
Progress and Challenges in the Use of a Liver-on-a-Chip for Hepatotropic Infectious Diseases



Abstract
1. Introduction
2. Necessity of Personalized Medicine for Hepatotropic Infectious Diseases
2.1. Hepatitis B Virus (HBV) Infection
2.2. Hepatitis C Virus (HCV) Infection
2.3. Malaria
3. Drug-Induced Liver Injury (DILI) in Personalized Medicine
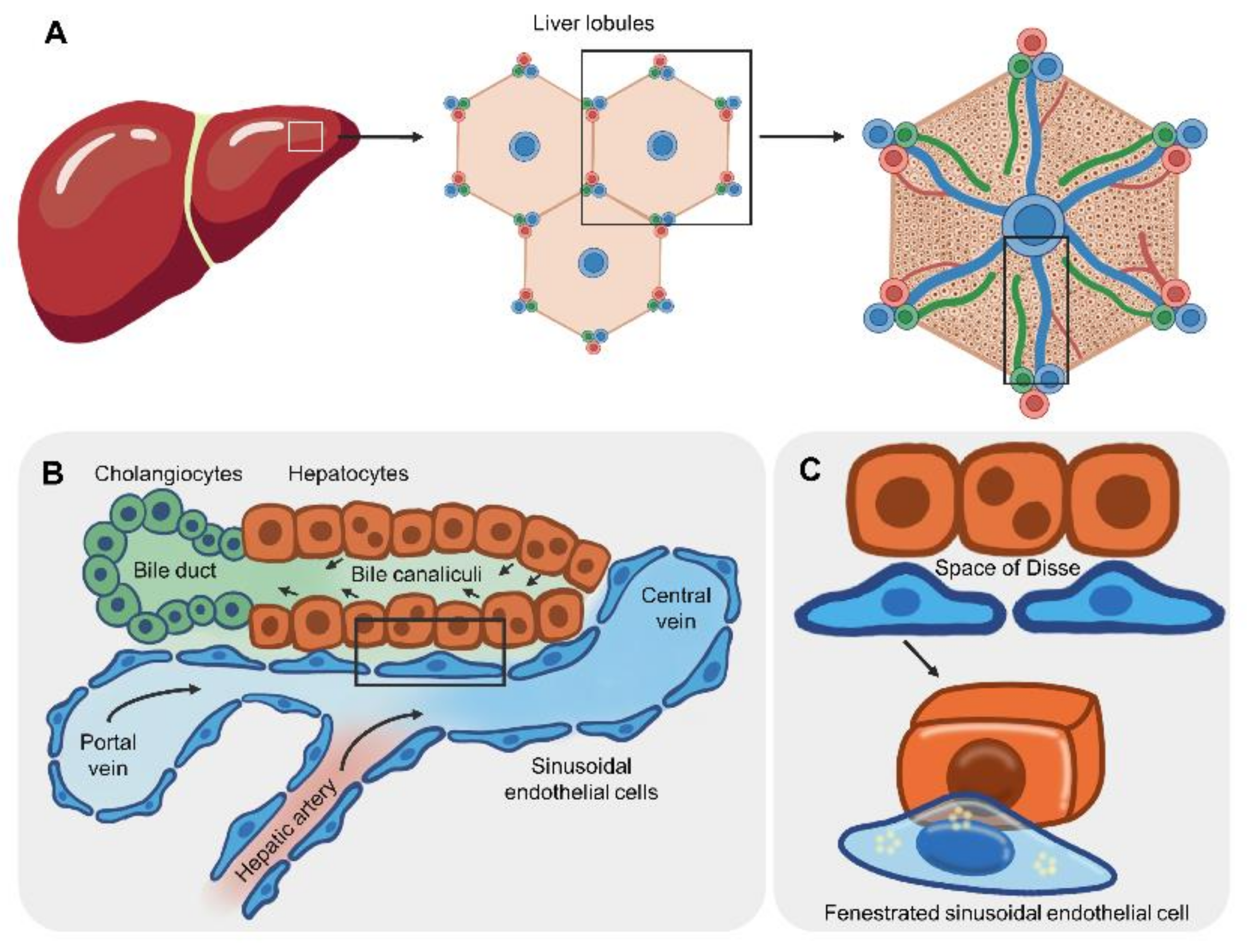
4. Basis of the Liver Microarchitecture for the Liver-on-a-Chip Design
5. Liver-on-a-Chip for Hepatotropic Infectious Diseases
6. Biosensor Assays of Liver Function
7. Combination of a Biosensor and Organ-on-a-Chip
7.1. Cellular Barrier Function
7.2. Cell–Cell Communication via Paracrine Signaling
7.3. Cell Viability
8. Source of Hepatocytes for the Personalized Liver-on-a-Chip
8.1. Liver Biopsy
8.2. Trans.-Differentiation
8.2.1. Mesenchymal Stem Cells (MSCs)
8.2.2. Fibroblasts
8.2.3. Hematopoietic Cells
Hematopoietic Stem and Progenitor Cells
Monocytes
8.3. Human Induced Pluripotent Stem Cells (iPSCs)
9. Opportunities and Challenges in the Use of the Liver-on-a-Chip in Personalized Medicine
9.1. Artificial Porous Layer
9.2. Endothelial Cells
9.3. Multiple Cell Types
9.4. Integration of Biosensors
9.4.1. Cellular Barrier
Basis of TEER
Applications of TEER as a Biosensor
The Use of the TEER-Based Organ-on-a-Chip
9.4.2. Characterization of Cells Using the MEA
10. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mozzi, A.; Pontremoli, C.; Sironi, M. Genetic susceptibility to infectious diseases: Current status and future perspectives from genome-wide approaches. Infect. Genet. Evol. 2018, 66, 286–307. [Google Scholar] [CrossRef]
- Kwok, A.J.; Mentzer, A.; Knight, J.C. Host genetics and infectious disease: New tools, insights and translational opportunities. Nat. Rev. Genet. 2021, 22, 137–153. [Google Scholar] [CrossRef]
- Low, S.K.; Takahashi, A.; Mushiroda, T.; Kubo, M. Genome-wide association study: A useful tool to identify common genetic variants associated with drug toxicity and efficacy in cancer pharmacogenomics. Clin. Cancer Res. 2014, 20, 2541–2552. [Google Scholar] [CrossRef]
- Petros, Z.; Makonnen, E.; Aklillu, E. Genome-wide association studies for idiosyncratic drug-induced hepatotoxicity: Looking back-looking forward to next-generation innovation. OMICS 2017, 21, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in il28b predicts hepatitis c treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Mallal, S.; Phillips, E.; Carosi, G.; Molina, J.M.; Workman, C.; Tomazic, J.; Jagel-Guedes, E.; Rugina, S.; Kozyrev, O.; Cid, J.F.; et al. Hla-b*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 2008, 358, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Higgins, E.; Al Shehri, T.; McAleer, M.A.; Conlon, N.; Feighery, C.; Lilic, D.; Irvine, A.D. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (stat1) mutation. J. Allergy Clin. Immunol. 2015, 135, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 2018, 172, 373–386 e310. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, G.; Mato-Berciano, A.; Pascual-Sabater, S.; Rovira-Rigau, M.; Cuatrecasas, M.; Fondevila, C.; Sanchez-Cabus, S.; Begthel, H.; Boj, S.F.; Clevers, H.; et al. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine 2020, 56, 102786. [Google Scholar] [CrossRef]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Clevers, H. Organoids: Modeling development and the stem cell niche in a dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef]
- Ham, O.; Jin, Y.B.; Kim, J.; Lee, M.O. Blood vessel formation in cerebral organoids formed from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2020, 521, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.S.; Kim, J.S.; Yeo, H.C.; Bae, C.M.; Han, H.J.; Baek, K.; Chang, W.; Lim, K.S.; Yun, S.P.; Shin, I.S.; et al. A simple metastatic brain cancer model using human embryonic stem cell-derived cerebral organoids. FASEB J. 2020, 34, 16464–16475. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.S.; Ki, S.J.; Thanavel, R.; Kim, J.J.; Lee, M.O.; Kim, J.; Jung, C.R.; Han, T.S.; Cho, H.S.; Ryu, C.M.; et al. Maturation of human intestinal organoids in vitro facilitates colonization by commensal lactobacilli by reinforcing the mucus layer. FASEB J. 2020, 34, 9899–9910. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.H.; Han, H.W.; Lee, S.E.; Hong, S.H.; Cho, S.H.; Kim, S.C.; Koo, S.K.; Kim, J.H. Modelling toxoplasma gondii infection in human cerebral organoids. Emerg. Microbes. Infect. 2020, 9, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef]
