Lab-on-a-Chip Technologies for the Single Cell Level: Separation, Analysis, and Diagnostics


Abstract
1. Introduction
What We Can Detect:
2. Methodologies
- Cell separation methods
- Combined separation and analysis on chip
- Molecular analysis of single cells
2.1. Cell Separation Methods
2.1.1. Dielectrophoresis (DEP)
2.1.2. Deterministic Lateral Displacement (DLD)
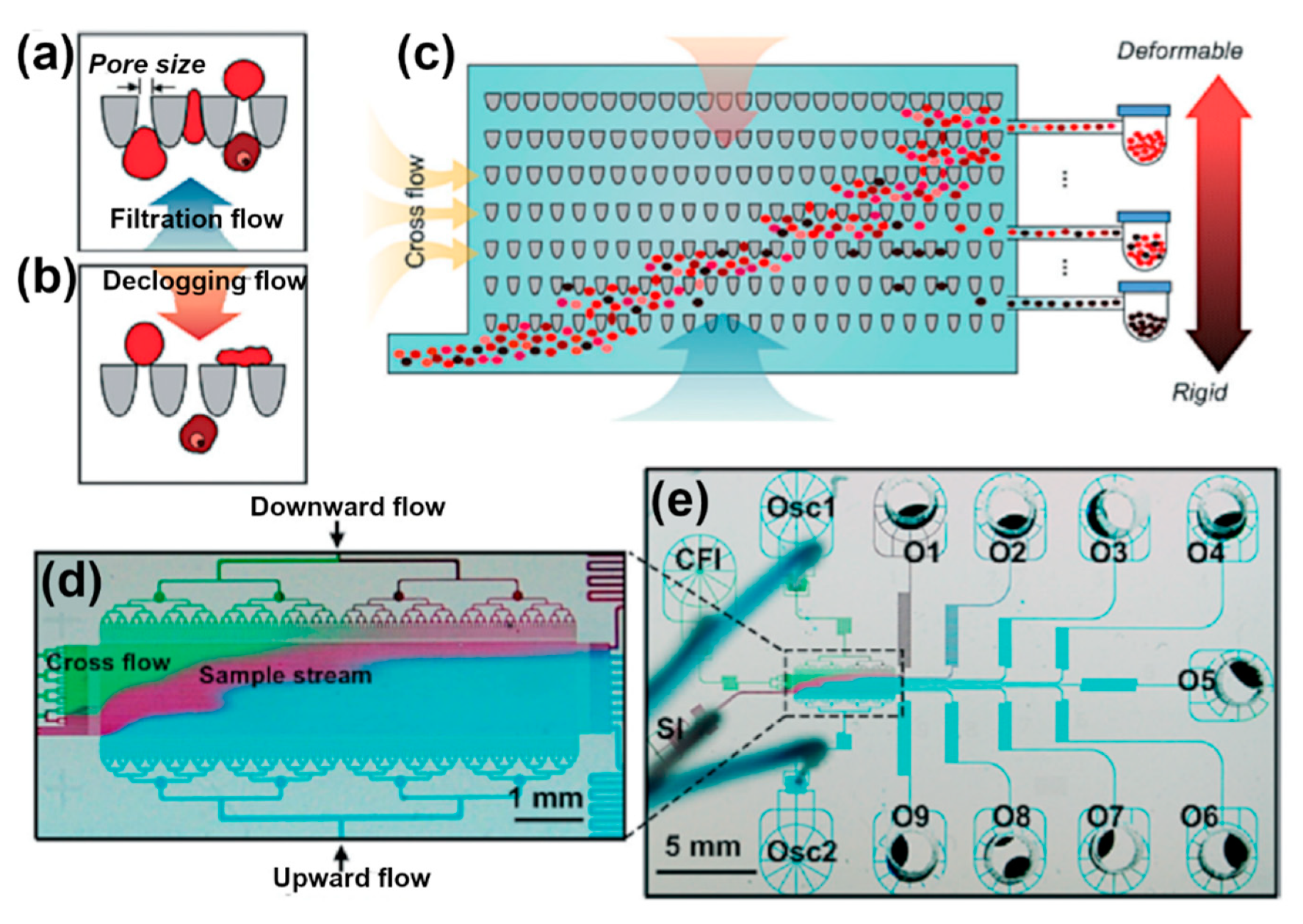
2.1.3. Deformability-Based
2.1.4. Margination and Dean-Flow
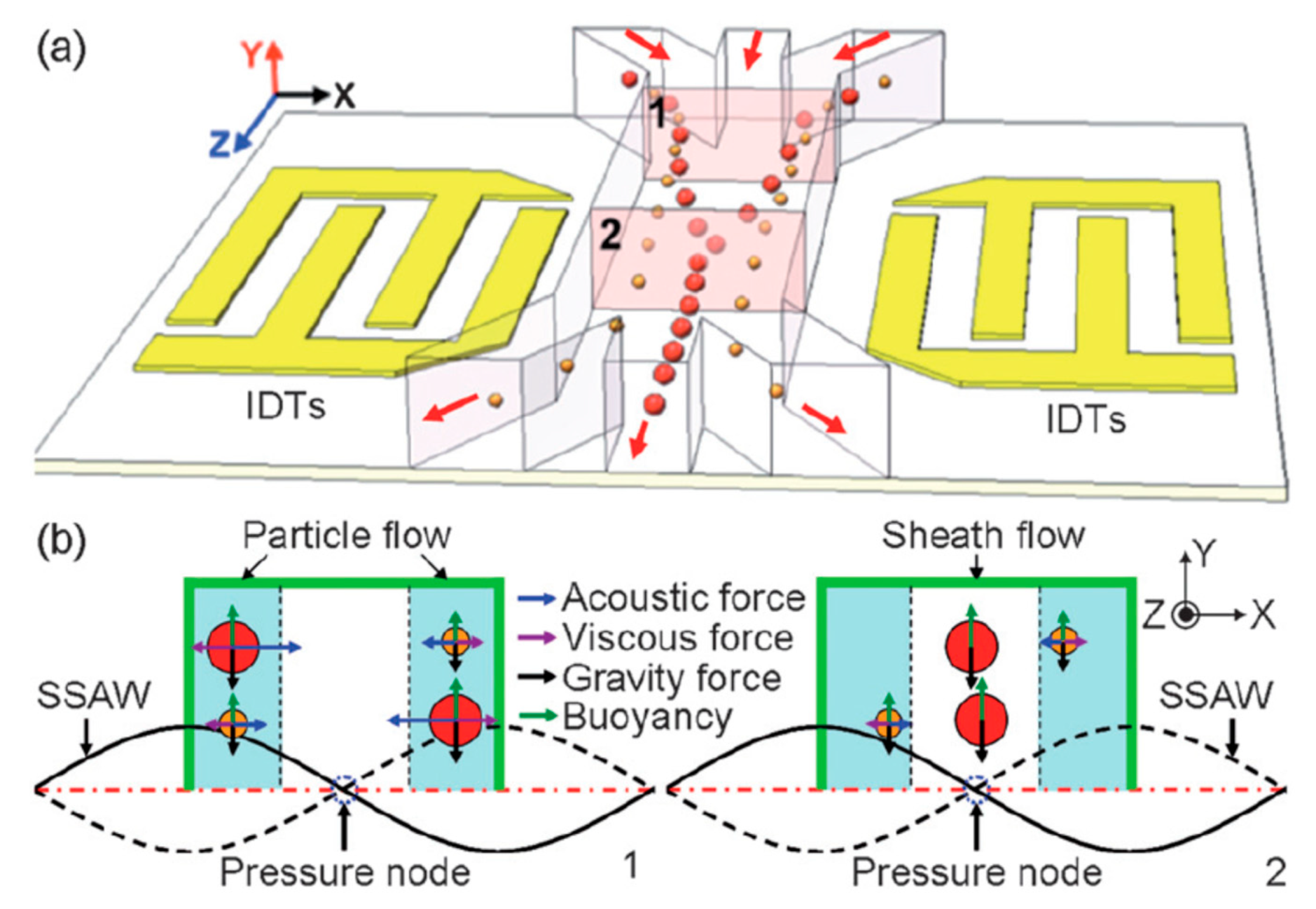
2.1.5. Surface Acoustic Waves (SAW)
2.1.6. Optical Trap and Tweezers
2.2. Combined Separation and Analysis on Chip
2.2.1. Droplet Microfluidics
2.2.2. Optical Density/ Refractive Index
2.2.3. Paper Microfluidics
2.3. Molecular Analysis of Single Cells
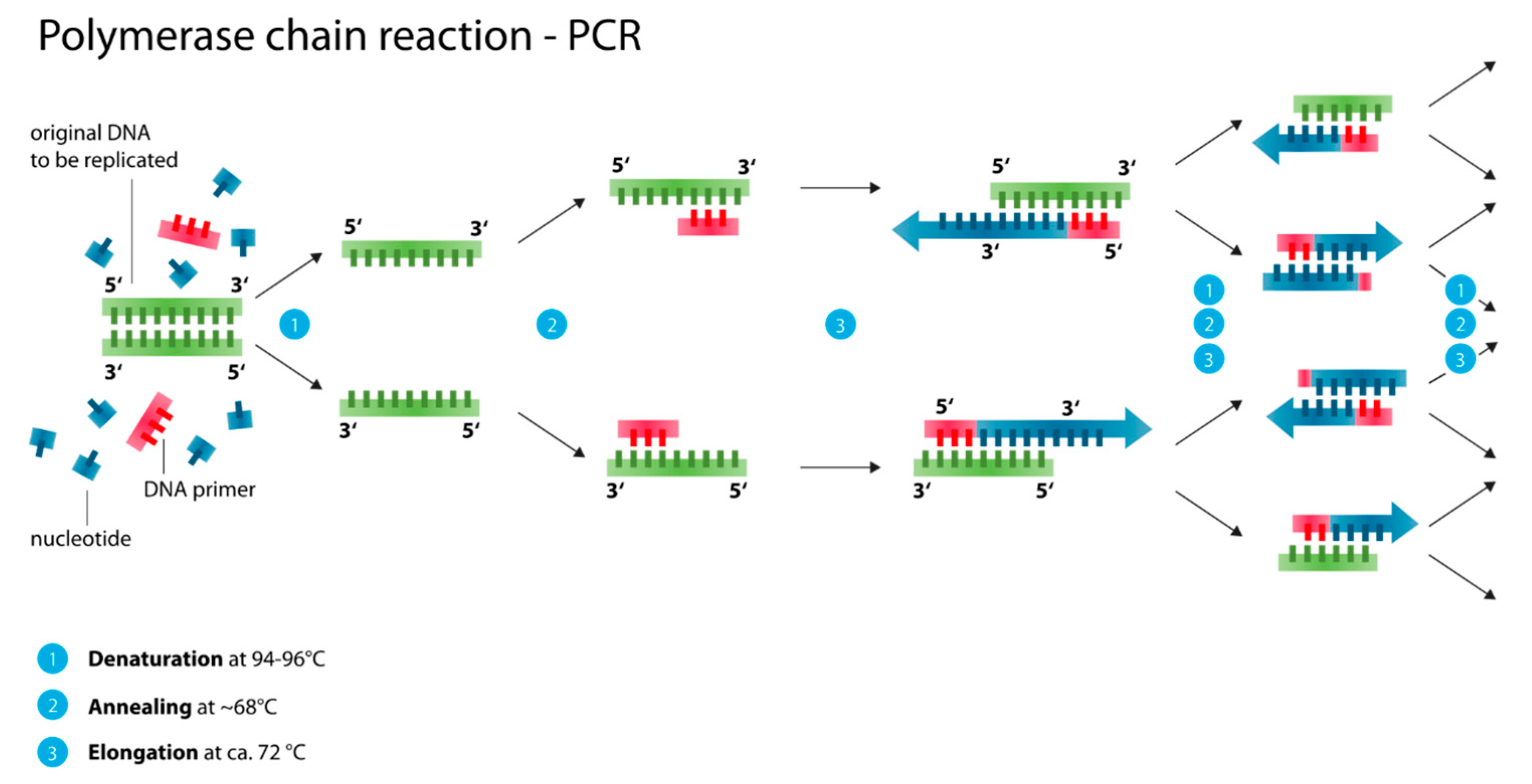
2.3.1. Polymerase Chain Reaction (PCR, Nested PCR, qPCR/ RT-PCR)
2.3.2. Loop-Mediated Isothermal Amplification (LAMP, NAAT, LAMPport, NINA-LAMP)
2.3.3. Proteomics, Metabolomics, Transcriptomics and Polyomics
3. Implementation
3.1. Point-of-Care Diagnostics (POC)
3.2. Biosensors in the Developping World and the Need for ASSURED
4. Discussion
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Nematbakhsh, Y.; Lim, C.T. Cell biomechanics and its applications in human disease diagnosis. Acta Mech. Sin. 2015, 31, 268–273. [Google Scholar] [CrossRef]
- Kozminsky, M.; Wang, Y.; Nagrath, S. The incorporation of microfluidics into circulating tumor cell isolation for clinical applications. Curr. Opin. Chem. Eng. 2016, 11, 59–66. [Google Scholar] [CrossRef]
- Okano, H.; Konishi, T.; Suzuki, T.; Suzuki, T.; Ariyasu, S.; Aoki, S.; Abe, R.; Hayase, M. Enrichment of circulating tumor cells in tumor-bearing mouse blood by a deterministic lateral displacement microfluidic device. Biomed. Microdevices 2015, 17, 1–11. [Google Scholar] [CrossRef]
- Au, S.H.; Edd, J.; Stoddard, A.E.; Wong, K.H.K.; Fachin, F.; Maheswaran, S.; Haber, D.A.; Stott, S.L.; Kapur, R.; Toner, M. Microfluidic isolation of circulating tumor cell clusters by size and asymmetry. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, W.; Huang, F.; Feng, H.; Shu, W.; Xu, X.; Chen, Y. High throughput capture of circulating tumor cells using an integrated microfluidic system. Biosens. Bioelectron. 2013, 47, 113–119. [Google Scholar] [CrossRef]
- Aghaamoo, M.; Aghilinejad, A.; Chen, X.; Xu, J. On the design of deterministic dielectrophoresis for continuous separation of circulating tumor cells from peripheral blood cells. Electrophoresis 2019, 40, 1486–1493. [Google Scholar] [CrossRef]
- Kong, T.F.; Ye, W.; Peng, W.K.; Hou, H.W.; Preiser, P.R.; Nguyen, N.-T.; Han, J. Enhancing malaria diagnosis through microfluidic cell enrichment and magnetic resonance relaxometry detection. Sci. Rep. 2015, 5, 11425. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Cooper, J.M.; Regnault, C.; Holm, S.H.; Beech, J.P.; Tegenfeldt, J.O.; Hochstetter, A. Microfluidics-Based Approaches to the Isolation of African Trypanosomes. Pathogens 2017, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Holm, S.H. Microfluidic Cell and Particle Sorting Using Deterministic Lateral Displacement; Lund University: Lund, Sweden, 2018. [Google Scholar]
- Clark, D.J.; Moore, C.M.; Flanagan, M.; Van Bocxlaer, K.; Piperaki, E.-T.; Yardley, V.; Croft, S.L.; Tyson, J.; Whitehouse, S.P.; O’Halloran, J.; et al. An efficient and novel technology for the extraction of parasite genomic DNA from whole blood or culture. Biotechniques 2019, 68, 1–6. [Google Scholar] [CrossRef]
- Reboud, J.; Xu, G.; Garrett, A.; Adriko, M.; Yang, Z.; Tukahebwa, E.M.; Rowell, C.; Cooper, J.M. Paper-based microfluidics for DNA diagnostics of malaria in low resource underserved rural communities. Proc. Natl. Acad. Sci. USA 2019, 116, 4834–4842. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.S.; Honrado, C.; Spencer, D.; Horton, B.; Bridle, H.L.; Morgan, H. Analysis of Parasitic Protozoa at the Single-cell Level using Microfluidic Impedance Cytometry. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Shin, Y.; Tan, J.K.S.; Lim, Y.B.; Lim, C.T.; Kim, S. High-throughput malaria parasite separation using a viscoelastic fluid for ultrasensitive PCR detection. Lab Chip 2016, 16, 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Warkiani, M.E.; Tay, A.K.P.; Khoo, B.L.; Xiaofeng, X.; Han, J.; Lim, C.T. Malaria detection using inertial microfluidics. Lab Chip 2015, 15, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Kolluri, N.; Klapperich, C.M.; Cabodi, M. Towards lab-on-a-chip diagnostics for malaria elimination. Lab Chip 2017, 18, 75–94. [Google Scholar] [CrossRef]
- Gong, M.M.; Nosrati, R.; San Gabriel, M.C.; Zini, A.; Sinton, D. Direct DNA Analysis with Paper-Based Ion Concentration Polarization. J. Am. Chem. Soc. 2015, 137, 13913–13919. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, R.; Gong, M.M.; Gabriel, M.C.S.; Pedraza, C.E.; Zini, A.; Sinton, D. Paper-based quantification of male fertility potential. Clin. Chem. 2016, 62, 458–465. [Google Scholar] [CrossRef]
- Coppola, M.A.; Klotz, K.L.; Kim, K.A.; Cho, H.Y.; Kang, J.; Shetty, J.; Howards, S.S.; Flickinger, C.J.; Herr, J.C. SpermCheck® Fertility, an immunodiagnostic home test that detects normozoospermia and severe oligozoospermia. Hum. Reprod. 2010, 25, 853–861. [Google Scholar] [CrossRef]
- Matsuura, K.; Chen, K.H.; Tsai, C.H.; Li, W.; Asano, Y.; Naruse, K.; Cheng, C.M. Paper-based diagnostic devices for evaluating the quality of human sperm. Microfluid. Nanofluid. 2014, 16, 857–867. [Google Scholar] [CrossRef]
- Nosrati, R.; Gong, M.M.; San Gabriel, M.C.; Zini, A.; Sinton, D. Paper-based sperm DNA integrity analysis. Anal. Methods 2016, 8, 6260–6264. [Google Scholar] [CrossRef]
- Birch, C.M.; Hou, H.W.; Han, J.; Niles, J.C. Identification of malaria parasite-infected red blood cell surface aptamers by inertial microfluidic SELEX (I-SELEX). Sci. Rep. 2015, 5, 1–16. [Google Scholar] [CrossRef]
- Song, J.; Liu, C.; Bais, S.; Mauk, M.G.; Bau, H.H.; Greenberg, R.M. Molecular Detection of Schistosome Infections with a Disposable Microfluidic Cassette. PLoS Negl. Trop. Dis. 2015, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.M.; Rusch, S.; Temiz, Y.; Beck, H.P.; Karlen, W.; Delamarche, E. Immuno-gold silver staining assays on capillary-driven microfluidics for the detection of malaria antigens. Biomed. Microdevices 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Beech, J.P.; Ho, B.D.; Garriss, G.; Oliveira, V.; Henriques-Normark, B.; Tegenfeldt, J.O. Separation of pathogenic bacteria by chain length. Anal. Chim. Acta 2018, 1000, 223–231. [Google Scholar] [CrossRef]
- Tottori, N.; Nisisako, T.; Park, J.; Yanagida, Y.; Hatsuzawa, T. Separation of viable and nonviable mammalian cells using a deterministic lateral displacement microfluidic device. Biomicrofluidics 2016, 10, 014125. [Google Scholar] [CrossRef]
