

A Dialysis Membrane-Integrated Microfluidic Device for Controlled Drug Retention and Nutrient Supply



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
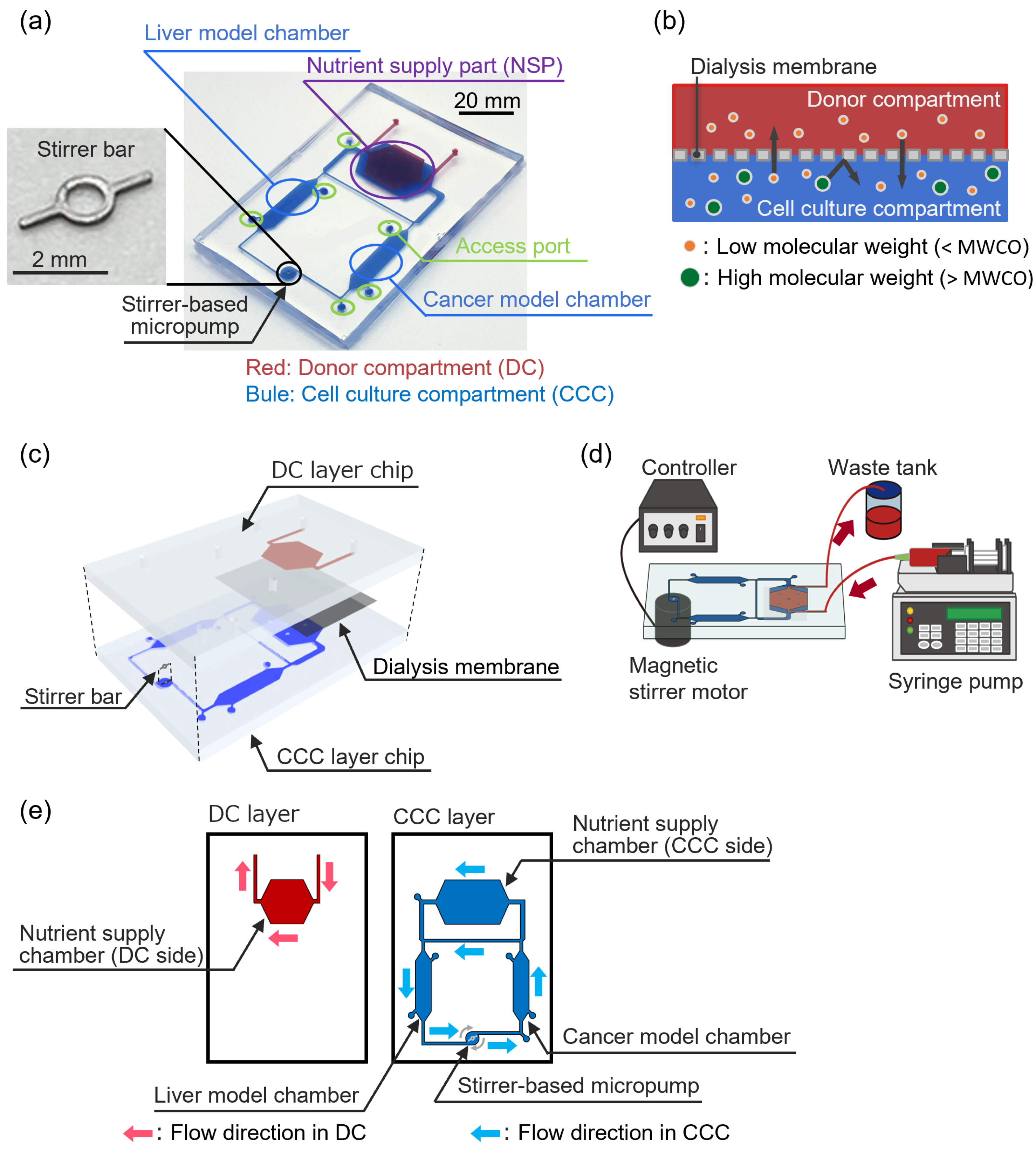
2.1. Dialysis Membrane-Integrated Microfluidic Device (DMiMD)
2.2. Cell Culture
2.3. Cell Density Evaluation
2.4. Quantification of Fluorescent Substances
2.5. Statistical Analysis
3. Results and Discussion
3.1. Evaluation of Molecular Selectivity Using the DMiMD
3.2. Evaluation of Nutrient Supply Performance Using the DMiMD
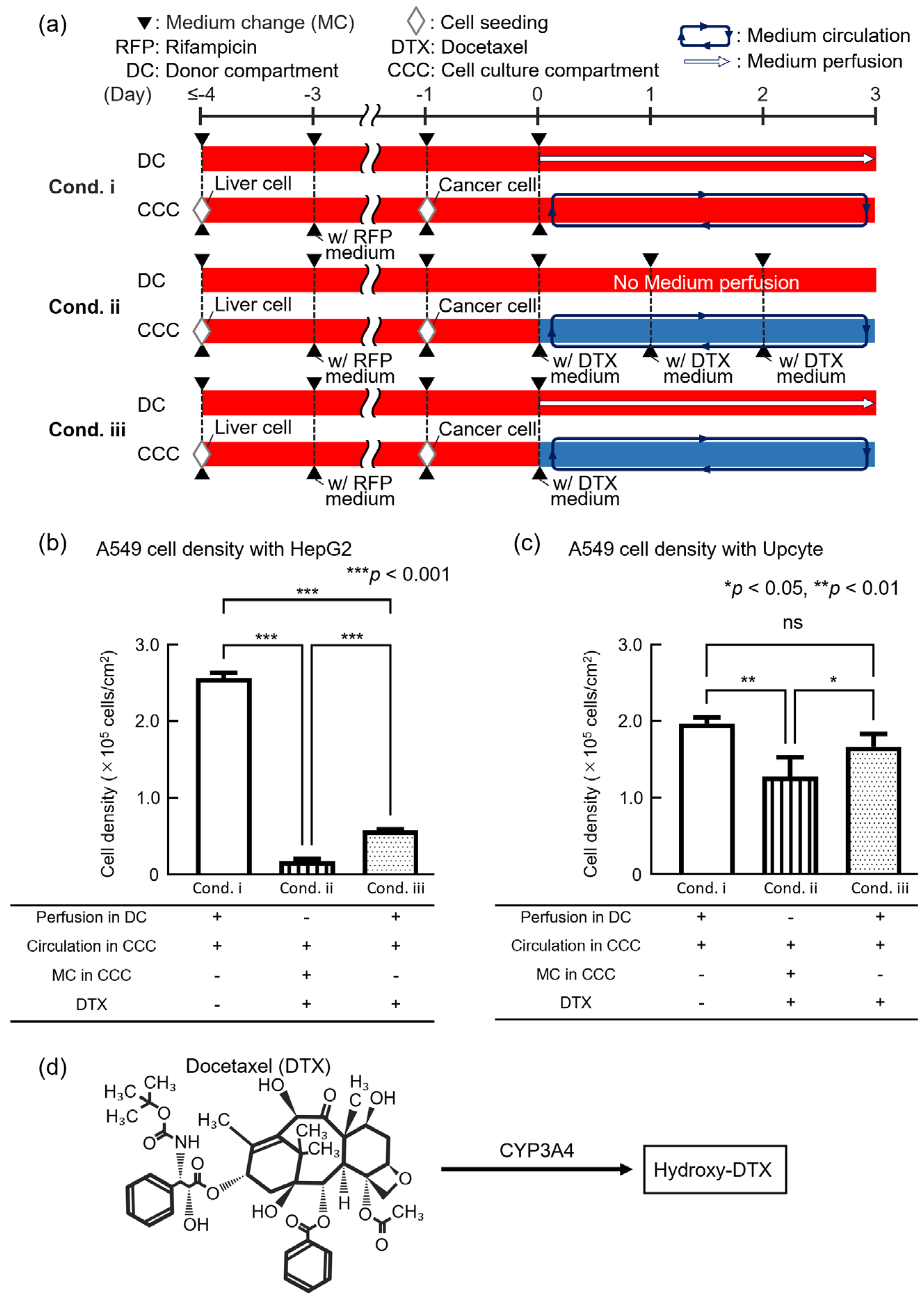
3.3. Evaluation of Anticancer Drug Efficacy Using the DMiMD
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Berlo, D.; van de Steeg, E.; Amirabadi, H.E.; Masereeuw, R. The Potential of Multi-Organ-on-Chip Models for Assessment of Drug Disposition as Alternative to Animal Testing. Curr. Opin. Toxicol. 2021, 27, 8–17. [Google Scholar] [CrossRef]
- Breslin, S.; O’Driscoll, L. Three-Dimensional Cell Culture: The Missing Link in Drug Discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/Body-on-a-Chip Based on Microfluidic Technology for Drug Discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Nishikawa, M.; Kutsuzawa, N.; Tokito, F.; Kobayashi, T.; Kurniawan, D.A.; Shioda, H.; Cao, W.; Shinha, K.; Nakamura, H.; et al. Advancements in Microphysiological Systems: Exploring Organoids and Organ-on-a-Chip Technologies in Drug Development -Focus on Pharmacokinetics Related Organs. Drug Metab. Pharmacokinet. 2025, 60, 101046. [Google Scholar] [CrossRef]
- Ito, Y.; Kawauchi, I.; Yanagita, Y.; Sakai, Y.; Nishikawa, M.; Arakawa, H.; Kadoguchi, M.; Tamai, I.; Esashika, K.; Takahashi, J.; et al. Microphysiological Systems for Realizing Microenvironment That Mimics Human Physiology—Functional Material and Its Standardization Applied to Microfluidics. Emergent Mater. 2024, 8, 1139–1151. [Google Scholar] [CrossRef]