- Ma, L.D.; Wang, Y.T.; Wang, J.R.; Wu, J.L.; Meng, X.S.; Hu, P.; Mu, X.; Liang, Q.L.; Luo, G.A. Design and fabrication of a liver-on-a-chip platform for convenient, highly efficient, and safe in situ perfusion culture of 3d hepatic spheroids. Lab. Chip 2018, 18, 2547–2562. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef]
- Jang, K.J.; Suh, K.Y. A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells. Lab. Chip 2010, 10, 36–42. [Google Scholar] [CrossRef]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3d cardiac microtissues. Lab. Chip 2016, 16, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab. Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Sugiura, S.; Shin, K.; Onuki-Nagasaki, R.; Ishida, S.; Kikuchi, K.; Kakiki, M.; Kanamori, T. A multi-throughput multi-organ-on-a-chip system on a plate formatted pneumatic pressure-driven medium circulation platform. Lab. Chip 2017, 18, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.; Miller, P. Trial watch: Phase ii and phase iii attrition rates 2011-2012. Nat. Rev. Drug. Discov. 2013, 12, 569. [Google Scholar] [CrossRef]
- Hwang, T.J.; Carpenter, D.; Lauffenburger, J.C.; Wang, B.; Franklin, J.M.; Kesselheim, A.S. Failure of investigational drugs in late-stage clinical development and publication of trial results. JAMA Intern. Med. 2016, 176, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.L.; Baker, T.K. The future of drug safety testing: Expanding the view and narrowing the focus. Drug. Discov. Today. 2009, 14, 162–167. [Google Scholar] [CrossRef]
- World Health Organization. Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- World Health Organization. Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- World Health Organization. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Ginzberg, D.; Wong, R.J.; Gish, R. Global hbv burden: Guesstimates and facts. Hepatol. Int. 2018, 12, 315–329. [Google Scholar] [CrossRef]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis b virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. Review article: Novel therapies for hepatitis b virus cure-advances and perspectives. Aliment. Pharmacol. Ther. 2016, 44, 213–222. [Google Scholar] [CrossRef]
- Song, J.E.; Kim, D.Y. Diagnosis of hepatitis b. Ann. Transl Med. 2016, 4, 338. [Google Scholar] [CrossRef]
- Akcay, I.M.; Katrinli, S.; Ozdil, K.; Doganay, G.D.; Doganay, L. Host genetic factors affecting hepatitis b infection outcomes: Insights from genome-wide association studies. World J. Gastroenterol. 2018, 24, 3347–3360. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S. Personalized treatment of hepatitis b. Clin. Mol. Hepatol. 2015, 21, 1–6. [Google Scholar] [CrossRef]
- Ortega-Prieto, A.M.; Skelton, J.K.; Wai, S.N.; Large, E.; Lussignol, M.; Vizcay-Barrena, G.; Hughes, D.; Fleck, R.A.; Thursz, M.; Catanese, M.T.; et al. 3d microfluidic liver cultures as a physiological preclinical tool for hepatitis b virus infection. Nat. Commun. 2018, 9, 682. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.; Aghemo, A.; Colombo, M. Individualized hepatocellular carcinoma risk: The challenges for designing successful chemoprevention strategies. World J. Gastroenterol. 2013, 19, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis c: Efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. [Google Scholar] [CrossRef]
- Aghemo, A.; De Francesco, R. New horizons in hepatitis c antiviral therapy with direct-acting antivirals. Hepatology 2013, 58, 428–438. [Google Scholar] [CrossRef]
- Burstow, N.J.; Mohamed, Z.; Gomaa, A.I.; Sonderup, M.W.; Cook, N.A.; Waked, I.; Spearman, C.W.; Taylor-Robinson, S.D. Hepatitis c treatment: Where are we now? Int. J. General Med. 2017, 10, 39–52. [Google Scholar] [CrossRef]
- Raj, V.S.; Hundie, G.B.; Schurch, A.C.; Smits, S.L.; Pas, S.D.; Le Pogam, S.; Janssen, H.L.A.; de Knegt, R.J.; Osterhaus, A.; Najera, I.; et al. Identification of hcv resistant variants against direct acting antivirals in plasma and liver of treatment naive patients. Sci. Rep. 2017, 7, 4688. [Google Scholar] [CrossRef]