- Beeman, A.Q.; Njus, Z.L.; Pandey, S.; Tylka, G.L. Chip technologies for screening chemical and biological agents against plant-parasitic nematodes. Phytopathology 2016, 106, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hou, X.; Wu, X.; Liang, T.; Zhang, X.; Wang, D.; Teng, F.; Dai, J.; Duan, H.; Guo, S.; et al. SARS-CoV-2 proteome microarray for mapping COVID-19 antibody interactions at amino acid resolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jenkins, C.; Orsburn, B. In silico approach to accelerate the development of mass spectrometry-based proteomics methods for detection of viral proteins: Application to COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Al-Motawa, M.; Abbas, H.; Wijten, P.; de la Fuente, A.; Xue, M.; Rabbani, N.; Thornalley, P.J. Vulnerabilities of the SARS-CoV-2 virus to proteotoxicity—Opportunity for repurposed chemotherapy of COVID-19 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K. Dark proteome of Newly Emerged SARS-CoV-2 in Comparison with Human and Bat Coronaviruses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liang, Q.; Li, J.; Guo, M.; Tian, X.; Liu, C.; Wang, X.; Yang, X.; Wu, P.; Xiao, Z.; Qu, Y.; et al. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; O’Meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Hüttenhain, R.; et al. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gong, M.M.; Sinton, D. Turning the Page: Advancing Paper-Based Microfluidics for Broad Diagnostic Application. Chem. Rev. 2017, 117, 8447–8480. [Google Scholar] [CrossRef]
- Roberts, S.F.; Fischhoff, M.A.; Sakowski, S.A.; Feldman, E.L. Perspective: Transforming science into medicine: How clinician-scientists can build bridges across research’s “valley of Death”. Acad. Med. 2012, 87, 266–270. [Google Scholar] [CrossRef]
- Bottazzi, M.; Dumonteil, E.; Valenzuela, J.; Betancourt-Cravioto, M.; Tapia-Conyer, R.; Hotez, P. Bridging the innovation gap for neglected tropical diseases in México: Capacity building for the development of a new generation of antipoverty vaccines. Bol. Med. Hosp. Infant. Mex. 2011, 68, 138–146. [Google Scholar]
- Sarkar, S.; Sabhachandani, P.; Konry, T. Isothermal Amplification Strategies for Detection in Microfluidic Devices. Trends Biotechnol. 2017, 35, 186–189. [Google Scholar] [CrossRef]
- Reece, A.; Xia, B.; Jiang, Z.; Noren, B.; McBride, R.; Oakey, J. Microfluidic techniques for high throughput single cell analysis. Curr. Opin. Biotechnol. 2016, 40, 90–96. [Google Scholar] [CrossRef]
- Esfandyarpour, R.; DiDonato, M.J.; Yang, Y.; Durmus, N.G.; Harris, J.S.; Davis, R.W. Multifunctional, inexpensive, and reusable nanoparticle-printed biochip for cell manipulation and diagnosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1306–E1315. [Google Scholar] [CrossRef]
- Zhang, S.; Tis, T.B.; Wei, Q. Smartphone-Based Clinical Diagnostics. Precision Medicine for Investigators, Practitioners and Providers; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Civin, C.I.; Ward, T.; Skelley, A.M.; Gandhi, K.; Peilun Lee, Z.; Dosier, C.R.; D’Silva, J.L.; Chen, Y.; Kim, M.J.; Moynihan, J.; et al. Automated leukocyte processing by microfluidic deterministic lateral displacement. Cytom. Part A 2016, 89, 1073–1083. [Google Scholar] [CrossRef]
- Pollak, J.J.; Houri-Yafin, A.; Salpeter, S.J. Computer Vision Malaria Diagnostic Systems—Progress and Prospects. Front. Public Health 2017, 5, 1–5. [Google Scholar] [CrossRef]
- Lee, W.G.; Kim, Y.G.; Chung, B.G.; Demirci, U.; Khademhosseini, A. Nano/Microfluidics for diagnosis of infectious diseases in developing countries. Adv. Drug Deliv. Rev. 2010, 62, 449–457. [Google Scholar] [CrossRef]
- Phuakrod, A.; Sripumkhai, W.; Jeamsaksiri, W.; Pattamang, P.; Juntasaro, E.; Thienthong, T.; Foongladda, S.; Brindley, P.J.; Wongkamchai, S. Diagnosis of feline filariasis assisted by a novel semi-automated microfluidic device in combination with high resolution melting real-time PCR. Parasites Vectors 2019, 12, 1–9. [Google Scholar] [CrossRef]
- Saeed, M.A.; Jabbar, A. “Smart Diagnosis” of Parasitic Diseases by Use of Smartphones. J. Clin. Microbiol. 2017, 56, e01469-17. [Google Scholar] [CrossRef]
- Ruiz-Vega, G.; Arias-Alpízar, K.; De Serna, E.; Borgheti-Cardoso, L.N.; Sulleiro, E.; Molina, I.; Fernàndez-Busquets, X.; Sánchez, A.; Campo, F.J.; Baldrich, E. Electrochemical POC device for fast malaria quantitative diagnosis in whole blood by using magnetic beads, poly-HRP and microfluidic paper electrodes. Biosens. Bioelectron. 2019, 150, 111925. [Google Scholar] [CrossRef]
- Choi, J.; Cho, S.J.; Kim, Y.T.; Shin, H. Development of a film-based immunochromatographic microfluidic device for malaria diagnosis. Biomed. Microdevices 2019, 21, 86. [Google Scholar] [CrossRef]
- Mao, R.; Ge, G.; Wang, Z.; Hao, R.; Zhang, G.; Yang, Z.; Lin, B.; Ma, Y.; Liu, H.; Du, Y. A multiplex microfluidic loop-mediated isothermal amplification array for detection of malaria-related parasites and vectors. Acta Trop. 2018, 178, 86–92. [Google Scholar] [CrossRef]
- Sema, M.; Alemu, A.; Bayih, A.G.; Getie, S.; Getnet, G.; Guelig, D.; Burton, R.; LaBarre, P.; Pillai, D.R. Evaluation of non-instrumented nucleic acid amplification by loop-mediated isothermal amplification (NINA-LAMP) for the diagnosis of malaria in Northwest Ethiopia. Malar. J. 2015, 14, 1–9. [Google Scholar] [CrossRef]
- Juul, S.; Nielsen, C.J.F.; Labouriau, R.; Roy, A.; Tesauro, C.; Jensen, P.W.; Harmsen, C.; Kristoffersen, E.L.; Chiu, Y.L.; Frohlich, R.; et al. Droplet microfluidics platform for highly sensitive and quantitative detection of malaria-causing plasmodium parasites based on enzyme activity measurement. ACS Nano 2012, 6, 10676–10683. [Google Scholar] [CrossRef]
- Fraser, L.A.; Kinghorn, A.B.; Dirkzwager, R.M.; Liang, S.; Cheung, Y.W.; Lim, B.; Shiu, S.C.C.; Tang, M.S.L.; Andrew, D.; Manitta, J.; et al. A portable microfluidic Aptamer-Tethered Enzyme Capture (APTEC) biosensor for malaria diagnosis. Biosens. Bioelectron. 2018, 100, 591–596. [Google Scholar] [CrossRef]
- Hochstetter, A. Motility, Manipulation and Controlling of Unicellular Organisms; Universität Basel: Basel, Switzerland, 2014. [Google Scholar]
- Hochstetter, A.; Pfohl, T. Motility, Force Generation, and Energy Consumption of Unicellular Parasites. Trends Parasitol. 2016, 32, 531–541. [Google Scholar] [CrossRef]
- Stellamanns, E.; Uppaluri, S.; Hochstetter, A.; Heddergott, N.; Engstler, M.; Pfohl, T. Optical trapping reveals propulsion forces, power generation and motility efficiency of the unicellular parasites Trypanosoma brucei brucei. Sci. Rep. 2015, 4, 6515. [Google Scholar] [CrossRef]
- Menachery, A.; Kremer, C.; Wong, P.E.; Carlsson, A.; Neale, S.L.; Barrett, M.P.; Cooper, J.M. Counterflow dielectrophoresis for trypanosome enrichment and detection in blood. Sci. Rep. 2012, 2, 1–5. [Google Scholar] [CrossRef]
- Wan, L.; Gao, J.; Chen, T.; Dong, C.; Li, H.; Wen, Y.Z.; Lun, Z.R.; Jia, Y.; Mak, P.I.; Martins, R.P. LampPort: A handheld digital microfluidic device for loop-mediated isothermal amplification (LAMP). Biomed. Microdevices 2019, 21, 9. [Google Scholar] [CrossRef]
- Hamula, C.L.A.; Zhang, H.; Li, F.; Wang, Z.; Chris Le, X.; Li, X.-F. Selection and analytical applications of aptamers binding microbial pathogens. TrAC Trends Anal. Chem. 2011, 30, 1587–1597. [Google Scholar] [CrossRef]
- Bourquin, Y.; Syed, A.; Reboud, J.; Ranford-Cartwright, L.C.; Barrett, M.P.; Cooper, J.M. Rare-cell enrichment by a rapid, label-free, ultrasonic isopycnic technique for medical diagnostics. Angew. Chem. Int. Ed. 2014, 53, 5587–5590. [Google Scholar] [CrossRef]
- Nasseri, B.; Soleimani, N.; Rabiee, N.; Kalbasi, A.; Karimi, M.; Hamblin, M.R. Point-of-care microfluidic devices for pathogen detection. Biosens. Bioelectron. 2018, 117, 112–128. [Google Scholar] [CrossRef]