- Kopec, A.K.; Yokokawa, R.; Khan, N.; Horii, I.; Finley, J.E.; Bono, C.P.; Donovan, C.; Roy, J.; Harney, J.; Burdick, A.D.; et al. Microphysiological Systems in Early Stage Drug Development: Perspectives on Current Applications and Future Impact. J. Toxicol. Sci. 2021, 46, 99–114. [Google Scholar] [CrossRef]
- Kim, J.J.; Bae, M.; Cho, D. Multi-Organ Microphysiological Systems Targeting Specific Organs for Recapitulating Disease Phenotypes via Organ Crosstalk. Small Sci. 2024, 4, 2400314. [Google Scholar] [CrossRef]
- Petreus, T.; Cadogan, E.; Hughes, G.; Smith, A.; Pilla Reddy, V.; Lau, A.; O’Connor, M.J.; Critchlow, S.; Ashford, M.; Oplustil O’Connor, L. Tumour-on-Chip Microfluidic Platform for Assessment of Drug Pharmacokinetics and Treatment Response. Commun. Biol. 2021, 4, 1001. [Google Scholar] [CrossRef]
- Singh, D.; Deosarkar, S.P.; Cadogan, E.; Flemington, V.; Bray, A.; Zhang, J.; Reiserer, R.S.; Schaffer, D.K.; Gerken, G.B.; Britt, C.M.; et al. A Microfluidic System That Replicates Pharmacokinetic (PK) Profiles In Vitro Improves Prediction of In Vivo Efficacy in Preclinical Models. PLoS Biol. 2022, 20, e3001624. [Google Scholar] [CrossRef]
- Guerrero, Y.A.; Desai, D.; Sullivan, C.; Kindt, E.; Spilker, M.E.; Maurer, T.S.; Solomon, D.E.; Bartlett, D.W. A Microfluidic Perfusion Platform for In Vitro Analysis of Drug Pharmacokinetic-Pharmacodynamic (PK-PD) Relationships. AAPS J. 2020, 22, 53. [Google Scholar] [CrossRef]
- Eswari, J.S.; Naik, S. A Critical Analysis on Various Technologies and Functionalized Materials for Manufacturing Dialysis Membranes. Mater. Sci. Energy Technol. 2020, 3, 116–126. [Google Scholar] [CrossRef]
- Imura, Y.; Yoshimura, E.; Sato, K. Microcirculation System with a Dialysis Part for Bioassays Evaluating Anticancer Activity and Retention. Anal. Chem. 2013, 85, 1683–1688. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Ikeda, T.; Nakayama, H.; Sakai, Y.; Fujii, T. An On-Chip Small Intestine-Liver Model for Pharmacokinetic Studies. J. Lab. Autom. 2015, 20, 265–273. [Google Scholar] [CrossRef]
- Koirala, R.P.; Dawanse, S.; Pantha, N. Diffusion of Glucose in Water: A Molecular Dynamics Study. J. Mol. Liq. 2022, 345, 117826. [Google Scholar] [CrossRef]