- Liu, C.H.; Liu, C.J.; Lin, C.L.; Liang, C.C.; Hsu, S.J.; Yang, S.S.; Hsu, C.S.; Tseng, T.C.; Wang, C.C.; Lai, M.Y.; et al. Pegylated interferon-alpha-2a plus ribavirin for treatment-naive asian patients with hepatitis c virus genotype 1 infection: A multicenter, randomized controlled trial. Clin. Infect. Dis. 2008, 47, 1260–1269. [Google Scholar] [CrossRef]
- Yan, K.K.; Guirgis, M.; Dinh, T.; George, J.; Dev, A.; Lee, A.; Zekry, A. Treatment responses in asians and caucasians with chronic hepatitis c infection. World J. Gastroenterol. 2008, 14, 3416–3420. [Google Scholar] [CrossRef]
- Muir, A.J.; Bornstein, J.D.; Killenberg, P.G.; Atlantic Coast Hepatitis Treatment, G. Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis c in blacks and non-hispanic whites. N. Engl. J. Med. 2004, 350, 2265–2271. [Google Scholar] [CrossRef]
- Rodriguez-Torres, M.; Jeffers, L.J.; Sheikh, M.Y.; Rossaro, L.; Ankoma-Sey, V.; Hamzeh, F.M.; Martin, P.; Latino Study, G. Peginterferon alfa-2a and ribavirin in latino and non-latino whites with hepatitis c. N. Engl. J. Med. 2009, 360, 257–267. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Lawitz, E.J.; Shiffman, M.L.; Muir, A.J.; Galler, G.W.; McCone, J.; Nyberg, L.M.; Lee, W.M.; Ghalib, R.H.; Schiff, E.R.; et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis c infection. N. Engl. J. Med. 2009, 361, 580–593. [Google Scholar] [CrossRef]
- Chen, L.; Borozan, I.; Feld, J.; Sun, J.; Tannis, L.L.; Coltescu, C.; Heathcote, J.; Edwards, A.M.; McGilvray, I.D. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis c viral infection. Gastroenterology 2005, 128, 1437–1444. [Google Scholar] [CrossRef]
- Hwang, Y.; Chen, E.Y.; Gu, Z.J.; Chuang, W.L.; Yu, M.L.; Lai, M.Y.; Chao, Y.C.; Lee, C.M.; Wang, J.H.; Dai, C.Y.; et al. Genetic predisposition of responsiveness to therapy for chronic hepatitis c. Pharmacogenomics 2006, 7, 697–709. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, H.; Borg, B.B.; Su, X.; Rhodes, S.L.; Yang, K.; Tong, X.; Tang, G.; Howell, C.D.; Rosen, H.R.; et al. A functional snp of interferon-gamma gene is important for interferon-alpha-induced and spontaneous recovery from hepatitis c virus infection. Proc. Natl. Acad. Sci. USA 2007, 104, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Morgan, T.R.; Bonkovsky, H.L.; Naishadham, D.; Pfeiffer, R.M.; Wright, E.C.; Hutchinson, A.A.; Crenshaw, A.T.; Bashirova, A.; Carrington, M.; et al. Variants in interferon-alpha pathway genes and response to pegylated interferon-alpha2a plus ribavirin for treatment of chronic hepatitis c virus infection in the hepatitis c antiviral long-term treatment against cirrhosis trial. Hepatology 2009, 49, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, R.J.; Dalgard, O. Individualized treatment of chronic hepatitis c with pegylated interferon and ribavirin. Pharmgenomics Pers. Med. 2010, 3, 1–13. [Google Scholar] [PubMed]
- Berg, T.; Naumann, U.; Stoehr, A.; Sick, C.; John, C.; Teuber, G.; Schiffelholz, W.; Mauss, S.; Lohmann, K.; Konig, B.; et al. Real-world effectiveness and safety of glecaprevir/pibrentasvir for the treatment of chronic hepatitis c infection: Data from the german hepatitis c-registry. Aliment. Pharmacol. Ther. 2019, 49, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Mera, J.; Joshi, K.; Thornton, K.; Box, T.; Scott, J.; Sedillo, M.; Deming, P.; David, C.; Essex, W.; Manch, R.; et al. Retrospective study demonstrating high rates of sustained virologic response after treatment with direct-acting antivirals among american indian/alaskan natives. Open. Forum. Infect. Dis. 2019, 6, ofz128. [Google Scholar] [CrossRef]
- Xia, H.; Lu, C.; Wang, Y.; Zaongo, S.D.; Hu, Y.; Wu, Y.; Yan, Z.; Ma, P. Efficacy and safety of direct-acting antiviral therapy in patients with chronic hepatitis c virus infection: A real-world single-center experience in tianjin, china. Front. Pharmacol. 2020, 11, 710. [Google Scholar] [CrossRef]