- Shembekar, N.; Chaipan, C.; Utharala, R.; Merten, C.A. Droplet-based microfluidics in drug discovery, transcriptomics and high-throughput molecular genetics. Lab Chip 2016, 16, 1314–1331. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Y.; Li, X.; Shi, Y.; Hu, B.; An, Y.; Zhu, Z.; Hong, G.; Yang, C.J. Frequency-enhanced transferrin receptor antibody-labelled microfluidic chip (FETAL-Chip) enables efficient enrichment of circulating nucleated red blood cells for non-invasive prenatal diagnosis. Lab Chip 2018, 18, 2749–2756. [Google Scholar] [CrossRef]
- Mauk, M.G.; Song, J.; Liu, C.; Bau, H.H. Simple approaches to minimally-instrumented, microfluidic-based point-of-care Nucleic Acid Amplification Tests. Biosensors 2018, 8, 17. [Google Scholar] [CrossRef]
- Pham, N.M.; Karlen, W.; Beck, H.P.; Delamarche, E. Malaria and the “last” parasite: How can technology help? Malar. J. 2018, 17, 1–16. [Google Scholar] [CrossRef]
- Regnault, C.; Dheeman, D.; Hochstetter, A. Microfluidic Devices for Drug Assays. High-Throughput 2018, 7, 18. [Google Scholar] [CrossRef]
- Hochstetter, A. Presegmentation Procedure Generates Smooth-Sided Microfluidic Devices: Unlocking Multiangle Imaging for Everyone? ACS Omega 2019, 4, 20972–20977. [Google Scholar] [CrossRef] [PubMed]
- Nordenfelt, P.; Cooper, J.M.; Hochstetter, A. Matrix-masking to balance nonuniform illumination in microscopy. Opt. Express 2018, 26, 17279–17288. [Google Scholar] [CrossRef] [PubMed]
- Hochstetter, A.; Stellamanns, E.; Deshpande, S.; Uppaluri, S.; Engstler, M.; Pfohl, T. Microfluidics-based single cell analysis reveals drug-dependent motility changes in trypanosomes. Lab Chip 2015, 15, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.; Antfolk, M.; Brodin, B.; Tenje, M. In Vitro Blood-Brain Barrier Models—An Overview of Established Models and New Microfluidic Approaches. J. Pharm. Sci. 2015, 104, 2727–2746. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Lee, S.-R.; Ko, J.; Son, K.; Tahk, D.; Ahn, J.; Im, C.; Jeon, N.L. A Low Permeability Microfluidic Blood-Brain Barrier Platform with Direct Contact between Perfusable Vascular Network and Astrocytes. Sci. Rep. 2017, 7, 8083. [Google Scholar] [CrossRef] [PubMed]
- Abhyankar, V.V.; Wu, M.; Koh, C.-Y.; Hatch, A.V. A Reversibly Sealed, Easy Access, Modular (SEAM) Microfluidic Architecture to Establish In Vitro Tissue Interfaces. PLoS ONE 2016, 11, e0156341. [Google Scholar] [CrossRef]
- Van Midwoud, P.M.; Janse, A.; Merema, M.T.; Groothuis, G.M.; Verpoorte, E. Comparison of biocompatibility and adsorption properties of different plastics for advanced microfluidic cell and tissue culture models. Anal. Chem. 2012, 84, 3938–3944. [Google Scholar] [CrossRef]
- Herland, A.; Van Der Meer, A.D.; FitzGerald, E.A.; Park, T.E.; Sleeboom, J.J.F.; Ingber, D.E. Distinct contributions of astrocytes and pericytes to neuroinflammation identified in a 3D human blood-brain barrier on a chip. PLoS ONE 2016, 11, e0150360. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2015, 36, 862–890. [Google Scholar] [CrossRef]
- Adriani, G.; Ma, D.; Pavesi, A.; Goh, E.L.K.; Kamm, R.D. Modeling the Blood-Brain Barrier in a 3D Triple Co-Culture Microfluidic System. In Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Milan, Italy, 25–29 August 2015; pp. 338–341. [Google Scholar]
- Wang, Y.I.; Abaci, H.E.; Shuler, M.L. Microfluidic blood-brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng. 2017, 114, 184–194. [Google Scholar] [CrossRef]
- Adriani, G.; Ma, D.; Pavesi, A.; Kamm, R.D.; Goh, E.L.K. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood-brain barrier. Lab Chip 2017, 17, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-C.; Huang, S.-B.; Wu, P.-C.; Shieh, D.-B.; Lee, G.-B. A microfluidic cell culture platform for real-time cellular imaging. Biomed. Microdevices 2009, 11, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Park, J.; Shin, C.; Do, Y.; Cho, Y.-K. Three dimensional multicellular co-cultures and anti-cancer drug assays in rapid prototyped multilevel microfluidic devices. Biomed. Microdevices 2013, 15, 627–634. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, Q.; Nie, J.; Fu, J.; He, Y. From Microfluidic Paper-Based Analytical Devices to Paper-Based Biofluidics with Integrated Continuous Perfusion. ACS Biomater. Sci. Eng. 2017, 3, 601–607. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Q.; Liu, W.; He, Z.; Lin, J.M. Recent advances in microfluidic 3D cellular scaffolds for drug assays. TrAC Trends Anal. Chem. 2017, 87, 19–31. [Google Scholar] [CrossRef]
- Hung, P.J.; Lee, P.J.; Sabounchi, P.; Lin, R.; Lee, L.P. Continuous perfusion microfluidic cell culture array for high-throughput cell-based assays. Biotechnol. Bioeng. 2005, 89, 1–8. [Google Scholar] [CrossRef]
- Zhang, B.; Green, J.V.; Murthy, S.K.; Radisic, M. Label-free enrichment of functional cardiomyocytes using microfluidic deterministic lateral flow displacement. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Shao, J.; Wu, L.; Wu, J.; Zheng, Y.; Zhao, H.; Lou, X.; Jin, Q.; Zhao, J. A microfluidic chip for permeability assays of endothelial monolayer. Biomed. Microdevices 2010, 12, 81–88. [Google Scholar] [CrossRef]
- Xu, Z.; Gao, Y.; Hao, Y.; Li, E.; Wang, Y.; Zhang, J.; Wang, W.; Gao, Z.; Wang, Q. Application of a microfluidic chip-based 3D co-culture to test drug sensitivity for individualized treatment of lung cancer. Biomaterials 2013, 34, 4109–4117. [Google Scholar] [CrossRef]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. USA 2009, 106, 14195–14200. [Google Scholar] [CrossRef]
- Frenz, L.; Blank, K.; Brouzes, E.; Griffiths, A.D. Reliable microfluidic on-chip incubation of droplets in delay-lines. Lab Chip 2009, 9, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Adekanmbi, E.O.; Srivastava, S.K. Dielectrophoretic applications for disease diagnostics using lab-on-a-chip platforms. Lab Chip 2016, 16, 2148–2167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, P.; Huang, P.-H.; Xie, Y.; Mai, J.D.; Wang, L.; Nguyen, N.-T.; Huang, T.J. Rare cell isolation and analysis in microfluidics. Lab Chip 2014, 14, 626. [Google Scholar] [CrossRef] [PubMed]
- Beech, J.P.; Jönsson, P.; Tegenfeldt, J.O. Tipping the balance of deterministic lateral displacement devices using dielectrophoresis. Lab Chip 2009, 9, 2698–2706. [Google Scholar] [CrossRef] [PubMed]
- Beech, J.P.; Keim, K.; Ho, B.D.; Guiducci, C.; Tegenfeldt, J.O. Active Posts in Deterministic Lateral Displacement Devices. Adv. Mater. Technol. 2019. [Google Scholar] [CrossRef]
- Calero, V.; Garcia-Sanchez, P.; Honrado, C.; Ramos, A.; Morgan, H. AC electrokinetic biased deterministic lateral displacement for tunable particle separation. Lab Chip 2019, 19, 1386–1396. [Google Scholar] [CrossRef]
- Smith, A.J.; O’Rorke, R.D.; Kale, A.; Rimsa, R.; Tomlinson, M.J.; Kirkham, J.; Davies, A.G.; Wälti, C.; Wood, C.D. Rapid cell separation with minimal manipulation for autologous cell therapies. Sci. Rep. 2017, 7, 41872. [Google Scholar] [CrossRef]
- Sharifi Noghabi, H.; Soo, M.; Khamenehfar, A.; Li, P.C.H. Dielectrophoretic trapping of single leukemic cells using the conventional and compact optical measurement systems. Electrophoresis 2019, 40, 1478–1485. [Google Scholar] [CrossRef]
- Faraghat, S.A.; Hoettges, K.F.; Steinbach, M.K.; van der Veen, D.R.; Brackenbury, W.J.; Henslee, E.A.; Labeed, F.H.; Hughes, M.P. High-throughput, low-loss, low-cost, and label-free cell separation using electrophysiology-activated cell enrichment. Proc. Natl. Acad. Sci. USA 2017, 114, 4591–4596. [Google Scholar] [CrossRef]
