossRef] [PubMed]
- Kapanidis, A.N.; Strick, T. Biology, one molecule at a time. Trends Biochem. Sci. 2009, 34, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Kovari, D.T.; Yan, Y.; Finzi, L.; Dunlap, D. Tethered Particle Motion: An easy technique for probing DNA topology and interactions with transcription factors. In Single Molecule Analysis: Methods and Protocols, Methos in Molecular Biology; Peterman, E.J.G., Ed.; Springer + Business Media LLC: New York, NY, USA, 2018; Volume 1665, pp. 5–17. [Google Scholar]
- Newby Lambert, M.; Vocker, E.; Blumberg, S.; Redemann, S.; Gajraj, A.; Meiners, J.C.; Walter, N.G. Mg2+-induced compaction of single RNA molecules monitored by tethered particle microscopy. Biophys. J. 2006, 90, 3672–3685. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, L.; Lui, B.H.; Blumberg, S.; Beausang, J.F.; Nelson, P.C.; Phillips, R. Calibration of tethered particle motion experiments. In Mathematics of DNA Structure, Function and Interactions (Dedicated to Nick Cozzarelli); Springer: New York, NY, USA, 2007. [Google Scholar]
- Manghi, M.; Tardin, C.; Baglio, J.; Rousseau, P.; Salome, L.; Destainville, N. Probing DNA conformational changes with high temporal resolution by tethered particle motion. Phys. Biol. 2010, 7, 046003. [Google Scholar] [CrossRef] [PubMed]
- Manzo, C.; Finzi, L. Quantitative analysis of DNA-looping kinetics from tethered particle motion experiments. Methods Enzymol. 2010, 475, 199–220. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.C.; Zurla, C.; Brogioli, D.; Beausang, J.F.; Finzi, L.; Dunlap, D. Tethered particle motion as a diagnostic of DNA tether length. J. Phys. Chem. B 2006, 110, 17260–17267. [Google Scholar] [CrossRef] [PubMed]
- Pouget, N.; Dennis, C.; Turlan, C.; Grigoriev, M.; Chandler, M.; Salome, L. Single-particle tracking for DNA tether length monitoring. Nucleic Acids Res. 2004, 32, e73. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, S.; Gajraj, A.; Pennington, M.W.; Meiners, J.C. Three-dimensional characterization of tethered microspheres by total internal reflection fluorescence microscopy. Biophys. J. 2005, 89, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Garcia, H.G.; Blumberg, S.; Towles, K.B.; Beausang, J.F.; Nelson, P.C.; Phillips, R. Concentration and length dependence of DNA looping in transcriptional regulation. PLoS ONE 2009, 4, e5621. [Google Scholar] [CrossRef] [PubMed]
- Segall, D.E.; Nelson, P.C.; Phillips, R. Volume-exclusion effects in tethered-particle experiments: Bead size matters. Phys. Rev. Lett. 2006, 96, 088306. [Google Scholar] [CrossRef] [PubMed]
- Beausang, J.F.; Zurla, C.; Manzo, C.; Dunlap, D.; Finzi, L.; Nelson, P.C. DNA looping kinetics analyzed using diffusive hidden Markov model. Biophys. J. 2007, 92, L64–L66. [Google Scholar] [CrossRef] [PubMed]
- Zurla, C.; Franzini, A.; Galli, G.; Dunlap, D.D.; Lewis, D.E.A.; Adhya, S.; Finzi, L. Novel tethered particle motion analysis of CI protein-mediated looping in the regulation of bacteriophage lambda. J. Phys. Condens. Matter 2006, 18, S225–S234. [Google Scholar] [CrossRef]
- Schafer, D.A.; Gelles, J.; Sheetz, M.P.; Landick, R. Transcription by single molecules of RNA polymerase observed by light microscopy. Nature 1991, 352, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Li, H.W. Direct observation of RecBCD helicase as single-stranded DNA translocases. J. Am. Chem. Soc. 2013, 135, 8920–8925. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Artsimovitch, I.; Landick, R.; Gelles, J. Nonequilibrium mechanism of transcription termination from observations of single RNA polymerase molecules. Proc. Natl. Acad. Sci. USA 1999, 96, 13124–13129. [Google Scholar] [CrossRef]
- Yin, H.; Landick, R.; Gelles, J. Tethered particle motion method for studying transcript elongation by a single RNA polymerase molecule. Biophys. J. 1994, 67, 2468–2478. [Google Scholar] [CrossRef]