- Cusato, J.; De Nicolo, A.; Boglione, L.; Favata, F.; Ariaudo, A.; Mornese Pinna, S.; Carcieri, C.; Guido, F.; Avataneo, V.; Cariti, G.; et al. Pharmacogenetics of the anti-hcv drug sofosbuvir: A preliminary study. J. Antimicrob. Chemother. 2018, 73, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Cusato, J.; Allegra, S.; De Nicolo, A.; Boglione, L.; Fatiguso, G.; Cariti, G.; Ciancio, A.; Smedile, A.; Strona, S.; Troshina, G.; et al. Abcb11 and abcb1 gene polymorphisms impact on telaprevir pharmacokinetic at one month of therapy. Biomed. Pharmacother 2015, 69, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Geddawy, A.; Ibrahim, Y.F.; Elbahie, N.M.; Ibrahim, M.A. Direct acting anti-hepatitis c virus drugs: Clinical pharmacology and future direction. J. Transl. Int. Med. 2017, 5, 8–17. [Google Scholar] [CrossRef] [PubMed]
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735. [Google Scholar] [CrossRef]
- Price, R.N.; Tjitra, E.; Guerra, C.A.; Yeung, S.; White, N.J.; Anstey, N.M. Vivax malaria: Neglected and not benign. Am. J. Trop. Med. Hyg. 2007, 77, 79–87. [Google Scholar] [CrossRef]
- Wells, T.N.; Burrows, J.N.; Baird, J.K. Targeting the hypnozoite reservoir of plasmodium vivax: The hidden obstacle to malaria elimination. Trends Parasitol. 2010, 26, 145–151. [Google Scholar] [CrossRef]
- Lacerda, M.V.G.; Llanos-Cuentas, A.; Krudsood, S.; Lon, C.; Saunders, D.L.; Mohammed, R.; Yilma, D.; Batista Pereira, D.; Espino, F.E.J.; Mia, R.Z.; et al. Single-dose tafenoquine to prevent relapse of plasmodium vivax malaria. N. Engl. J. Med. 2019, 380, 215–228. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Lacerda, M.V.G.; Hien, T.T.; Velez, I.D.; Namaik-Larp, C.; Chu, C.S.; Villegas, M.F.; Val, F.; Monteiro, W.M.; Brito, M.A.M.; et al. Tafenoquine versus primaquine to prevent relapse of plasmodium vivax malaria. N. Engl. J. Med. 2019, 380, 229–241. [Google Scholar] [CrossRef]
- Pybus, B.S.; Marcsisin, S.R.; Jin, X.; Deye, G.; Sousa, J.C.; Li, Q.; Caridha, D.; Zeng, Q.; Reichard, G.A.; Ockenhouse, C.; et al. The metabolism of primaquine to its active metabolite is dependent on cyp 2d6. Malar. J. 2013, 12, 212. [Google Scholar] [CrossRef]
- Marcsisin, S.R.; Reichard, G.; Pybus, B.S. Primaquine pharmacology in the context of cyp 2d6 pharmacogenomics: Current state of the art. Pharmacol. Ther. 2016, 161, 1–10. [Google Scholar] [CrossRef]
- Bennett, J.W.; Pybus, B.S.; Yadava, A.; Tosh, D.; Sousa, J.C.; McCarthy, W.F.; Deye, G.; Melendez, V.; Ockenhouse, C.F. Primaquine failure and cytochrome p-450 2d6 in plasmodium vivax malaria. N. Engl. J. Med. 2013, 369, 1381–1382. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Simon, S.D.; Pearce, R.E.; Bradford, L.D.; Kennedy, M.J.; Leeder, J.S. The cyp2d6 activity score: Translating genotype information into a qualitative measure of phenotype. Clin. Pharmacol. Ther. 2008, 83, 234–242. [Google Scholar] [CrossRef]
- Ingram, R.J.; Crenna-Darusallam, C.; Soebianto, S.; Noviyanti, R.; Baird, J.K. The clinical and public health problem of relapse despite primaquine therapy: Case review of repeated relapses of plasmodium vivax acquired in papua new guinea. Malar. J. 2014, 13, 488. [Google Scholar] [CrossRef][Green Version]
- Baird, J.K.; Louisa, M.; Noviyanti, R.; Ekawati, L.; Elyazar, I.; Subekti, D.; Chand, K.; Gayatri, A.; Instiaty, M.D.; Soebianto, S.; et al. Association of impaired cytochrome p450 2d6 activity genotype and phenotype with therapeutic efficacy of primaquine treatment for latent plasmodium vivax malaria. JAMA Netw. Open 2018, 1, e181449. [Google Scholar]
- Brasil, L.W.; Rodrigues-Soares, F.; Santoro, A.B.; Almeida, A.C.G.; Kuhn, A.; Ramasawmy, R.; Lacerda, M.V.G.; Monteiro, W.M.; Suarez-Kurtz, G. Cyp2d6 activity and the risk of recurrence of plasmodium vivax malaria in the brazilian amazon: A prospective cohort study. Malar. J. 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Dowd, S.; Gatton, M.L.; Auliff, A.; Edstein, M.D.; Cheng, Q. Cytochrome p450 2d6 profiles and their relationship with outcomes of primaquine anti-relapse therapy in australian defence force personnel deployed to papua new guinea and east timor. Malar. J. 2019, 18, 140. [Google Scholar] [CrossRef]
- Baird, J.K.; Battle, K.E.; Howes, R.E. Primaquine ineligibility in anti-relapse therapy of plasmodium vivax malaria: The problem of g6pd deficiency and cytochrome p-450 2d6 polymorphisms. Malar. J. 2018, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M.; Acute Liver Failure Study, G. Drug-induced acute liver failure: Results of a u.S. Multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef]