- Huang, L.R.; Cox, E.C.; Austin, R.H.; Sturm, J.C. Continuous particle separation through deterministic lateral displacement. Science 2004, 304, 987–990. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, F.; Du, J.; Shu, W.; Feng, H.; Xu, X.; Chen, Y. Rapid isolation of cancer cells using microfluidic deterministic lateral displacement structure. Biomicrofluidics 2013, 7, 11801. [Google Scholar] [CrossRef] [PubMed]
- Loutherback, K.; D’Silva, J.; Liu, L.; Wu, A.; Austin, R.H.; Sturm, J.C. Deterministic separation of cancer cells from blood at 10 mL/min. AIP Adv. 2012, 2, 42107. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.A.; Inglis, D.W.; Morton, K.J.; Lawrence, D.A.; Huang, L.R.; Chou, S.Y.; Sturm, J.C.; Austin, R.H. Deterministic hydrodynamics: Taking blood apart. Proc. Natl. Acad. Sci. USA 2006, 103, 14779–14784. [Google Scholar] [CrossRef] [PubMed]
- Krüger, T.; Holmes, D.; Coveney, P.V. Deformability-based red blood cell separation in deterministic lateral displacement devices—A simulation study. Biomicrofluidics 2014, 8, 054114. [Google Scholar] [CrossRef]
- Joensson, H.N.; Uhlén, M.; Svahn, H.A. Droplet size based separation by deterministic lateral displacement- separating droplets by cell-induced shrinking. Lab Chip 2011, 11, 1305–1310. [Google Scholar] [CrossRef]
- Inglis, D.W.; Herman, N.; Vesey, G. Highly accurate deterministic lateral displacement device and its application to purification of fungal spores. Biomicrofluidics 2010, 4. [Google Scholar] [CrossRef]
- Zeming, K.K.; Salafi, T.; Chen, C.H.; Zhang, Y. Asymmetrical Deterministic Lateral Displacement Gaps for Dual Functions of Enhanced Separation and Throughput of Red Blood Cells. Sci. Rep. 2016, 6, 22934. [Google Scholar] [CrossRef]
- Wunsch, B.H.; Smith, J.T.; Gifford, S.M.; Wang, C.; Brink, M.; Bruce, R.L.; Austin, R.H.; Stolovitzky, G.; Astier, Y. Nanoscale lateral displacement arrays for the separation of exosomes and colloids down to 20 nm. Nat. Nanotechnol. 2016, 11, 936–940. [Google Scholar] [CrossRef]
- Hou, S.; Chen, J.-F.; Song, M.; Zhu, Y.; Jan, Y.J.; Chen, S.H.; Weng, T.-H.; Ling, D.-A.; Chen, S.-F.; Ro, T.; et al. Imprinted NanoVelcro Microchips for Isolation and Characterization of Circulating Fetal Trophoblasts: Toward Noninvasive Prenatal Diagnostics. ACS Nano 2017, 11, 8167–8177. [Google Scholar] [CrossRef]
- Pariset, E.; Pudda, C.; Boizot, F.; Verplanck, N.; Berthier, J.; Thuaire, A.; Agache, V. Anticipating Cutoff Diameters in Deterministic Lateral Displacement (DLD) Microfluidic Devices for an Optimized Particle Separation. Small 2017, 13. [Google Scholar] [CrossRef]
- Vernekar, R.; Krüger, T. Breakdown of deterministic lateral displacement efficiency for non-dilute suspensions: A numerical study. Med. Eng. Phys. 2015, 37, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Holm, S.H.; Zhang, Z.; Beech, J.P.; Gompper, G.; Fedosov, D.A.; Tegenfeldt, J.O. Microfluidic Particle Sorting in Concentrated Erythrocyte Suspensions. Phys. Rev. Appl. 2019, 12, 014051. [Google Scholar] [CrossRef]
- Alizadehrad, D.; Krüger, T.; Engstler, M.; Stark, H. Simulating the Complex Cell Design of Trypanosoma brucei and Its Motility. PLoS Comput. Biol. 2015, 11, e1003967. [Google Scholar] [CrossRef] [PubMed]
- D’Avino, G. Non-Newtonian deterministic lateral displacement separator: Theory and simulations. Rheol. Acta 2013, 52, 221–236. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, J.; Yuan, D.; Li, W. Hybrid microfluidics combined with active and passive approaches for continuous cell separation. Electrophoresis 2017, 38, 238–249. [Google Scholar] [CrossRef]
- Zhang, Z.; Chien, W.; Henry, E.; Fedosov, D.A.; Gompper, G. Sharp-edged geometric obstacles in microfluidics promote deformability-based sorting of cells. Phys. Rev. Fluids 2019, 4, 1–18. [Google Scholar] [CrossRef]
- Imai, Y.; Kondo, H.; Ishikawa, T.; Teck Lim, C.; Yamaguchi, T. Modeling of hemodynamics arising from malaria infection. J. Biomech. 2010, 43, 1386–1393. [Google Scholar] [CrossRef]
- Guo, Q.; Duffy, S.P.; Matthews, K.; Deng, X.; Santoso, A.T.; Islamzada, E.; Ma, H. Deformability based sorting of red blood cells improves diagnostic sensitivity for malaria caused by Plasmodium falciparum. Lab Chip 2016, 16, 645–654. [Google Scholar] [CrossRef]
- Park, E.S.; Jin, C.; Guo, Q.; Ang, R.R.; Duffy, S.P.; Matthews, K.; Azad, A.; Abdi, H.; Todenhöfer, T.; Bazov, J.; et al. Continuous Flow Deformability-Based Separation of Circulating Tumor Cells Using Microfluidic Ratchets. Small 2016, 12, 1909–1919. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z.; Liu, H.; Zhang, Z.; Lin, C.; Wang, B. Hybrid magnetic and deformability based isolation of circulating tumor cells using microfluidics. AIP Adv. 2019, 9. [Google Scholar] [CrossRef]
- Chen, H. A Triplet Parallelizing Spiral Microfluidic Chip for Continuous Separation of Tumor Cells. Sci. Rep. 2018, 8, 4042. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Yeh, L.-H.; Li, F.-C.; Mauroy, B.; Joo, S. Deformability-Based Electrokinetic Particle Separation. Micromachines 2016, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.W.; Bhagat, A.A.S.; Lin Chong, A.G.; Mao, P.; Wei Tan, K.S.; Han, J.; Lim, C.T. Deformability based cell margination—A simple microfluidic design for malaria-infected erythrocyte separation. Lab Chip 2010, 10, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Nakaaki, K.; Kondo, H.; Ishikawa, T.; Teck Lim, C.; Yamaguchi, T. Margination of red blood cells infected by Plasmodium falciparum in a microvessel. J. Biomech. 2011, 44, 1553–1558. [Google Scholar] [CrossRef]
- Xiang, N.; Wang, J.; Li, Q.; Han, Y.; Huang, D.; Ni, Z. Precise Size-Based Cell Separation via the Coupling of Inertial Microfluidics and Deterministic Lateral Displacement. Anal. Chem. 2019, 91, 10328–10334. [Google Scholar] [CrossRef]
- Morsi, S.A.; Alexander, A.J. An investigation of particle trajectories in two-phase flow systems. J. Fluid Mech. 1972, 55, 193–208. [Google Scholar] [CrossRef]
- Ho, B.P.; Leal, L.G. Inertial migration of rigid spheres in two-dimensional unidirectional flows. J. Fluid Mech. 1974, 65, 365–400. [Google Scholar] [CrossRef]
- Liu, C.; Xue, C.; Sun, J.; Hu, G. A generalized formula for inertial lift on a sphere in microchannels. Lab Chip 2016, 16, 884–892. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, D.; Zhao, Q.; Teo, A.J.T.; Yan, S.; Ooi, C.H.; Li, W.; Nguyen, N.-T. Fundamentals of Differential Particle Inertial Focusing in Symmetric Sinusoidal Microchannels. Anal. Chem. 2019, 91, 4077–4084. [Google Scholar] [CrossRef]
- Sun, J.; Li, M.; Liu, C.; Zhang, Y.; Liu, D.; Liu, W.; Hu, G.; Jiang, X. Double spiral microchannel for label-free tumor cell separation and enrichment. Lab Chip 2012, 12, 3952–3960. [Google Scholar] [CrossRef]
- Bhagat, A.A.S.; Kuntaegowdanahalli, S.S.; Papautsky, I. Continuous particle separation in spiral microchannels using dean flows and differential migration. Lab Chip 2008, 8, 1906–1914. [Google Scholar] [CrossRef]
- Kemna, E.W.M.; Schoeman, R.M.; Wolbers, F.; Vermes, I.; Weitz, D.A.; Van Den Berg, A. High-yield cell ordering and deterministic cell-in-droplet encapsulation using Dean flow in a curved microchannel. Lab Chip 2012, 12, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Ookawara, S.; Street, D.; Ogawa, K. Numerical study on development of particle concentration profiles in a curved microchannel. Chem. Eng. Sci. 2006, 61, 3714–3724. [Google Scholar] [CrossRef]