- Van der Valk, R.A.; Laurens, N.; Dame, R.T. Tethered particle motion analysis of the DNA binding properties of architectural proteins. In The Bacterial Nucleoid: Methods and Protocols, Methods in Molecular Biology; Espeli, O., Ed.; Springer Science+Business Media LLC: New York, NY, USA, 2017; Volume 1624, pp. 127–142. [Google Scholar]
- Brunet, A.; Chevalier, S.; Destainville, N.; Manghi, M.; Rousseau, P.; Salhi, M.; Salome, L.; Tardin, C. Probing a label-free local bend in DNA by single molecule tethered particle motion. Nucleic Acids Res. 2015, 43, e72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Driessen, R.P.; Sitters, G.; Laurens, N.; Moolenaar, G.F.; Wuite, G.J.; Goosen, N.; Dame, R.T. Effect of temperature on the intrinsic flexibility of DNA and its interaction with architectural proteins. Biochemistry 2014, 53, 6430–6438. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; van de Meent, J.W.; Phillips, R.; Wiggins, C.H.; Linden, M. Multiple LacI-mediated loops revealed by Bayesian statistics and tethered particle motion. Nucleic Acids Res. 2014, 42, 10265–10277. [Google Scholar] [CrossRef] [PubMed]
- Laurens, N.; Bellamy, S.R.; Harms, A.F.; Kovacheva, Y.S.; Halford, S.E.; Wuite, G.J. Dissecting protein-induced DNA looping dynamics in real time. Nucleic Acids Res. 2009, 37, 5454–5464. [Google Scholar] [CrossRef] [PubMed]
- Vanzi, F.; Broggio, C.; Sacconi, L.; Pavone, F.S. Lac repressor hinge flexibility and DNA looping: Single molecule kinetics by tethered particle motion. Nucleic Acids Res. 2006, 34, 3409–3420. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.F.; Liu, Z.N.; Chow, S.Y.; Lu, Y.H.; Li, H. Histone chaperone-mediated nucleosome assembly process. PLoS ONE 2015, 10, e0115007. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dodd, I.B.; Dunlap, D.D.; Shearwin, K.E.; Finzi, L. Single molecule analysis of DNA wrapping and looping by a circular 14mer wheel of the bacteriophage 186 CI repressor. Nucleic Acids Res. 2013, 41, 5746–5756. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mack, A.H.; Schlingman, D.J.; Salinas, R.D.; Regan, L.; Mochrie, S.G. Condensation transition and forced unravelling of DNA-histone H1 torroids: A multi-state free energy landscape. J. Phys. Condens. Matter 2015, 27, 064106. [Google Scholar] [CrossRef] [PubMed]
- Guerra, R.F.; Imperadori, L.; Mantovani, R.; Dunlap, D.D.; Finzi, L. DNA compaction by the nuclear factor-Y. Biophys. J. 2007, 93, 176–182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, Y.W.; Huang, T.; Tsai, C.T.; Chang, Y.Y.; Li, H.W.; Hsu, C.H.; Fan, H.F. Using single-molecule approaches to study archaeal DNA-binding protein Alba1. Biochemistry 2013, 52, 7714–7722. [Google Scholar] [CrossRef] [PubMed]
- Nir, G.; Lindner, M.; Dietrich, H.R.; Girshevitz, O.; Vorgias, C.E.; Garini, Y. HU protein induces incoherent DNA persistence length. Biophys. J. 2011, 100, 784–790. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pouget, N.; Turlan, C.; Destainville, N.; Salome, L.; Chandler, M. IS911 transpososome assembly as analysed by tethered particle motion. Nucleic Acids Res. 2006, 34, 4313–4323. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, D.D.; Zurla, C.; Manzo, C.; Finzi, L. Pobing DNA topology using tetered particle motion. In Single Molecule Analysis: Methods and Protocols, Methods in Molecular Biology; Peterman, E.J.G., White, G.J.L., Eds.; Springer Science+Business Media LLC: New York, NY, USA, 2011; Volume 783, pp. 295–312. [Google Scholar]
- Fan, H.F.; Li, H.W. Studying RecBCD helicase translocation along Chi-DNA using tethered particle motion with a stretching force. Biophys. J. 2009, 96, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yehoshua, S.; Ali, S.S.; Navarre, W.W.; Milstein, J.N. A biomechanical mechanism for initiating DNA packaging. Nucleic Acids Res. 2014, 42, 11921–11927. [Google Scholar] [CrossRef] [PubMed][Green Version]