- Sunil Kumar, N.; Remalayam, B.; Thomas, V.; Ramachandran, T.M.; Sunil Kumar, K. Outcomes and predictors of mortality in patients with drug-induced liver injury at a tertiary hospital in south india: A single-centre experience. J. Clin. Exp. Hepatol. 2021, 11, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Devarbhavi, H.; Aithal, G.; Treeprasertsuk, S.; Takikawa, H.; Mao, Y.; Shasthry, S.M.; Hamid, S.; Tan, S.S.; Philips, C.A.; George, J.; et al. Drug-induced liver injury: Asia pacific association of study of liver consensus guidelines. Hepatol. Int. 2021, 15, 258–282. [Google Scholar] [CrossRef]
- Hoofnagle, J.H.; Bjornsson, E.S. Drug-induced liver injury-types and phenotypes. N. Engl. J. Med. 2019, 381, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.W.; Watkins, P.B. Are patients with elevated liver tests at increased risk of drug-induced liver injury? Gastroenterology 2004, 126, 1477–1480. [Google Scholar] [CrossRef]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.; Lee, W.; Stolz, A.; Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.; Barnhart, H.; et al. Features and outcomes of 899 patients with drug-induced liver injury: The dilin prospective study. Gastroenterology 2015, 148, 1340–1352 e1347. [Google Scholar] [CrossRef]
- Treem, W.R.; Palmer, M.; Lonjon-Domanec, I.; Seekins, D.; Dimick-Santos, L.; Avigan, M.I.; Marcinak, J.F.; Dash, A.; Regev, A.; Maller, E.; et al. Consensus guidelines: Best practices for detection, assessment and management of suspected acute drug-induced liver injury during clinical trials in adults with chronic viral hepatitis and adults with cirrhosis secondary to hepatitis b, c and nonalcoholic steatohepatitis. Drug Saf. 2021, 44, 133–165. [Google Scholar] [PubMed]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. Hla-b*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Molokhia, M.; Shen, Y.; Urban, T.J.; Aithal, G.P.; Andrade, R.J.; Day, C.P.; Ruiz-Cabello, F.; Donaldson, P.T.; Stephens, C.; et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple hla class i and ii alleles. Gastroenterology 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J.; Cirulli, E.T.; Gu, J.; Kleiner, D.; Ostrov, D.; Phillips, E.; Schutte, R.; Barnhart, H.; Chalasani, N.; Watkins, P.B.; et al. The role of hla-a*33:01 in patients with cholestatic hepatitis attributed to terbinafine. J. Hepatol. 2018, 69, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Wojciech Pawlina, M.H.R. Histology: A Text and Atlas, 8th ed.; Lippincott Wiliams & Wikins: Baltimore, MD, USA, 2006. [Google Scholar]
- Lautt, W. Hepatic circulation: Physiology and pathophysiology. In Colloquium Series on Integrated Systems Physiology; Morgan & Claypool Publishers: San Rafael, CA, USA, 2009. [Google Scholar]
- Stanger, B.Z. Cellular homeostasis and repair in the mammalian liver. Annu. Rev. Physiol. 2015, 77, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef]
- Sodunke, T.R.; Bouchard, M.J.; Noh, H.M. Microfluidic platform for hepatitis b viral replication study. Biomed. Microdevices 2008, 10, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Rawat, S.; Duchemin, N.; Bouchard, M.; Noh, M. Human liver sinusoid on a chip for hepatitis b virus replication study. Micromachines 2017, 8, 27. [Google Scholar] [CrossRef]
- Song, M.J.; Yun, D.H.; Min, N.K.; Hong, S.I. Electrochemical biosensor array for liver diagnosis using silanization technique on nanoporous silicon electrode. J. Biosci. Bioeng. 2007, 103, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.H.; Chang, Y.T.; Liu, K.L.; Chang, H.Y.; Yew, T.R. Electrical impedimetric biosensors for liver function detection. Biosens. Bioelectron. 2011, 28, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Chiu, P.H.; Wang, Y.H.; Yang, C.F.; Feng, S.W. Electrochemical formation of crooked gold nanorods and gold networked structures by the additive organic solvent. J. Colloid. Interface Sci. 2007, 306, 56–65. [Google Scholar] [CrossRef]
- Prill, S.; Bavli, D.; Levy, G.; Ezra, E.; Schmalzlin, E.; Jaeger, M.S.; Schwarz, M.; Duschl, C.; Cohen, M.; Nahmias, Y. Real-time monitoring of oxygen uptake in hepatic bioreactor shows cyp450-independent mitochondrial toxicity of acetaminophen and amiodarone. Arch. Toxicol. 2016, 90, 1181–1191. [Google Scholar] [CrossRef]
- Moman, R.N.; Gupta, N.; Varacallo, M. Physiology, albumin. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459198/ (accessed on 15 June 2021).