- Zuvin, M.; Mansur, N.; Birol, S.Z.; Trabzon, L.; Sayı Yazgan, A. Human breast cancer cell enrichment by Dean flow driven microfluidic channels. Microsyst. Technol. 2016, 22, 645–652. [Google Scholar] [CrossRef]
- Zhou, J.; Giridhar, P.V.; Kasper, S.; Papautsky, I. Modulation of aspect ratio for complete separation in an inertial microfluidic channel. Lab Chip 2013, 13, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Mach, A.J.; di Carlo, D. Continuous scalable blood filtration device using inertial microfluidics. Biotechnol. Bioeng. 2010, 107, 302–311. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, Y.; Wang, M.; Chung, A.J. Continuous inertial microparticle and blood cell separation in straight channels with local microstructures. Lab Chip 2016, 16, 532–542. [Google Scholar] [CrossRef]
- Shi, J.; Huang, H.; Stratton, Z.; Huang, Y.; Huang, T.J. Continuous particle separation in a microfluidic channel via standing surface acoustic waves (SSAW). Lab Chip 2009, 9, 3354–3359. [Google Scholar] [CrossRef]
- Destgeer, G.; Lee, K.H.; Jung, J.H.; Alazzam, A.; Sung, H.J. Continuous separation of particles in a PDMS microfluidic channel via travelling surface acoustic waves (TSAW). Lab Chip 2013, 13, 4210–4216. [Google Scholar] [CrossRef]
- Wiklund, M.; Green, R.; Ohlin, M. Acoustofluidics 14: Applications of acoustic streaming in microfluidic devices. Lab Chip 2012, 12, 2438–2451. [Google Scholar] [CrossRef]
- Patel, M.V.; Tovar, A.R.; Lee, A.P. Lateral cavity acoustic transducer as an on-chip cell/particle microfluidic switch. Lab Chip 2012, 12, 139–145. [Google Scholar] [CrossRef]
- Tovar, A.R.; Patel, M.V.; Lee, A.P. Lateral air cavities for microfluidic pumping with the use of acoustic energy. Microfluid. Nanofluid. 2011, 10, 1269–1278. [Google Scholar] [CrossRef]
- Xu, Y.; Hashmi, A.; Yu, G.; Lu, X.; Kwon, H.J.; Chen, X.; Xu, J. Microbubble array for on-chip worm processing. Appl. Phys. Lett. 2013, 102. [Google Scholar] [CrossRef]
- Ozcelik, A.; Rufo, J.; Guo, F.; Gu, Y.; Li, P.; Lata, J.; Huang, T.J. Acoustic tweezers for the life sciences. Nat. Methods 2018, 15, 1021–1028. [Google Scholar] [CrossRef]
- Ashkin, A.; Dziedzic, J.M. Optical Levitation by Radiation Pressure. Appl. Phys. Lett. 1971, 19, 283. [Google Scholar] [CrossRef]
- Flynn, R.A.; Shao, B.; Chachisvilis, M.; Ozkan, M.; Esener, S.C. Two-beam optical traps: Refractive index and size measurements of microscale objects. Biomed. Microdevices 2005, 7, 93–97. [Google Scholar] [CrossRef]
- Berg-Sørensen, K.; Marie, R.; Dziegiel, M.H.; Nielsen, K.S.; Rungling, T.B. Deformation of Single Cells—Optical Two-Beam Traps and More. In International Society for Optics and Photonics, Proceedings of the Complex Light and Optical Forces XIII, San Francisco, CA, USA, 2–7 February 2019; Andrews, D.L., Galvez, E.J., Glückstad, J., Eds.; SPIE: Bellingham, WA, USA, 2019; Volume 10935, p. 39. [Google Scholar]
- Zhang, H.; Liu, K.-K. Optical tweezers for single cells. J. R. Soc. Interface 2008, 5, 671–690. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, M. Investigation of inclined dual-fiber optical tweezers for 3D manipulation and force sensing. Opt. Express 2009, 17, 13624–13638. [Google Scholar] [CrossRef]
- Grier, D.G. A revolution in optical manipulation. Nature 2003, 424, 810–816. [Google Scholar] [CrossRef]
- Padgett, M.; Di Leonardo, R. Holographic optical tweezers and their relevance to lab on chip devices. Lab Chip 2011, 11, 1196–1205. [Google Scholar] [CrossRef]
- Santucci, S.C.; Cojoc, D.; Amenitsch, H.; Marmiroli, B.; Sartori, B.; Burghammer, M.; Schoeder, S.; DiCola, E.; Reynolds, M.; Riekel, C. Optical tweezers for synchrotron radiation probing of trapped biological and soft matter objects in aqueous environments. Anal. Chem. 2011, 83, 4863–4870. [Google Scholar] [CrossRef] [PubMed]
- Schonbrun, E.; Piestun, R.; Jordan, P.; Cooper, J.; Wulff, K.D.; Courtial, J.; Padgett, M. 3D interferometric optical tweezers using a single spatial light modulator. Opt. Express 2005, 13, 3777. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, G.; Jordan, P.; Courtial, J.; Padgett, M.; Cooper, J.; Laczik, Z. Assembly of 3-dimensional structures using programmable holographic optical tweezers. Opt. Express 2004, 12, 5475–5480. [Google Scholar] [CrossRef]
- Leach, J.; Sinclair, G.; Jordan, P.; Courtial, J.; Padgett, M.; Cooper, J.; Laczik, Z. 3D manipulation of particles into crystal structures using holographic optical tweezers. Opt. Express 2004, 12, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Preece, D.; Keen, S.; Botvinick, E.; Bowman, R.; Padgett, M.; Leach, J. Independent polarisation control of multiple optical traps. Opt. Express 2008, 16, 15897. [Google Scholar] [CrossRef]
- Hesseling, C.; Woerdemann, M.; Hermerschmidt, A.; Denz, C. Controlling ghost traps in holographic optical tweezers. Opt. Lett. 2011, 36, 3657–3659. [Google Scholar] [CrossRef]
- Dalal, A.; Chowdhury, A.; Dasgupta, R.; Majumder, S.K. Improved generation of periodic optical trap arrays using noniterative algorithm. Opt. Eng. 2017, 56. [Google Scholar] [CrossRef]
- Tang, X.; Nan, F.; Han, F.; Yan, Z. Simultaneously shaping the intensity and phase of light for optical nanomanipulation. arXiv 2019, arXiv:1910.08244. [Google Scholar]
- Perozziello, G.; Candeloro, P.; Laura Coluccio, M.; Di Fabrizio, E. Optofluidics for handling and analysis of single living cells. Nanofluid 2017, 4, 18–23. [Google Scholar] [CrossRef][Green Version]
- Jorgolli, M.; Nevill, T.; Winters, A.; Chen, I.; Chong, S.; Lin, F.F.; Mock, M.; Chen, C.; Le, K.; Tan, C.; et al. Nanoscale integration of single cell biologics discovery processes using optofluidic manipulation and monitoring. Biotechnol. Bioeng. 2019, 116, 2393–2411. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.; Huang, H.; Sun, D. A switching controller for high speed cell transportation by using a robot-aided optical tweezers system. Automatica 2018, 89, 308–315. [Google Scholar] [CrossRef]
- Xie, M.; Shakoor, A.; Wu, C. Manipulation of Biological Cells Using a Robot-Aided Optical Tweezers System. Micromachines 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Carey, T.R.; Cotner, K.L.; Li, B.; Sohn, L.L. Developments in label-free microfluidic methods for single-cell analysis and sorting. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1529. [Google Scholar] [CrossRef]
- Wang, M.M.; Tu, E.; Raymond, D.E.; Yang, J.M.; Zhang, H.; Hagen, N.; Dees, B.; Mercer, E.M.; Forster, A.H.; Kariv, I.; et al. Microfluidic sorting of mammalian cells by optical force switching. Nat. Biotechnol. 2005, 23, 83–87. [Google Scholar] [CrossRef]
- Sajeesh, P.; Sen, A.K. Particle separation and sorting in microfluidic devices: A review. Microfluid. Nanofluidics 2014, 17, 1–52. [Google Scholar] [CrossRef]
- Cao, B.; Kelbauskas, L.; Chan, S.; Shetty, R.M.; Smith, D.; Meldrum, D.R. Rotation of single live mammalian cells using dynamic holographic optical tweezers. Opt. Lasers Eng. 2017, 92, 70–75. [Google Scholar]
- Keloth, A.; Anderson, O.; Risbridger, D.; Paterson, L. Single cell isolation using optical tweezers. Micromachines 2018, 9, 434. [Google Scholar]