- Kimura, H.; Yamamoto, T.; Sakai, H.; Sakai, Y.; Fujii, T. An Integrated Microfluidic System for Long-Term Perfusion Culture and on-Line Monitoring of Intestinal Tissue Models. Lab Chip 2008, 8, 741–746. [Google Scholar] [CrossRef]
- McDonald, J.C.; Whitesides, G.M. Poly(Dimethylsiloxane) as a Material for Fabricating Microfluidic Devices. Acc. Chem. Res. 2002, 35, 491–499. [Google Scholar] [CrossRef]
- Kim, A.A.; Kustanovich, K.; Baratian, D.; Ainla, A.; Shaali, M.; Jeffries, G.D.M.; Jesorka, A. SU-8 Free-Standing Microfluidic Probes. Biomicrofluidics 2017, 11, 014112. [Google Scholar] [CrossRef]
- Borók, A.; Laboda, K.; Bonyár, A. PDMS Bonding Technologies for Microfluidic Applications: A Review. Biosensors 2021, 11, 292. [Google Scholar] [CrossRef]
- Mazzari, A.L.D.A.; Milton, F.; Frangos, S.; Carvalho, A.C.B.; Silveira, D.; De Assis Rocha Neves, F.; Prieto, J.M. In Vitro Effects of Four Native Brazilian Medicinal Plants in CYP3A4 mRNA Gene Expression, Glutathione Levels, and P-Glycoprotein Activity. Front. Pharmacol. 2016, 7, 265. [Google Scholar] [CrossRef]
- Usui, T.; Saitoh, Y.; Komada, F. Induction of CYP3As in HepG2 Cells by Several Drugs. Association between Induction of CYP3A4 and Expression of Glucocorticoid Receptor. Biol. Pharm. Bull. 2003, 26, 510–517. [Google Scholar] [CrossRef]
- Shankland, E.; Livesey, J.; Wiseman, R.; Krohn, K. Multinuclear NMR Studies of an Actively Dividing Artificial Tumor. Physiol. Res. 2002, 51, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.-H.; Zeng, R.-F.; Fang, S.; Dai, Q.-S.; Li, H.-P.; Long, J.-T. Liposome-Based Co-Delivery of siRNA and Docetaxel for the Synergistic Treatment of Lung Cancer. Int. J. Pharm. 2014, 474, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, A.; Martínez-Alcázar, M.P.; Durán, I.; Calvo, E.; Valenzuela, B.; Barbas, C.; García, A. Simultaneous Online SPE–HPLC–MS/MS Analysis of Docetaxel, Temsirolimus and Sirolimus in Whole Blood and Human Plasma. J. Chromatogr. B 2013, 921–922, 35–42. [Google Scholar] [CrossRef]