- Lee, S.; Sung, D.B.; Kang, S.; Parameswaran, S.; Choi, J.H.; Lee, J.S.; Han, M.S. Development of human serum albumin selective fluorescent probe using thieno[3,2-b]pyridine-5(4h)-one fluorophore derivatives. Sensors 2019, 19, 5298. [Google Scholar] [CrossRef] [PubMed]
- Maoz, B.M.; Herland, A.; Henry, O.Y.F.; Leineweber, W.D.; Yadid, M.; Doyle, J.; Mannix, R.; Kujala, V.J.; FitzGerald, E.A.; Parker, K.K.; et al. Organs-on-chips with combined multi-electrode array and transepithelial electrical resistance measurement capabilities. Lab. Chip 2017, 17, 2294–2302. [Google Scholar] [CrossRef]
- Lind, J.U.; Busbee, T.A.; Valentine, A.D.; Pasqualini, F.S.; Yuan, H.; Yadid, M.; Park, S.J.; Kotikian, A.; Nesmith, A.P.; Campbell, P.H.; et al. Instrumented cardiac microphysiological devices via multimaterial three-dimensional printing. Nat. Mater. 2017, 16, 303–308. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Zhou, Q.; Patel, D.; Kwa, T.; Haque, A.; Matharu, Z.; Stybayeva, G.; Gao, Y.; Diehl, A.M.; Revzin, A. Liver injury-on-a-chip: Microfluidic co-cultures with integrated biosensors for monitoring liver cell signaling during injury. Lab. Chip 2015, 15, 4467–4478. [Google Scholar] [CrossRef]
- Chang, Y.; Li, H. Hepatic antifibrotic pharmacotherapy: Are we approaching success? J. Clin. Transl. Hepatol. 2020, 8, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Gehre, C.; Flechner, M.; Kammerer, S.; Kupper, J.H.; Coleman, C.D.; Puschel, G.P.; Uhlig, K.; Duschl, C. Real time monitoring of oxygen uptake of hepatocytes in a microreactor using optical microsensors. Sci. Rep. 2020, 10, 13700. [Google Scholar] [CrossRef]
- Afshari, A.; Shamdani, S.; Uzan, G.; Naserian, S.; Azarpira, N. Different approaches for transformation of mesenchymal stem cells into hepatocyte-like cells. Stem Cell Res. Ther. 2020, 11, 54. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells-current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef] [PubMed]
- Banas, A.; Teratani, T.; Yamamoto, Y.; Tokuhara, M.; Takeshita, F.; Quinn, G.; Okochi, H.; Ochiya, T. Adipose tissue-derived mesenchymal stem cells as a source of human hepatocytes. Hepatology 2007, 46, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Aurich, H.; Sgodda, M.; Kaltwasser, P.; Vetter, M.; Weise, A.; Liehr, T.; Brulport, M.; Hengstler, J.G.; Dollinger, M.M.; Fleig, W.E.; et al. Hepatocyte differentiation of mesenchymal stem cells from human adipose tissue in vitro promotes hepatic integration in vivo. Gut 2009, 58, 570–581. [Google Scholar] [CrossRef]
- Kogiso, T.; Nagahara, H.; Otsuka, M.; Shiratori, K.; Dowdy, S.F. Transdifferentiation of human fibroblasts into hepatocyte-like cells by defined transcriptional factors. Hepatol. Int. 2013, 7, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Lysy, P.A.; Smets, F.; Sibille, C.; Najimi, M.; Sokal, E.M. Human skin fibroblasts: From mesodermal to hepatocyte-like differentiation. Hepatology 2007, 46, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Mukhopadhyay, A. In vitro transdifferentiation of adult hematopoietic stem cells: An alternative source of engraftable hepatocytes. J. Hepatol. 2008, 49, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Jaiswal, A.K.; Mukhopadhyay, A. Hepatocyte nuclear factor-4alpha induces transdifferentiation of hematopoietic cells into hepatocytes. J. Biol. Chem. 2010, 285, 4725–4731. [Google Scholar] [CrossRef]
- Sellamuthu, S.; Manikandan, R.; Thiagarajan, R.; Babu, G.; Dinesh, D.; Prabhu, D.; Arulvasu, C. In vitro trans-differentiation of human umbilical cord derived hematopoietic stem cells into hepatocyte like cells using combination of growth factors for cell based therapy. Cytotechnology 2011, 63, 259–268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jung, Y.J.; Ryu, K.H.; Cho, K.A.; Woo, S.Y.; Seoh, J.Y.; Cho, S.J.; Joo, S.Y.; Yoo, K.; Ho-Seoung, H. In vitro hepatic differentiation of human umbilical cord blood and bone marrow cells. Pediatr. Hematol. Oncol. 2008, 25, 481–491. [Google Scholar] [CrossRef]
- Benesic, A.; Rahm, N.L.; Ernst, S.; Gerbes, A.L. Human monocyte-derived cells with individual hepatocyte characteristics: A novel tool for personalized in vitro studies. Lab. Investig. 2012, 92, 926–936. [Google Scholar] [CrossRef]
- Benesic, A.; Leitl, A.; Gerbes, A.L. Monocyte-derived hepatocyte-like cells for causality assessment of idiosyncratic drug-induced liver injury. Gut 2016, 65, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Lampert, A.; Bennett, D.L.; McDermott, L.A.; Neureiter, A.; Eberhardt, E.; Winner, B.; Zenke, M. Human sensory neurons derived from pluripotent stem cells for disease modelling and personalized medicine. Neurobiol. Pain 2020, 8, 100055. [Google Scholar] [CrossRef]
- Paik, D.T.; Chandy, M.; Wu, J.C. Patient and disease-specific induced pluripotent stem cells for discovery of personalized cardiovascular drugs and therapeutics. Pharmacol. Rev. 2020, 72, 320–342. [Google Scholar] [CrossRef] [PubMed]
- Truong, V.; Viken, K.; Geng, Z.; Barkan, S.; Johnson, B.; Ebeling, M.C.; Montezuma, S.R.; Ferrington, D.A.; Dutton, J.R. Automating human induced pluripotent stem cell culture and differentiation of ipsc-derived retinal pigment epithelium for personalized drug testing. SLAS Technol. 2021, 26, 287–299. [Google Scholar] [PubMed]