- Rappaz, B.; Marquet, P.; Cuche, E.; Emery, Y.; Depeursinge, C.; Magistretti, P.J. Measurement of the integral refractive index and dynamic cell morphometry of living cells with digital holographic microscopy. Opt. Express 2005, 13, 9361. [Google Scholar] [CrossRef]
- Curl, C.L.; Bellair, C.J.; Harris, T.; Allman, B.E.; Harris, P.J.; Stewart, A.G.; Roberts, A.; Nugent, K.A.; Delbridge, L.M.D. Refractive index measurement in viable cells using quantitative phase-amplitude microscopy and confocal microscopy. Cytom. Part A 2005, 65, 88–92. [Google Scholar] [CrossRef]
- Kaminski, T.S.; Garstecki, P. Controlled droplet microfluidic systems for multistep chemical and biological assays. Chem. Soc. Rev. 2017, 46, 6210–6226. [Google Scholar] [CrossRef]
- Liu, C.; Xu, X.; Li, B.; Situ, B.; Pan, W.; Hu, Y.; An, T.; Yao, S.; Zheng, L. Single-Exosome-Counting Immunoassays for Cancer Diagnostics. Nano Lett. 2018, 18, 4226–4232. [Google Scholar] [CrossRef] [PubMed]
- Fornell, A.; Cushing, K.; Nilsson, J.; Tenje, M. Binary particle separation in droplet microfluidics using acoustophoresis. Appl. Phys. Lett. 2018, 112. [Google Scholar] [CrossRef]
- Huh, D.; Bahng, J.H.; Ling, Y.; Wei, H.-H.; Kripfgans, O.D.; Fowlkes, J.B.; Grotberg, J.B.; Takayama, S. Gravity-Driven Microfluidic Particle Sorting Device with Hydrodynamic Separation Amplification. Anal. Chem. 2007, 79, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Seemann, R.; Brinkmann, M.; Pfohl, T.; Herminghaus, S. Droplet based microfluidics. Rep. Prog. Phys. 2012, 75, 016601. [Google Scholar] [CrossRef]
- Chabert, M.; Viovy, J.-L. Microfluidic high-throughput encapsulation and hydrodynamic self-sorting of single cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3191–3196. [Google Scholar] [CrossRef]
- Tan, Y.C.; Ho, Y.L.; Lee, A.P. Droplet coalescence by geometrically mediated flow in microfluidic channels. Microfluid. Nanofluid. 2007, 3, 495–499. [Google Scholar] [CrossRef]
- Fidalgo, L.M.; Whyte, G.; Bratton, D.; Kaminski, C.F.; Abell, C.; Huck, W.T.S. From Microdroplets to Microfluidics: Selective Emulsion Separation in Microfluidic Devices. Angew. Chem. Int. Ed. 2008, 47, 2042–2045. [Google Scholar] [CrossRef]
- Ding, R.; Lloyd Ung, W.; Heyman, J.A.; Weitz, D.A. Sensitive and predictable separation of microfluidic droplets by size using in-line passive filter. Biomicrofluidics 2017, 11, 014114. [Google Scholar] [CrossRef]
- Kim, S.; Ganapathysubramanian, B.; Anand, R.K. Concentration Enrichment, Separation, and Cation Exchange in Nanoliter-Scale Water-in-Oil Droplets. J. Am. Chem. Soc. 2020, 142, 3196–3204. [Google Scholar] [CrossRef]
- Huang, C.; Han, S.I.; Han, A. In-Droplet Cell Separation Based on Different Dielectrophoretic Response. In Proceedings of the 2019 20th International Conference on Solid-State Sensors, Actuators and Microsystems and Eurosensors XXXIII, Transducers 2019 and Eurosensors XXXIII, Berlin, Germany, 23–27 June 2019; pp. 492–495. [Google Scholar]
- Wang, Y.; Zhao, Y.; Cho, S.K. Efficient in-droplet separation of magnetic particles for digital microfluidics. J. Micromech. Microeng. 2007, 17, 2148–2156. [Google Scholar] [CrossRef]
- Brouzes, E.; Kruse, T.; Kimmerling, R.; Strey, H.H. Rapid and continuous magnetic separation in droplet microfluidic devices. Lab Chip 2015, 15, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sung, K.-J.; Lin, X.N.; Burns, M.A. Bead mediated separation of microparticles in droplets. PLoS ONE 2017, 12, e0173479. [Google Scholar] [CrossRef] [PubMed]
- Sista, R.S.; Ng, R.; Nuffer, M.; Basmajian, M.; Coyne, J.; Elderbroom, J.; Hull, D.; Kay, K.; Krishnamurthy, M.; Roberts, C.; et al. Digital Microfluidic Platform to Maximize Diagnostic Tests with Low Sample Volumes from Newborns and Pediatric Patients. Diagnostics 2020, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.J.; Liu, A.Q.; Lim, C.S.; Ayi, T.C.; Yap, P.H. Determining refractive index of single living cell using an integrated microchip. Sens. Actuators A Phys. 2007, 133, 349–354. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chin, L.K.; Ser, W.; Ayi, T.C.; Yap, P.H.; Bourouina, T.; Leprince-Wang, Y. An optofluidic imaging system to measure the biophysical signature of single waterborne bacteria. Lab Chip 2014, 14, 4237–4243. [Google Scholar] [CrossRef] [PubMed]
- Song, W.Z.; Zhang, X.M.; Liu, A.Q.; Lim, C.S.; Yap, P.H.; Hosseini, H.M.M. Refractive index measurement of single living cells using on-chip Fabry-Ṕrot cavity. Appl. Phys. Lett. 2006, 89, 15–18. [Google Scholar] [CrossRef]
- Song, W.Z.; Liu, A.Q.; Swaminathan, S.; Lim, C.S.; Yap, P.H.; Ayi, T.C. Determination of single living cell’s dry/water mass using optofluidic chip. Appl. Phys. Lett. 2007, 91, 1–4. [Google Scholar] [CrossRef]
- Chin, L.K.; Liu, A.Q.; Lim, C.S.; Zhang, X.M.; Ng, J.H.; Hao, J.Z.; Takahashi, S. Differential single living cell refractometry using grating resonant cavity with optical trap. Appl. Phys. Lett. 2007, 91, 1–4. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chin, L.K.; Ser, W.; Chen, H.F.; Hsieh, C.M.; Lee, C.H.; Sung, K.B.; Ayi, T.C.; Yap, P.H.; Liedberg, B.; et al. Cell refractive index for cell biology and disease diagnosis: Past, present and future. Lab Chip 2016, 16, 634–644. [Google Scholar] [CrossRef]
- Prevedel, R.; Diz-Muñoz, A.; Ruocco, G.; Antonacci, G. Brillouin microscopy—A revolutionary tool for mechanobiology? arXiv 2019, arXiv:1901.02006. [Google Scholar]
- Bateman, J.B.; Wagman, J.; Carstensen, E.L. Refraction and absorption of light in bacterial suspensions. Kolloid-Zeitschrift Zeitschrift für Polymere 1966, 208, 44–58. [Google Scholar] [CrossRef]
- Balaev, A.E.; Dvoretski, K.N.; Doubrovski, V.A. Refractive Index of Escherichia Coli Cells. In Proceedings of the SPIE 4707, Saratov Fall Meeting 2001: Optical Technologies in Biophysics and Medicine III, Saratov, Russia, 16 July 2002. [Google Scholar]
- Tuminello, P.S.; Arakawa, E.T.; Khare, B.N.; Wrobel, J.M.; Querry, M.R.; Milham, M.E. Optical properties of Bacillus subtilis spores from 02 to 25 µm. Appl. Opt. 1997, 36, 2818. [Google Scholar] [CrossRef]
- Joseph, S. Refractometry of fungi. J. Microsc. 1983, 131, 163–172. [Google Scholar] [CrossRef]
- Hsu, W.-C.; Su, J.-W.; Tseng, T.-Y.; Sung, K.-B. Tomographic diffractive microscopy of living cells based on a common-path configuration. Opt. Lett. 2014, 39, 2210. [Google Scholar] [CrossRef]
- Charrière, F.; Marian, A.; Montfort, F.; Kuehn, J.; Colomb, T.; Cuche, E.; Marquet, P.; Depeursinge, C. Cell refractive index tomography by digital holographic microscopy. Opt. Lett. 2006, 31, 178. [Google Scholar] [CrossRef] [PubMed]
- Lue, N.; Popescu, G.; Ikeda, T.; Badizadegan, K.; Dasari, R.R.; Feld, M.S. Live cell refractometry using hilbert phase microscopy. Opt. InfoBase Conf. Pap. 2006, 113, 13327–13330. [Google Scholar]
- Sung, Y.; Lue, N.; Hamza, B.; Martel, J.; Irimia, D.; Dasari, R.R.; Choi, W.; Yaqoob, Z.; So, P. Three-dimensional holographic refractive-index measurement of continuously flowing cells in a microfluidic channel. Phys. Rev. Appl. 2014, 1, 1–8. [Google Scholar]
- Su, J.W.; Hsu, W.C.; Chou, C.Y.; Chang, C.H.; Sung, K. Bin Digital holographic microtomography for high-resolution refractive index mapping of live cells. J. Biophotonics 2013, 6, 416–424. [Google Scholar] [CrossRef]
- Chu, Y.C.; Chang, W.Y.; Chen, K.H.; Chen, J.H.; Tsai, B.C.; Hsu, K.Y. Full-field refractive index measurement with simultaneous phase-shift interferometry. Optik 2014, 125, 3307–3310. [Google Scholar] [CrossRef]