- May, P.F.J.; Pinkney, J.N.M.; Zawadzki, P.; Evans, G.W.; Sherratt, D.J.; Kapanidis, A.N. Tethered fluorophore motion: Studying large DNA conformational changes by single-fluorophore imaging. Biophys. J. 2014, 107, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Ucuncuoglu, S.; Schneider, D.A.; Weeks, E.R.; Dunlap, D.; Finzi, L. Multiplexed, Tethered Particle Microscopy for Studies of DNA-Enzyme Dynamics. Methods Enzymol. 2017, 582, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Plenat, T.; Tardin, C.; Rousseau, P.; Salome, L. High-throughput single-molecule analysis of DNA-protein interactions by tethered particle motion. Nucleic Acids Res. 2012, 40, e89. [Google Scholar] [CrossRef] [PubMed]
- Fournes, F.; Crozat, E.; Pages, C.; Tardin, C.; Salome, L.; Cornet, F.; Rousseau, P. FtsK translocation permits discrimination between an endogenous and an imported Xer/dif recombination complex. Proc. Natl. Acad. Sci. USA 2016, 113, 7882–7887. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, M.; Ma, C.H.; Kachroo, A.H.; Rowley, P.A.; Guga, P.; Fan, H.F.; Voziyanov, Y. An overview of tyrosine site-specific recombination: From a Flp perspective. Microbiol. Spectr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Stark, W.M. The serine recombinases. Microbiol. Spectr. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.F. Real-time single-molecule tethered particle motion experiments reveal the kinetics and mechanisms of Cre-mediated site-specific recombination. Nucleic Acids Res. 2012, 40, 6208–6222. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.F.; Cheng, Y.S.; Ma, C.H.; Jayaram, M. Single molecule TPM analysis of the catalytic pentad mutants of Cre and Flp site-specific recombinases: Contributions of the pentad residues to the pre-chemical steps of recombination. Nucleic Acids Res. 2015, 43, 3237–3255. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.F.; Hsieh, T.S.; Ma, C.H.; Jayaram, M. Single-molecule analysis of ϕC31 integrase-mediated site-specific recombination by tethered particle motion. Nucleic Acids Res. 2016, 44, 10804–10823. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.F.; Ma, C.H.; Jayaram, M. Real-time single-molecule tethered particle motion analysis reveals mechanistic similarities and contrasts of Flp site-specific recombinase with Cre and l Int. Nucleic Acids Res. 2013, 41, 7031–7047. [Google Scholar] [CrossRef] [PubMed]
- Mumm, J.P.; Landy, A.; Gelles, J. Viewing single lambda site-specific recombination events from start to finish. EMBO J. 2006, 4586–4595. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Sun, M.; Ghosh, P.; Hatfull, G.F.; Grindley, N.D.; Marko, J.F. Single-molecule analysis reveals the molecular bearing mechanism of DNA strand exchange by a serine recombinase. Proc. Natl. Acad. Sci. USA 2011, 108, 7419–7424. [Google Scholar] [CrossRef] [PubMed]
- Diagne, C.T.; Salhi, M.; Crozat, E.; Salome, L.; Cornet, F.; Rousseau, P.; Tardin, C. TPM analyses reveal that FtsK contributes both to the assembly and the activation of the XerCD-dif recombination synapse. Nucleic Acids Res. 2014, 42, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Keenholtz, R.A.; Grindley, N.D.; Hatfull, G.F.; Marko, J.F. Crossover-site sequence and DNA torsional stress control strand interchanges by the Bxb1 site-specific serine recombinase. Nucleic Acids Res. 2016, 44, 8921–8932. [Google Scholar] [CrossRef] [PubMed][Green Version]
- May, P.F.; Zawadzki, P.; Sherratt, D.J.; Kapanidis, A.N.; Arciszewska, L.K. Assembly, translocation, and activation of XerCD-dif recombination by FtsK translocase analyzed in real-time by FRET and two-color tethered fluorophore motion. Proc. Natl. Acad. Sci. USA 2015, 112, E5133–E5141. [Google Scholar] [CrossRef] [PubMed]