- Tajiri, S.; Yamanaka, S.; Fujimoto, T.; Matsumoto, K.; Taguchi, A.; Nishinakamura, R.; Okano, H.J.; Yokoo, T. Regenerative potential of induced pluripotent stem cells derived from patients undergoing haemodialysis in kidney regeneration. Sci. Rep. 2018, 8, 14919. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem. Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Kulkeaw, K.; Tubsuwan, A.; Tongkrajang, N.; Whangviboonkij, N. Generation of human liver organoids from pluripotent stem cell-derived hepatic endoderms. PeerJ 2020, 8, e9968. [Google Scholar] [CrossRef]
- Nishizawa, M.; Chonabayashi, K.; Nomura, M.; Tanaka, A.; Nakamura, M.; Inagaki, A.; Nishikawa, M.; Takei, I.; Oishi, A.; Tanabe, K.; et al. Epigenetic variation between human induced pluripotent stem cell lines is an indicator of differentiation capacity. Cell Stem Cell 2016, 19, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Noor, N.; Shapira, A.; Edri, R.; Gal, I.; Wertheim, L.; Dvir, T. 3d printing of personalized thick and perfusable cardiac patches and hearts. Adv. Sci. 2019, 6, 1900344. [Google Scholar] [CrossRef]
- King, O.; Sunyovszki, I.; Terracciano, C.M. Vascularisation of pluripotent stem cell-derived myocardium: Biomechanical insights for physiological relevance in cardiac tissue engineering. Pflugers. Arch. 2021, 473. [Google Scholar] [CrossRef]
- Besser, R.R.; Bowles, A.C.; Alassaf, A.; Carbonero, D.; Maciel, R.; Saporta, M.; Agarwal, A. A chemically defined common medium for culture of c2c12 skeletal muscle and human induced pluripotent stem cell derived spinal spheroids. Cell Mol. Bioeng. 2020, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Schwach, V.; Slaats, R.H.; Passier, R. Human pluripotent stem cell-derived cardiomyocytes for assessment of anticancer drug-induced cardiotoxicity. Front. Cardiovasc. Med. 2020, 7, 50. [Google Scholar] [CrossRef]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating nonalcoholic fatty liver disease in a liver-on-a-chip microfluidic device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef]
- Banaeiyan, A.A.; Theobald, J.; Paukstyte, J.; Wolfl, S.; Adiels, C.B.; Goksor, M. Design and fabrication of a scalable liver-lobule-on-a-chip microphysiological platform. Biofabrication 2017, 9, 015014. [Google Scholar] [CrossRef]
- Ho, C.T.; Lin, R.Z.; Chen, R.J.; Chin, C.K.; Gong, S.E.; Chang, H.Y.; Peng, H.L.; Hsu, L.; Yew, T.R.; Chang, S.F.; et al. Liver-cell patterning lab chip: Mimicking the morphology of liver lobule tissue. Lab. Chip 2013, 13, 3578–3587. [Google Scholar] [CrossRef] [PubMed]
- Prodanov, L.; Jindal, R.; Bale, S.S.; Hegde, M.; McCarty, W.J.; Golberg, I.; Bhushan, A.; Yarmush, M.L.; Usta, O.B. Long-term maintenance of a microfluidic 3d human liver sinusoid. Biotechnol. Bioeng. 2016, 113, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Delalat, B.; Cozzi, C.; Rasi Ghaemi, S.; Polito, G.; Kriel, F.H.; Michl, T.D.; Harding, F.J.; Priest, C.; Barillaro, G.; Voelcker, N.H. Microengineered bioartificial liver chip for drug toxicity screening. Adv. Func. Mat. 2018, 28, 1801825. [Google Scholar] [CrossRef]
- Segarra, M.; Aburto, M.R.; Acker-Palmer, A. Blood-brain barrier dynamics to maintain brain homeostasis. Trends Neurosci. 2021, 44, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.L.; Qurashi, M.; Shetty, S. The role of sinusoidal endothelial cells in the axis of inflammation and cancer within the liver. Front. Physiol. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. Teer measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Eigenmann, D.E.; Xue, G.; Kim, K.S.; Moses, A.V.; Hamburger, M.; Oufir, M. Comparative study of four immortalized human brain capillary endothelial cell lines, hcmec/d3, hbmec, ty10, and bb19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS 2013, 10, 33. [Google Scholar] [CrossRef]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Kamiloglu, S.; Grootaert, C.; Capanoglu, E.; Ozkan, C.; Smagghe, G.; Raes, K.; Van Camp, J. Anti-inflammatory potential of black carrot (daucus carota l.) polyphenols in a co-culture model of intestinal caco-2 and endothelial ea.Hy926 cells. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Bernas, M.J.; Cardoso, F.L.; Daley, S.K.; Weinand, M.E.; Campos, A.R.; Ferreira, A.J.; Hoying, J.B.; Witte, M.H.; Brites, D.; Persidsky, Y.; et al. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat. Protoc. 2010, 5, 1265–1272. [Google Scholar] [CrossRef]
- Duff, T.; Carter, S.; Feldman, G.; McEwan, G.; Pfaller, W.; Rhodes, P.; Ryan, M.; Hawksworth, G. Transepithelial resistance and inulin permeability as endpoints in in vitro nephrotoxicity testing. Altern. Lab. Anim. 2002, 30 (Suppl. 2), 53–59. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; O’Kane, R.L.; Simpson, I.A.; Vina, J.R. Structure of the blood-brain barrier and its role in the transport of amino acids. J. Nutr. 2006, 136, 218S–226S. [Google Scholar] [CrossRef] [PubMed]