- Moh, K.J.; Yuan, X.-C.; Bu, J.; Zhu, S.W.; Gao, B.Z. Surface plasmon resonance imaging of cell-substrate contacts with radially polarized beams. Opt. Express 2008, 16, 20734. [Google Scholar] [CrossRef][Green Version]
- Lee, J.Y.; Lee, C.W.; Lin, E.H.; Wei, P.K. Single live cell refractometer using nanoparticle coated fiber tip. Appl. Phys. Lett. 2008, 93, 173110–173113. [Google Scholar] [CrossRef]
- Phillips, K.G.; Jacques, S.L.; McCarty, O.J.T. Measurement of Single Cell Refractive Index, Dry Mass, Volume, and Density Using a Transillumination Microscope. Phys. Rev. Lett. 2012, 109, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Phillips, S.T.; Butte, M.J.; Whitesides, G.M. Patterned Paper as a Platform for Inexpensive, Low-Volume, Portable Bioassays. Angew. Chem. Int. Ed. 2007, 46, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Nilghaz, A.; Guan, L.; Tan, W.; Shen, W. Advances of Paper-Based Microfluidics for Diagnostics—The Original Motivation and Current Status. ACS Sens. 2016, 1, 1382–1393. [Google Scholar] [CrossRef]
- He, Y.; Wu, Y.; Fu, J.-Z.; Wu, W.-B. Fabrication of paper-based microfluidic analysis devices: A review. RSC Adv. 2015, 5, 78109–78127. [Google Scholar] [CrossRef]
- Martinez, A.W.; Phillips, S.T.; Carrilho, E.; Thomas, S.W.; Sindi, H.; Whitesides, G.M. Simple Telemedicine for Developing Regions: Camera Phones and Paper-Based Microfluidic Devices for Real-Time, Off-Site Diagnosis. Anal. Chem. 2008, 80, 3699–3707. [Google Scholar] [CrossRef]
- Lisowski, P.; Zarzycki, P.K. Microfluidic paper-based analytical devices (μPADs) and micro total analysis systems (μTAS): Development, applications and future trends. Chromatographia 2013, 76, 1201–1214. [Google Scholar] [CrossRef]
- Su, W.; Gao, X.; Jiang, L.; Qin, J. Microfluidic platform towards point-of-care diagnostics in infectious diseases. J. Chromatogr. A 2015, 1377, 13–26. [Google Scholar] [CrossRef]
- Mu, X.; Zhang, L.; Chang, S.; Cui, W.; Zheng, Z. Multiplex microfluidic paper-based immunoassay for the diagnosis of hepatitis C virus infection. Anal. Chem. 2014, 86, 5338–5344. [Google Scholar] [CrossRef]
- Thom, N.K.; Yeung, K.; Pillion, M.B.; Phillips, S.T. “Fluidic batteries” as low-cost sources of power in paper-based microfluidic devices. Lab Chip 2012, 12, 1768–1770. [Google Scholar] [CrossRef]
- Sánchez-Ovejero, C.; Benito-Lopez, F.; Díez, P.; Casulli, A.; Siles-Lucas, M.; Fuentes, M.; Manzano-Román, R. Sensing parasites: Proteomic and advanced bio-detection alternatives. J. Proteomics 2016, 136, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Zole, E.; Ranka, R. Mitochondria, its DNA and telomeres in ageing and human population. Biogerontology 2018, 19, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.Y.; Chiang, Y.C.; Hsu, C.H.; Chen, J.J.; Hsu, C.C.; Chao, A.C.; Lin, Y.S. Peanut detection using droplet microfluidic polymerase chain reaction device. J. Sens. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Gascoyne, P.; Satayavivad, J.; Ruchirawat, M. Microfluidic approaches to malaria detection. Acta Trop. 2004, 89, 357–369. [Google Scholar] [CrossRef]
- Ryan, U.; Paparini, A.; Oskam, C. New Technologies for Detection of Enteric Parasites. Trends Parasitol. 2017, 33, 532–546. [Google Scholar] [CrossRef]
- Ahrberg, C.D.; Manz, A.; Chung, B.G. Polymerase chain reaction in microfluidic devices. Lab Chip 2016, 16, 3866–3884. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef]
- Haring, R.; Wallaschofski, H. Diving through the “-omics”: The case for deep phenotyping and systems epidemiology. OMICS 2012, 16, 231–234. [Google Scholar] [CrossRef]
- Nassar, S.; Raddassi, K.; Wu, T.; Wisnewski, A. Single-Cell Metabolomics by Mass Spectrometry for Drug Discovery: Moving Forward. Drug Des. Open Access 2017, 6, 1000149. [Google Scholar]
- Wang, D.; Bodovitz, S. Single cell analysis: The new frontier in “omics”. Trends Biotechnol. 2010, 28, 281–290. [Google Scholar] [CrossRef]
- Ogbaga, C.C.; Stepien, P.; Dyson, B.C.; Rattray, N.J.W.; Ellis, D.I.; Goodacre, R.; Johnson, G.N. Biochemical Analyses of Sorghum Varieties Reveal Differential Responses to Drought. PLoS ONE 2016. [Google Scholar] [CrossRef]
- Gika, H.G.; Theodoridis, G.A.; Wingate, J.E.; Wilson, I.D. Within-day reproducibility of an HPLC-MS-based method for metabonomic analysis: Application to human urine. J. Proteome Res. 2007, 6, 3291–3303. [Google Scholar] [CrossRef]
- Soga, T.; Ohashi, Y.; Ueno, Y.; Naraoka, H.; Tomita, M.; Nishioka, T. Quantitative Metabolome Analysis Using Capillary Electrophoresis Mass Spectrometry. J. Proteome Res. 2003, 2, 488–494. [Google Scholar] [CrossRef]
- Bissonnette, L.; Bergeron, M.G. POC Tests in Microbial Diagnostics: Current Status. Methods in Microbiology, 1st ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; Volume 42. [Google Scholar]
- Tay, A.; Pavesi, A.; Yazdi, S.R.; Lim, C.T.; Warkiani, M.E. Advances in microfluidics in combating infectious diseases. Biotechnol. Adv. 2016, 34, 404–421. [Google Scholar] [CrossRef]
- Sharma, S.; Zapatero-Rodríguez, J.; Estrela, P.; O’Kennedy, R. Point-of-Care Diagnostics in Low Resource Settings: Present Status and Future Role of Microfluidics. Biosensors 2015, 5, 577–601. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Trujillo-de Santiago, G.; Alvarez, M.M.; Schiff, S.J.; Boyden, E.S.; Khademhosseini, A. Expansion mini-microscopy: An enabling alternative in point-of-care diagnostics. Curr. Opin. Biomed. Eng. 2017, 1, 45–53. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Gorthi, S.S. Automated blood sample preparation unit (ABSPU) for portable microfluidic flow cytometry. SLAS Technol. 2017, 22, 73–80. [Google Scholar] [CrossRef]
- Yager, P.; Edwards, T.; Fu, E.; Helton, K.; Nelson, K.; Tam, M.R.; Weigl, B.H. Microfluidic diagnostic technologies for global public health. Nature 2006, 442, 412–418. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique (Abbreviation) | Applications |
|---|---|
| Dielectrophoresis (DEP) | Separation of blood, infected blood cells, parasites |
| Deterministic lateral displacement (DLD) | Separation of blood, infected blood cells, parasites, CTC, spores, DNA, viruses |
| Deformability-based separation | Separation of blood, infected blood cells, parasites |
| Margination & Dean Flow | Separation of blood, infected blood cells, parasites, CTC |
| Surface acoustic waves (SAW) | Separation of blood, parasites, CTC, multicellular organism |
| Optical tweezers (OT) | Separation of rare cells, parasites, CTC |
| Optical density/refractive index | Fast optical analysis of cell size, shape and optic density |
| Droplet microfluidics | Separation and storage of cells, mixing with targeted start of reactions, post-lysis analysis of cell contents |
| Paper microfluidics | Separation of cells, post-lysis analysis of cell contents |
| PCR-based techniques | Targeted enrichment of subcellular fragments to verify their presence or identity of the previously separated cells |
| LAMP-based techniques | Similar to PCR, but generally also employable in environments with very limited resources |
| Prote-/metabol-/transcript-/poly omics | Gather data on all the parts of a cell, their transcriptomes or their metabolites respectively. Elucidating contents, pathways and interactions, (e.g., host–viral interactions of SARS-CoV-2). |
| Pros | Cons |
|---|---|