- Pinkney, J.N.; Zawadzki, P.; Mazuryk, J.; Arciszewska, L.K.; Sherratt, D.J.; Kapanidis, A.N. Capturing reaction paths and intermediates in Cre-loxP recombination using single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2012, 109, 20871–20876. [Google Scholar] [CrossRef] [PubMed]
- Zawadzki, P.; May, P.F.; Baker, R.A.; Pinkney, J.N.; Kapanidis, A.N.; Sherratt, D.J.; Arciszewska, L.K. Conformational transitions during FtsK translocase activation of individual XerCD-dif recombination complexes. Proc. Natl. Acad. Sci. USA 2013, 110, 17302–17307. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.C. Site-specific DNA Inversion by serine recombinases. Microbiol. Spectr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.A. Serine resolvases. Microbiol. Spectr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Van Duyne, G.D. Cre recombinase. Microbiol. Spectr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dhar, G.; Heiss, J.K.; Johnson, R.C. Mechanical constraints on Hin subunit rotation imposed by the Fis/enhancer system and DNA supercoiling during site-specific recombination. Mol. Cell 2009, 34, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Kanaar, R.; van de Putte, P.; Cozzarelli, N.R. Gin-mediated recombination of catenated and knotted DNA substrates: Implications for the mechanism of interaction between cis-acting sites. Cell 1989, 58, 147–159. [Google Scholar] [CrossRef]
- Laxmikanthan, G.; Xu, C.; Brilot, A.F.; Warren, D.; Steele, L.; Seah, N.; Tong, W.; Grigorieff, N.; Landy, A.; Van Duyne, G.D. Structure of a Holliday junction complex reveals mechanisms governing a highly regulated DNA transaction. eLife 2016, 5, e14313. [Google Scholar] [CrossRef] [PubMed]
- Strater, N.; Sherratt, D.J.; Colloms, S.D. X-ray structure of aminopeptidase A from Escherichia coli and a model for the nucleoprotein complex in Xer site-specific recombination. EMBO J. 1999, 18, 4513–4522. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Evans, B.; Rosenfeldt, H.; Jayaram, M. Bending-incompetent variants of Flp recombinase mediate strand transfer in half-site recombinations: Role of DNA bending in recombination. Gene 1992, 119, 37–48. [Google Scholar] [PubMed]
- Schwartz, C.J.; Sadowski, P.D. FLP protein of 2 micron circle plasmid of yeast induces multiple bends in the FLP recognition target site. J. Mol. Biol. 1990, 216, 289–298. [Google Scholar] [CrossRef]
- Chen, Y.; Narendra, U.; Iype, L.E.; Cox, M.M.; Rice, P.A. Crystal structure of a Flp recombinase-Holliday junction complex: Assembly of an active oligomer by helix swapping. Mol. Cell 2000, 6, 885–897. [Google Scholar] [CrossRef]
- Conway, A.B.; Chen, Y.; Rice, P.A. Structural plasticity of the Flp-Holliday junction complex. J. Mol. Biol. 2003, 326, 425–434. [Google Scholar] [CrossRef]
- Sternberg, N.; Hamilton, D.; Austin, S.; Yarmolinsky, M.; Hoess, R. Site-specific recombination and its role in the life cycle of bacteriophage P1. Cold Spring Harb. Symp. Quant. Biol. 1981, 45, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Sau, S.; Ma, C.H.; Kachroo, A.H.; Rowley, P.A.; Chang, K.M.; Fan, H.F.; Jayaram, M. The partitioning and copy number control systems of the selfish yeast plasmid: An optimized molecular design for stable persistence in host cells. Microbiol. Spectr. 2014. [Google Scholar] [CrossRef]
- Futcher, A.B. Copy number amplification of the 2 micron circle plasmid of Saccharomyces cerevisiae. J. Theor. Biol. 1986, 119, 197–204. [Google Scholar] [CrossRef]
- Volkert, F.C.; Broach, J.R. Site-specific recombination promotes plasmid amplification in yeast. Cell 1986, 46, 541–550. [Google Scholar] [CrossRef]
- Ringrose, L.; Lounnas, V.; Ehrlich, L.; Buchholz, F.; Wade, R.; Stewart, A.F. Comparative kinetic analysis of FLP and Cre recombinases: Mathematical models for DNA binding and recombination. J. Mol. Biol. 1998, 284, 363–384. [Google Scholar] [CrossRef] [PubMed]