- Morse, D.L.; Gray, H.; Payne, C.M.; Gillies, R.J. Docetaxel Induces Cell Death through Mitotic Catastrophe in Human Breast Cancer Cells. Mol. Cancer Ther. 2005, 4, 1495–1504. [Google Scholar] [CrossRef]
- Engels, F.K.; Ten Tije, A.J.; Baker, S.D.; Lee, C.K.K.; Loos, W.J.; Vulto, A.G.; Verweij, J.; Sparreboom, A. Effect of Cytochrome P450 3A4 Inhibition on the Pharmacokinetics of Docetaxel. Clin. Pharmacol. Ther. 2004, 75, 448–454. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 Pharmacogenetics and Cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef]
- Loos, W. Clinical Pharmacokinetics of Unbound Docetaxel: Role of Polysorbate 80 and Serum Proteins. Clin. Pharmacol. Ther. 2003, 74, 364–371. [Google Scholar] [CrossRef]
- Tolosa, L.; Gómez-Lechón, M.J.; López, S.; Guzmán, C.; Castell, J.V.; Donato, M.T.; Jover, R. Human Upcyte Hepatocytes: Characterization of the Hepatic Phenotype and Evaluation for Acute and Long-Term Hepatotoxicity Routine Testing. Toxicol. Sci. 2016, 152, 214–229. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyashita, H.; Ito, Y.; Shinha, K.; Nakamura, H.; Kimura, H. A Dialysis Membrane-Integrated Microfluidic Device for Controlled Drug Retention and Nutrient Supply. Micromachines 2025, 16, 745. https://doi.org/10.3390/mi16070745
Miyashita H, Ito Y, Shinha K, Nakamura H, Kimura H. A Dialysis Membrane-Integrated Microfluidic Device for Controlled Drug Retention and Nutrient Supply. Micromachines. 2025; 16(7):745. https://doi.org/10.3390/mi16070745
Chicago/Turabian StyleMiyashita, Hajime, Yuya Ito, Kenta Shinha, Hiroko Nakamura, and Hiroshi Kimura. 2025. "A Dialysis Membrane-Integrated Microfluidic Device for Controlled Drug Retention and Nutrient Supply" Micromachines 16, no. 7: 745. https://doi.org/10.3390/mi16070745
APA StyleMiyashita, H., Ito, Y., Shinha, K., Nakamura, H., & Kimura, H. (2025). A Dialysis Membrane-Integrated Microfluidic Device for Controlled Drug Retention and Nutrient Supply. Micromachines, 16(7), 745. https://doi.org/10.3390/mi16070745