- Prabhakarpandian, B.; Shen, M.C.; Nichols, J.B.; Mills, I.R.; Sidoryk-Wegrzynowicz, M.; Aschner, M.; Pant, K. Sym-bbb: A microfluidic blood brain barrier model. Lab. Chip 2013, 13, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Booth, R.; Kim, H. Characterization of a microfluidic in vitro model of the blood-brain barrier (mubbb). Lab. Chip 2012, 12, 1784–1792. [Google Scholar] [CrossRef]
- Griep, L.M.; Wolbers, F.; de Wagenaar, B.; ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; van der Meer, A.D.; van den Berg, A. Bbb on chip: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef]
- Almog, R.; Daniel, R.; Vernick, S.; Ron, A.; Ben-Yoav, H.; Shacham-Diamand, Y. On-chip detection of cellular activity. Adv. Biochem. Eng. Biotechnol. 2010, 117, 179–191. [Google Scholar]
- Thuenauer, R.; Rodriguez-Boulan, E.; Romer, W. Microfluidic approaches for epithelial cell layer culture and characterisation. Analyst 2014, 139, 3206–3218. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, R.; Yamamoto, H.; Hayakawa, T.; Katsurabayashi, S.; Niwano, M.; Hirano-Iwata, A. Dependence and homeostasis of membrane impedance on cell morphology in cultured hippocampal neurons. Sci. Rep. 2018, 8, 9905. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Luong, J.H. On-line monitoring of cell growth and cytotoxicity using electric cell-substrate impedance sensing (ecis). Biotechnol. Prog. 2003, 19, 1000–1005. [Google Scholar] [CrossRef]
- Naumowicz, M.; Figaszewski, Z.A. Impedance analysis of phosphatidylcholine/alpha-tocopherol system in bilayer lipid membranes. J. Membr. Biol. 2005, 205, 29–36. [Google Scholar] [CrossRef]
- Naumowicz, M.; Figaszewski, Z.A. Impedance analysis of lipid domains in phosphatidylcholine bilayer membranes containing ergosterol. Biophys. J. 2005, 89, 3174–3182. [Google Scholar] [CrossRef] [PubMed]
- Denelavas, A.; Weibel, F.; Prummer, M.; Imbach, A.; Clerc, R.G.; Apfel, C.M.; Hertel, C. Real-time cellular impedance measurements detect Ca2+ channel-dependent oscillations of morphology in human h295r adrenoma cells. Biochim. Biophys. Acta 2011, 1813, 754–762. [Google Scholar] [CrossRef]
- Wegener, J.; Keese, C.R.; Giaever, I. Electric cell-substrate impedance sensing (ecis) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp. Cell Res. 2000, 259, 158–166. [Google Scholar] [CrossRef]
- Meissner, R.; Eker, B.; Kasi, H.; Bertsch, A.; Renaud, P. Distinguishing drug-induced minor morphological changes from major cellular damage via label-free impedimetric toxicity screening. Lab. Chip 2011, 11, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Asphahani, F.; Thein, M.; Veiseh, O.; Edmondson, D.; Kosai, R.; Veiseh, M.; Xu, J.; Zhang, M. Influence of cell adhesion and spreading on impedance characteristics of cell-based sensors. Biosens. Bioelectron. 2008, 23, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Asphahani, F.; Thein, M.; Wang, K.; Wood, D.; Wong, S.S.; Xu, J.; Zhang, M. Real-time characterization of cytotoxicity using single-cell impedance monitoring. Analyst 2012, 137, 3011–3019. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functions | Recognition Compartment | Surface for Immobilizationand Transduction | Ref. |
|---|---|---|---|
| Enzymes |
| [88] |
| Human serum albumin (HSA) | Antibody |
| [89] |
| Electrochemical sensing |
| [90] | |
| Oxygen | Phosphorescence probe | Microbeads | [91] |
| Commercial Name of TEER Device | Electrodes for the Apical-Basolateral Compartment | Culture Well Format | Applications and References |
|---|---|---|---|
| Epithelial Volt-ohmmeter (EVOM) | A pair of two electrodes (chopstick electrode) | Transwell | BBB using immortalized human brain capillary endothelial cell lines [133] |
| REMS Auto-Sampler (World Precision Instruments) | Chopstick electrode located on the robotic arm and the REMS electrode interface | Transwell | (1) BBB using murine brain microvascular endothelial cells [134] (2) Gastrointestinal model using human colon adenocarcinoma cell line Caco-2 and endothelial cell line EA.hy926 [135] |
| EndOhm (World Precision Instruments) | Concentric electrodes on the top and bottom | Single chamber | Renal epithelial barrier using Madin Darby canine kidney (MDCK) strain 1 cells and porcine epithelial kidney cells (LLC-PK1) [137] |
| ECISA pplied BioPhysics | A gold electrode-containing film | Single chamber | BBB using human brain microvascular endothelial cells [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulkeaw, K.; Pengsart, W. Progress and Challenges in the Use of a Liver-on-a-Chip for Hepatotropic Infectious Diseases. Micromachines 2021, 12, 842. https://doi.org/10.3390/mi12070842
Kulkeaw K, Pengsart W. Progress and Challenges in the Use of a Liver-on-a-Chip for Hepatotropic Infectious Diseases. Micromachines. 2021; 12(7):842. https://doi.org/10.3390/mi12070842
Chicago/Turabian StyleKulkeaw, Kasem, and Worakamol Pengsart. 2021. "Progress and Challenges in the Use of a Liver-on-a-Chip for Hepatotropic Infectious Diseases" Micromachines 12, no. 7: 842. https://doi.org/10.3390/mi12070842
APA StyleKulkeaw, K., & Pengsart, W. (2021). Progress and Challenges in the Use of a Liver-on-a-Chip for Hepatotropic Infectious Diseases. Micromachines, 12(7), 842. https://doi.org/10.3390/mi12070842