- Gibb, B.; Gupta, K.; Ghosh, K.; Sharp, R.; Chen, J.; Van Duyne, G.D. Requirements for catalysis in the Cre recombinase active site. Nucleic Acids Res. 2010, 5817–5832. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grainge, I.; Buck, D.; Jayaram, M. Geometry of site alignment during int family recombination: Antiparallel synapsis by the Flp recombinase. J. Mol. Biol. 2000, 298, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Grainge, I.; Buck, D.; Jayaram, M. Symmetric DNA sites are functionally asymmetric within Flp and Cre site-specific DNA recombination synapses. J. Mol. Biol. 2002, 320, 515–527. [Google Scholar] [CrossRef]
- Guo, F.; Gopaul, D.N.; van Duyne, G.D. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 1997, 389, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.H.; Liu, Y.T.; Savva, C.G.; Rowley, P.A.; Cannon, B.; Fan, H.F.; Russell, R.; Holzenburg, A.; Jayaram, M. Organization of DNA partners and strand exchange mechanisms during Flp site-specific recombination analyzed by difference topology, single molecule FRET and single molecule TPM. J. Mol. Biol. 2014, 426, 793–815. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C. Phage-encoded serine integrases and other large serine recombinases. Microbiol. Spectr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Sharp, R.; Yuan, J.B.; Li, H.; Van Duyne, G.D. Coiled-coil interactions mediate serine integrase directionality. Nucleic Acids Res. 2017, 45, 7339–7353. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Yuan, P.; Perry, K.; Sharp, R.; Van Duyne, G.D. Attachment site recognition and regulation of directionality by the serine integrases. Nucleic Acids Res. 2013, 41, 8341–8356. [Google Scholar] [CrossRef] [PubMed]
- Olorunniji, F.J.; Buck, D.E.; Colloms, S.D.; McEwan, A.R.; Smith, M.C.; Stark, W.M.; Rosser, S.J. Gated rotation mechanism of site-specific recombination by ϕC31 integrase. Proc. Natl. Acad. Sci. USA 2012, 109, 19661–19666. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Steiner, A.M.; Ghirlando, R.; Li, N.S.; Rowland, S.J.; Boocock, M.R.; Stark, W.M.; Piccirilli, J.A.; Rice, P.A. Sin resolvase catalytic activity and oligomerization state are tightly coupled. J. Mol. Biol. 2010, 404, 16–33. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olorunniji, F.J.; He, J.; Wenwieser, S.V.; Boocock, M.R.; Stark, W.M. Synapsis and catalysis by activated Tn3 resolvase mutants. Nucleic Acids Res. 2008, 36, 7181–7191. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A.; Smith, M.C.; Younger, E.; Smith, M.C. A motif in the C-terminal domain of fC31 integrase controls the directionality of recombination. Nucleic Acids Res. 2008, 36, 3879–3891. [Google Scholar] [CrossRef] [PubMed]
- Fogg, P.C.M.; Younger, E.; Fernando, B.D.; Khaleel, T.; Stark, W.M.; Smith, M.C.M. Recombination directionality factor gp3 binds ϕC31 integrase via the zinc domain, potentially affecting the trajectory of the coiled-coil motif. Nucleic Acids Res. 2018, 46, 1308–1320. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, H.-F.; Ma, C.-H.; Jayaram, M. Single-Molecule Tethered Particle Motion: Stepwise Analyses of Site-Specific DNA Recombination. Micromachines 2018, 9, 216. https://doi.org/10.3390/mi9050216
Fan H-F, Ma C-H, Jayaram M. Single-Molecule Tethered Particle Motion: Stepwise Analyses of Site-Specific DNA Recombination. Micromachines. 2018; 9(5):216. https://doi.org/10.3390/mi9050216
Chicago/Turabian StyleFan, Hsiu-Fang, Chien-Hui Ma, and Makkuni Jayaram. 2018. "Single-Molecule Tethered Particle Motion: Stepwise Analyses of Site-Specific DNA Recombination" Micromachines 9, no. 5: 216. https://doi.org/10.3390/mi9050216
APA StyleFan, H.-F., Ma, C.-H., & Jayaram, M. (2018). Single-Molecule Tethered Particle Motion: Stepwise Analyses of Site-Specific DNA Recombination. Micromachines, 9(5), 216. https://doi.org/10.3390/mi9050216