| Adaptable to different cells by frequency | Needs electricity |
| Potentially parallelizable | Low throughput, unless 3D- electrodes are used |
| Compatible with other techniques (e.g., DLD, SAW molecular analysis after cell lysis) | Cell frequencies need to be experimentally found |
| - | Separation is rather slow and hard to automate |
| Pros | Cons |
|---|---|
| Needs no electricity (e.g., using handheld syringes) | Prone to clogging |
| Tiny defects can be tolerated by redundancy | Trapped air bubbles can completely ruin separation |
| Can be parallelized | Low throughput due to tiny volume |
| Can be run constantly and separated cells can be collected for further processing in closed loop systems. | The separation process of DLD has not been fully understood so far and entails many different aspects that are thus far neglected. |
| Pros | Cons |
|---|---|
| Generally reusable setups | Defects in the matrix can undo separation |
| More resistant to clogging than DLD | Trapped air bubbles can trap deformable cells |
| Can theoretically be parallelized | Low throughput due to tiny volumes |
| Can run even without ratchets | Cannot be run constantly (due to oscillation), unless paired with other approaches e.g., DLD, immuno-magnetic, inertial, or electrokinetic sorting |
| Pros | Cons |
|---|---|
| Generally reusable setups | Comparably big footprint |
| Can theoretically be parallelized | Prone to clogging |
| Does not need high resolution lithography Can handle higher cell densities Bigger volumes can be handled | - |
| Can be run continuously Can be combined with other technologies (e.g., droplet MF) | - |
| Pros | Cons |
|---|---|
| Tunable to different cells by changing frequency | Needs electricity |
| Work also with bigger, multicellular organisms | Needs piezoelectric and other expensive material |
| Can be parallelized | Calculations of optimal settings for acoustic forces not yet fully elucidated |
| Can be run continuously | Small volumes |
| Can be combined with other techniques | Forces might not fully compete with motile cells |
| Pros | Cons |
|---|---|
| Contact-free micromanipulation | Needs electricity and lasers & optical setup |
| Works intracellularly | Relies on differences in refractive indices (e.g., DNA-rich parts like the nucleus) |
| Can be used to measure forces on the cellular level | Potential photodamage to sample and devices at wrong wavelengths and higher exposures |
| Can be combined with other techniques | Forces might not fully compete with motile cells |
| Strong local forces can be achieved An SLM-setup allows cell manipulation in 3D | However, only in “transparent” samples Hard and expensive to parallelize |
| Pros | Cons |
|---|---|
| High throughput and parallelization possible | Additional hydrophobic phase needed, plus either detergent (surfactant) or electrowetting |
| Reactions can be triggered and their process studied | Microfluidic devices are more complex and thus far cannot be brought outside a lab |
| Entire libraries of drug targets can be screened | - |
| Droplets can be stored in loops for long term studies | - |
| Can be combined with other techniques | - |
| Approach | Measurement Technique | Refractive Index Resolution | Spatial Resolution (nm) | Minimal Mass Density Change (g/mL)1 | Ref. |
|---|---|---|---|---|---|
| Bulk (Average refractive index of suspended cells) | Interference refractometer | NA | 0.0163 | [187] | |
| Light scattering | NA | 0.0542 | [188] | ||
| Light transmission and reflection | NA | 0.0542 | [187] | ||
| Optical densitometer | NA | 0.0163 | [189] | ||
| Single cell level | Fabry-Pérot resonant cavity | NA | 0.0163 | [182] | |
| Grating resonant cavity | NA | 0.0054 | [184] | ||
| Immersion refractometer | NA | 0.0054 | [190] | ||
| Laser resonant cavity | NA | 0.0217 | [180] | ||
| Light scattering | NA | 0.0542 | [140] | ||
| Mach-Zehnder Interferometer | NA | 0.0054 | [183] | ||
| (Sub-)cellular refractive index mapping | Common-path tomographic diffractive microscopy | NA | 0.0054 | [191] | |
| Confocal quantitative phase microscopy | NA | 0.0217 | [164] | ||
| Digital holographic microscopy | NA | 0.0016 | [163] | ||
| NA | 0.0542 | [192] | |||
| Hilbert phase microscopy | 1000 | 0.0108 | [193] | ||
| Microfluidic off-axis holography | 350 | 0.0217 | [194] | ||
| Phase-shifting interferometry | 250 | 0.0488 | [195] | ||
| NA | 0.0016 | [196] | |||
| Surface Plasmon nano-optical probe | 80 | 0.0002 | [197,198] | ||
| Tomographic bright-field imaging | 260 | 0.0434 | [199] |
| Pros | Cons |
|---|---|
| Very adaptable to target cells in general | Has to be combined with separation techniques |
| Generally can be used for high-throughout | Additional setups can be expensive, depending on the technique employed |
| Depending on the technique, the needed machinery might already be present for the setup | - |
| Generally damage-free to sample cells | - |
| Can be combined with other techniques | - |
| Pros | Cons |
|---|---|
| Can integrate separation and detection | No continuous phase or separation of cells |
| Can be used for complete diagnostic chips | Not usable for high throughput |
| Potentially cheap enough for disposable tests | Parallelization limited |
| Disposable tests can be combined with analytic cassettes to allow mass field testing with combined lab-based analysis | Slower than liquid–liquid microfluidics |
| Very mature technology with many applications | Mostly single use devices |
| Pros | Cons |
|---|---|
| Reliable | Needs electricity/highly controlled heat source |
| Can be combined for diagnostic chips | Needs computer hard and software for quantitative tests |
| Parallelizable and ready for high throughput | Sensitive to impurities and contamination |
| To some extent, test kits can be pre-treated with reactants to enable field-applicable testing | - |
| Can be combined with all above separation techniques, including paper microfluidics | - |
| Pros | Cons |
|---|---|
| Can give a complete picture of intracellular life | Needs electricity |
| Can be combined for diagnostic chips | Needs computer hard and software for qualitative and quantitative tests |
| Generally ready for high throughput | Sensitive to impurities and contamination |
| Can be used to find out changes (due to stimuli) in complex samples like tissues | Needs highly trained personnel |
| Can be combined with all above separation techniques, limited with paper microfluidics | Most commercial setups lack the flexibility to adapt the setup (i.e., change eluents) to improve separation of non-standard target molecules |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hochstetter, A. Lab-on-a-Chip Technologies for the Single Cell Level: Separation, Analysis, and Diagnostics. Micromachines 2020, 11, 468. https://doi.org/10.3390/mi11050468
Hochstetter A. Lab-on-a-Chip Technologies for the Single Cell Level: Separation, Analysis, and Diagnostics. Micromachines. 2020; 11(5):468. https://doi.org/10.3390/mi11050468
Chicago/Turabian StyleHochstetter, Axel. 2020. "Lab-on-a-Chip Technologies for the Single Cell Level: Separation, Analysis, and Diagnostics" Micromachines 11, no. 5: 468. https://doi.org/10.3390/mi11050468
APA StyleHochstetter, A. (2020). Lab-on-a-Chip Technologies for the Single Cell Level: Separation, Analysis, and Diagnostics. Micromachines, 11(5), 468. https://doi.org/10.3390/mi11050468